Article Text

Abstract
Objective Our goals were to evaluate the antitumour efficacy of Lactobacillus rhamnosus GG (LGG) in combination with immune checkpoint blockade (ICB) immunotherapies on tumour growth and to investigate the underlying mechanisms.
Design We used murine models of colorectal cancer and melanoma to evaluate whether oral administration of LGG improves the efficacy of ICB therapies. We performed the whole genome shotgun metagenome sequencing of intestinal contents and RNA sequencing of dendritic cells (DCs). In a series of in vitro and in vivo experiments, we further defined the immunological and molecular mechanisms of LGG-mediated antitumour immunity.
Results We demonstrate that oral administration of live LGG augmented the antitumour activity of anti-programmed cell death 1 (PD-1) immunotherapy by increasing tumour-infiltrating DCs and T cells. Moreover, the combination treatment shifted the gut microbial community towards enrichment in Lactobacillus murinus and Bacteroides uniformis, that are known to increase DC activation and CD8+tumour recruitment. Mechanistically, treatment with live LGG alone or in combination with anti-PD-1 antibody triggered type I interferon (IFN) production in DCs, enhancing the cross-priming of antitumour CD8+ T cells. In DCs, cyclic GMP-AMP synthase (cGAS)/stimulator of IFN genes (STING) was required for IFN-β induction in response to LGG, as evidenced by the significant decrease in IFN-β levels in cGAS or STING-deficient DCs. LGG induces IFN-β production via the cGAS/STING/TANK binding kinase 1/interferon regulatory factor 7 axis in DCs.
Conclusion Our findings have offered valuable insight into the molecular mechanisms of live LGG-mediated antitumour immunity and establish an empirical basis for developing oral administration of live LGG as a combination agent with ICB for cancer therapies.
- cancer immunobiology
- immunotherapy
- immunology
- gut immunology
- probiotics
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Lactobacillus rhamnosus GG (LGG) is one of the most well characterised and used probiotics.
LGG has been reported to increase inflammatory cytokine secretion levels by dendritic cells (DCs).
What are the new findings?
LGG synergised with immune checkpoint blockade (ICB) agents to improve the antitumour responses in murine cancer models.
Oral administration of live LGG augmented the antitumour activity of anti-programmed cell death 1 (PD-1) immunotherapy by increasing tumour-infiltrating DCs and T cells.
LGG triggered type I interferon (IFN) production in DCs, enhancing the cross-priming of antitumour CD8+ T cells.
Mechanistically, LGG induced IFN-β production via the cyclic GMP-AMP synthase/stimulator of IFN genes/TANK binding kinase 1/Interferon regulatory factor 7 axis in DCs.
Combination treatment with LGG and anti-PD-1 shifted the gut microbial community towards enrichment in Lactobacillus murinus and Bacteroides uniformis, that are known to increase DC activation and CD8+ tumour recruitment.
How might it impact on clinical practice in the foreseeable future?
These findings define the molecular mechanisms of live LGG-mediated antitumour immunity, which is beneficial to ongoing clinical trials.
These findings suggest that LGG can be used as a combination agent with ICB for cancer treatment.
Introduction
Cancer immunotherapies, such as use of immune checkpoint blockade (ICB), have led to remarkable advances in the treatment of a wide range of cancers, including many with historically poor responses to conventional therapy.1 Either alone or in combination, these strategies are centred on the blockade of immune inhibitory receptors, such as cytotoxic T-lymphocyte associated protein 4 (CTLA4), programmed cell death 1 (PD-1), programmed cell death-ligand 1 (PD-L1) and T cell immunoglobulin domain and mucin domain-3 (TIM-3).2–4 While most patients who respond to ICB agents maintain long-lasting disease control, one-third of patients relapse.5 It is anticipated that new-generation of combinatorial therapies will be required to improve the response to ICB therapies and efficiently treat a larger proportion of patients with cancer.6 7
Several studies have reported that oral administration of specific commensal bacteria in combination with anti-PD-1/PD-L1 antibodies can nearly abolish tumour growth.8 9 Lactobacillus rhamnosus GG (LGG) is one of the most well characterised and used probiotics.10 LGG, a part of the natural human commensal microflora, has exhibited possible benefits for the prevention of ulcerative colitis and moderation of diarrhoea.11 Importantly, LGG has also been suggested to modulate the inflammatory state during cancer development and transformation.12 13 For instance, administration of LGG in a hepatocellular carcinoma mouse model has been shown to impair tumour progression.14 Despite studies testing the usefulness of LGG in cancer treatment, it remains unknown whether LGG can synergize with ICB, such as anti-PD-1/PD-L1 monoclonal antibody to improve the antitumour responses.
Commensal probiotics interact with the host mucosal system, affecting systemic immunity.15 LGG has been reported to increase inflammatory cytokine secretion levels by dendritic cells (DCs), macrophages and monocytes, suggesting that their activation relies on increase of cytokine secretion.16 17 However, the molecular mechanisms for LGG’s antitumour immune activity are still unclear. Several other gram-positive bacteria were shown to induce type I interferon (IFN) through activation of the cyclic GMP-AMP synthase (cGAS)–stimulator of IFN genes (STING) axis.18 19 In this signalling cascade, cGAS acts as a secondary messenger to activate the endoplasmic reticulum-localised protein stimulator of IFN genes (STING); activated STING then proceeds to recruit TANK binding kinase 1 (TBK1) to phosphorylate the IFN regulatory factor, ultimately leading to type I IFN production. Through this manner, the cGAS-STING pathway plays a critical role in antitumour immunity by regulating type I IFN expression.20 Motivated by these emerging insights, we hypothesised that LGG might induce type I IFN production to promote innate immune responses against the tumour.
In this study, we evaluated the antitumour efficacy of LGG in combination with anti-PD-1, anti-PD-L1 or anti-TIM-3 immunotherapy on tumour growth using murine cancer models and investigated the underlying mechanisms. We found that combination with oral administration of live LGG markedly suppressed tumour growth. Mechanistically, administration of either live LGG or combination treatment with anti-PD-1 antibody increased the population and antitumour function of DCs, thereby promoting recruitment of activated CD8+ T cells to the tumour microenvironment. We also report a direct mechanism by which LGG induces type I IFN-β production through a cGAS/STING/TBK1/interferon regulatory factor 7 (IRF7)-dependent signal pathway.
Methods
In vivo animal studies
All mice were housed and used according to the animal experimental guidelines set by the Institute of Animal Care and Use Committee of The University of Chicago. All animals were maintained in pathogen-free conditions and cared for in accordance with the International Association for Assessment and Accreditation of Laboratory Animal Care policies and certification. Cgas −/−, Sting −/−, Sting f/f, Rag1 −/− and Ifnar −/− mice were purchased from Jackson Laboratory.
Tumour growth and treatments
MC38 cells (1×106) or B16F10 cells (2×105) were subcutaneously injected in the right flank of mice. Mice were pooled and randomly divided into different groups when the tumour reached a volume of approximately 100 mm3. IgG isotype control antibody, anti-PD-1 antibody, anit-PD-L1 antibody or anti-TIM-3 (200 µg per mouse) were injected intraperitoneally two times each week. LGG was orally administered twice 1 week. Tumours were measured two times a week for 3–4 weeks. Animals were euthanised when the tumour volume reached 2000 mm3. For CD8+ T cell depletion experiments, 200 µg of anti-CD8α antibody were delivered by intraperitoneal injection, 1 day before other treatments. For type I IFN blockade experiments, 200 µg of anti-IFNAR1 mAb were intraperitoneally injected 1 day before other treatments. For in vivo DC depletion, reduction of DCs was achieved by intraperitoneal injection of diphtheria toxin (DT) (50 ng) in CD11c-DTR transgenic mice, 24 hours prior to other treatments.
Statistical analysis
To estimate the statistical significance of differences between two groups, we used an unpaired Student’s t-tests to calculate two-tailed p values. A two-way analysis of variance (ANOVA) was performed when more than two groups were compared. Survival analysis was performed using Kaplan-Meier curves and evaluated with log-rank Mantel-Cox tests. Error bars indicate the SEM unless otherwise noted. P values are labelled in the figures. P values were denoted as follows: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Statistical analyses were performed by using GraphPad Prism (V.7.0).
Results
Oral administration of live LGG improves the response to anti-PD-1 in murine melanoma and colon cancer models
Oral supplementation of commensal bacteria has attracted attention recently for its potential to modulate the antitumour response following ICB immunotherapy.9 To evaluate whether oral administration of LGG actually improves the efficacy of ICB therapies, we treated MC38 tumour bearing mice with live LGG either alone or in combination with anti-PD-1 antibody. The LGG+anti-PD-1 (combination treatment) markedly inhibited tumour growth compared with either single agent alone, assessed by both tumour size and survival (figure 1A,B). To determine whether oral administration of live LGG enhances the response to other checkpoint blockade strategies, we also tested the combination of LGG with anti-PD-L1 antibody or anti-TIM-3 antibody. Mice treated with the combination of LGG and anti-PD-L1 or anti-TIM-3 displayed a comparable inhibition of tumour growth (online supplemental figure S1A,B). Due to its known primary resistance to ICB, the B16 melanoma tumour bearing mice were also evaluated with the combination treatment. The combination treatment significantly reduced tumour volume and prolonged mouse survival compared with any single treatment, although no such benefits were observed for mice treated with anti-PD-1 monotherapy (figure 1C,D). These results suggest that LGG can augment the antitumour responses following ICB.
Supplemental material

Oral administration of live Lactobacillus rhamnosus GG (LGG) improves the response to anti-programmed cell death 1 (PD-1) in melanoma and colon cancer mouse models. (A–F) C57BL/6 mice were subcutaneously inoculated with MC38 cells (A, B, E and F) or B16F10 cells (C and D). When tumour volumes reached approximately 100 mm3, tumours received respective treatments as indicated. (A) Tumour volume of MC38 tumour bearing-mice treated with IgG control antibody, anti-PD-1 antibody, oral administration of live LGG (2×109 CFU) or combined LGG and anti-PD-1 antibody (n=5). (B) Survival curves for each treatment group in (A); log-rank (Mantel-Cox) test survival comparison. (C) Tumour volumes of B16 tumour bearing-mice treated with IgG control antibody, anti-PD-1 antibody, oral administration of live LGG or combined LGG and anti-PD-1 antibody (n=4). (D) Survival curves for each treatment group as (C); log-rank (Mantel-Cox) test survival comparison (B and D). *p<0.05; ****p<0.0001. (E) Tumour volume of MC38 tumour bearing-mice treated with IgG control antibody, anti-PD-1 antibody, oral administration of heated-LGG or combined heated-LGG and anti-PD-1 antibody (n=5). (F) Tumour volumes of MC38 tumour bearing-mice treated with IgG control antibody, anti-PD-1 antibody, intratumoural of live LGG (2×107 CFU) or combined intratumoural of live LGG and anti-PD-1 antibody (n=5). Tumour volumes were measured and plotted individually. (A, C, E and F) Data are expressed as mean±SEM Tumour volume results were analysed by using two-way analysis of variance; *p<0.05; **p<0.01; ****p<0.0001.
It was noted that LGG monotherapy significantly suppressed the growth of both MC38 and B16 tumours (figure 1A,C, p=0.0283 and p=0.0235, respectively). These results are consistent with the published literature showing that LGG alone reduced the growth of liver tumours in mice.14 We thus examined whether LGG can directly affect tumour cell survival using a colony formation assay with MC38 cells in vitro. LGG had no direct effect on the proliferation of tumour cells (online supplemental figure S1C). Taken together, these results suggest that LGG directly induces antitumour immunity. Recent findings have demonstrated that inactivated LGG can influence immune regulation.21 However, gavaging the same dose of heat-killed LGG in this study failed to improve the response to anti-PD-1 antibody treatment in MC38 tumour bearing mice (figure 1E). These results reveal that oral delivery of live LGG, but not inactivated LGG, was required to facilitate the therapeutic effects of anti-PD-1 antibody.
It has been reported that several gut-derived microorganisms were detected and colonised inside human colorectal cancer22 and intratumoural injection of probiotics can enhance the antitumour immunity.23 Notably, intratumoural administration of live LGG in mice bearing MC38 tumours in this study was unable to control tumour growth and did not influence the response to anti-PD-1 immunotherapy (figure 1F). To further confirm whether LGG colonises in the tumour after systemic administration, we used a selective culture plate (de Man, Rogosa and Sharpe agar (MRS)) to detect LGG bacterial load in the MC38 tumour tissue after two doses of oral administration of live LGG (2.0×109 CFU/mouse). LGG were nearly undetectable in the tumour tissue (online supplemental figure S1D). These results indicate that oral administration of LGG was able to induce antitumour immunity and enhance the antitumour effects of ICB without localising in tumours.
Combination treatment with LGG and anti-PD-1 antibody modifies the composition of gut microbiota
Given that differential composition of gut microbiome has been reported to modulate response to cancer immunotherapy,9 24 we investigated whether the combination treatment modulates the gut microbiota. Whole genome shotgun metagenome sequencing was performed to define the microbiome profiles of MC38 tumour bearing mice. Unconstrained principal coordinate analysis of Bray-Curtis and Jaccard distance were carried out to assess the effect of LGG or anti-PD-1 treatment on microbial community assembly. We used permutational ANOVA (PERMANOVA), nested on cage to correct for possible contributions that may arise from co-housed mice in the same cage,25 26 to compare overall microbial community composition. Bray-Curtis distances indicated that the largest source of variation in the intestinal microbiota was proximity to LGG treatment in our experiments, while anti-PD-1 treatment displayed a smaller but still significant association with the structure of the microbial community (figure 2A). The intestinal microbiota of mice with anti-PD-1 treatment formed two clusters, which separated along the first coordinate axis indicating it was the LGG shifting the gut microbiota community (figure 2A and online supplemental figure S2A). We also observed that changes in microbiota after LGG or/and anti-PD-1 treatment were not influenced by cage effects. Next, we examined the taxonomic alpha diversity (Shannon, bacterial richness and Simpson index) within each sample. However, no changes were found among these groups (figure 2B and online supplemental figure S2B,C).

Combination treatment with Lactobacillus rhamnosus GG (LGG) and anti-programmedcell death 1 (PD-1) antibody modulates the composition of gut microbiota. (A) Unconstrained principal coordinate analysis (PCoA) (for principal coordinates PCo1 and PCo2) with Bray-Curtis distance, with permutational analysis of variance (PERMANOVA) p values nested on cage. The distribution of the first two most informative coordinates was performed in the corresponding marginal box plot. Red and blue dot represent without/with LGG treatment, respectively. Mixed linear model was tested for the first two coordinates of PCoA with Bray-Curtis distance (explaining 60.8% of the variance), evaluating the null hypothesis that the fixed effects LGG, anti-PD-1 and LGG×anti-PD-1 interactions had no effect on the coordinates. Significance was assessed using type III ANOVA with F tests for fixed effects. All p values were adjusted by using the Benjamini and Hochberg methods. *p<0.05; **p<0.01. (B) Shannon index of the intestinal microbiota from the indicated treatment groups. Statistics described the mixed linear model of Shannon index of the intestinal microbiota in LGG or/and anti-PD-1 treated mice, all comparisons and false discovery rate (FDR)-corrected p values were showed. Significance was assessed using type III ANOVA with F tests for fixed effects. All p values were adjusted by using the Benjamini and Hochberg methods. (C) Distribution of the gut microbiota at the phylum-level. The numbers of replicated samples in this figure are as follows: control (n=6), LGG (n=5), anti-PD-1 (n=7), anti-PD-1+LGG (n=7). (D) Relative abundance of Bacteroidetes (phylum level). Data bars represent means. Error bars represent SEM. FDR-corrected p values from the mixed linear model. *p<0.05. (E–G) Manhattan plot showing species enriched in either the combination treatment group, anti-PD-1 treatment (D) or LGG (E) or control (F) group. Each dot, triangle, quadrilateral or hexagonal star represents a single species. Species enriched in combination treatment group are represented by filled triangles; for anti-PD-1 treatment group: empty triangles; for LGG treatment group: hexagonal star; for IgG-control group: quadrilateral (FDR adjusted p<0.05 and log2fold change >1, Wilcoxon rank sum test). Species are arranged at the phylum level and coloured according to the phylum. CPM, counts per million.
The taxonomy profiles at the phylum level revealed that the gut microbiota community in the mice treated with LGG (either LGG monotherapy or the combination treatment) was dominated by Firmicutes and Bacteroidetes, whereas Firmicutes and Verrucomicrobia were the most abundant bacteria among the other treatment groups (figure 2C,D). Bacteroidetes and Firmicutes are typically enriched in the intestinal microbiota of healthy humans, whereas Verrucomicrobia is enriched in the gut of patients with pancreatic cancer.27 This consensus shift towards decreased Verrucomicrobia and increased Bacteroidetes in combination treatment group suggests the rebalanced gut microbiota to a healthier status. Hierarchical clustering based on the relative abundance of different orders showed that Bacteroidales, listed in top five most abundant orders in all four groups, was enriched in both LGG monotherapy and the combination treatment (online supplemental figure S2D). Moreover, it was significantly more abundant in the combination treatment relative to the anti-PD-1 monotherapy group (p<0.05) (online supplemental figure S2E). Thus, the combination treatment with LGG and anti-PD-1 antibody appear to influence the microbial community.
We also identified all the significantly enriched species according to their taxonomy using Manhattan plots. Compared with the control or anti-PD-1 monotherapy group, the enriched species in the combination treatment group belonged to a wide range of phyla, including Bacteroidetes, Actinobacteria, Proteobacteria, Cyanobacteria (false discovery rate (FDR) adjusted p<0.05, figure 2E,G). The combination treatment enriched the species belonged to Proteobacteria in contrast with the LGG monotherapy group (FDR adjusted p<0.05, figure 2F). To further confirm these, we also performed high-dimensional class comparisons using LefSe linear discriminant analysis to demonstrate the differentially abundant microbes. Among the enriched species, we noticed that Lactobacillus murinus and Bacteroides uniformis were enriched in the LGG treatment (online supplemental figure S2F). It has been reported that L. murinus is associated with the activation of murine intestinal DCs28 and B. uniformis is positively correlated with the frequency of IFN-γ+ CD8+ T cells in mesenteric lymph nodes (MLN) in mice, and therefore can trigger antitumour immunity.29 Collectively, LGG treatment enriches microbiota associated with antitumour immune activation.
Combination treatment with LGG and anti-PD-1 antibody promotes activation of cytotoxic CD8+ T cells
We next aimed to dissect how the combination treatment affects T cell responses and alters the tumour microenvironment. To determine whether antitumour effects of LGG depend on a T cell response, we administered LGG to Rag1 −/− mice bearing MC38 tumours. Notably, LGG either alone or in combination with anti-PD-1 antibody failed to slow tumour progression (figure 3A). We next examined whether the antitumour activity of live LGG or combination treatment requires CD8+ T cells by depleting CD8+ T cell using depletion antibody. The antitumour effect of LGG or combination treatment was abrogated in the absence of CD8+ T cells (figure 3B). These findings indicate that the therapeutic efficacy of combination treatment or LGG monotherapy depended on CD8+ T cells.
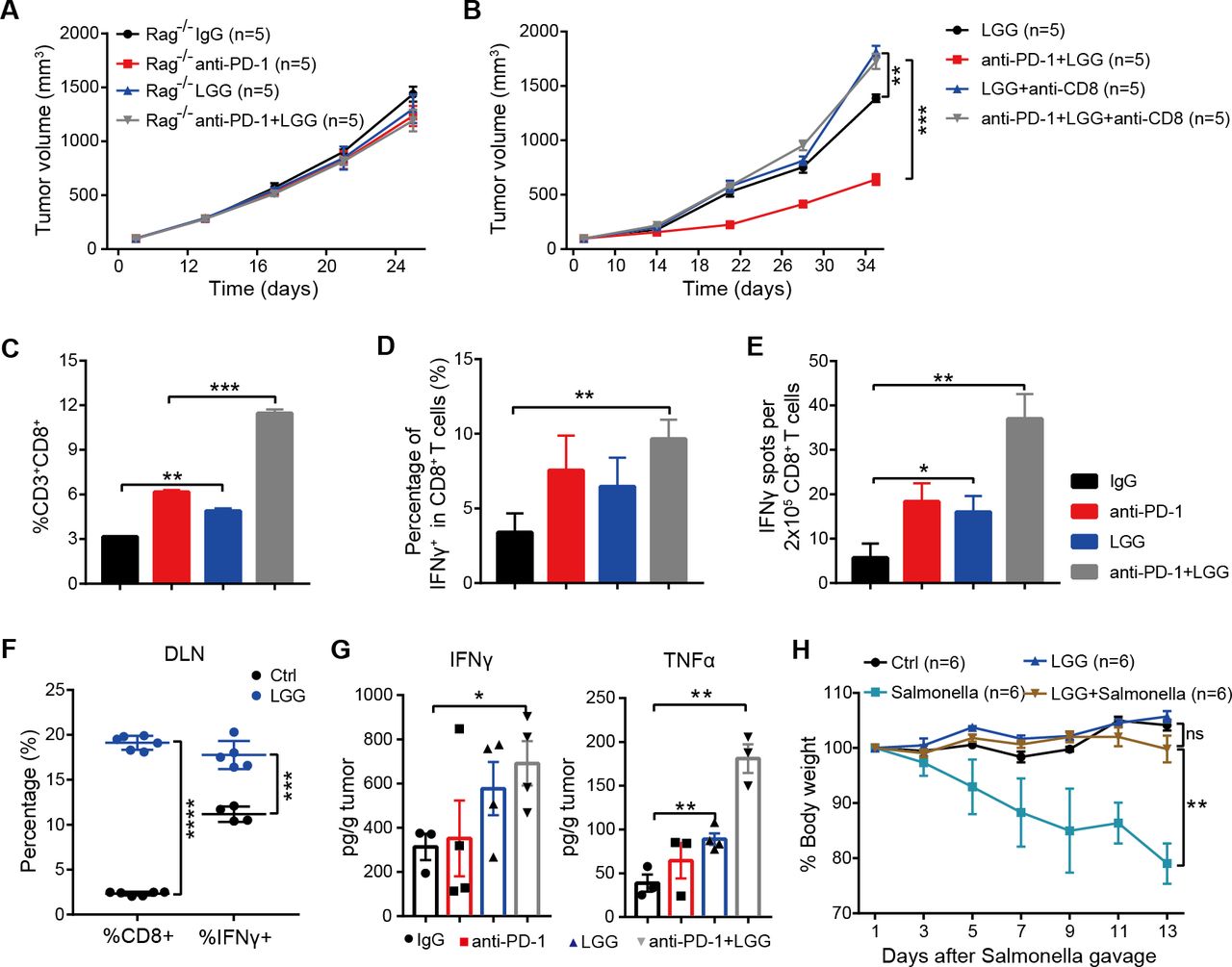
Combination treatment with Lactobacillus rhamnosus GG (LGG) and anti-programmedcell death 1 (PD-1) antibody promotes activation of cytotoxic CD8+ T cells. (A) Rag1 −/− mice were subcutaneously inoculated with MC38 cells and treated with either IgG control antibody, anti-PD-1 antibody, oral administration of LGG (2×109 CFU) or combination of LGG and anti-PD-1 antibody (n=5). (B) C57BL/6 mice were subcutaneously inoculated with MC38 cells and treated with the combination of LGG and anti-PD-1 antibody (n=5). Two hundred micrograms of anti-CD8 antibody was administered intraperitoneally (i.p.) 24 hours before other treatments. (A and B) Data are presented as mean±SEM. Tumour volume results were analysed by using two-way analysisof variance (ANOVA); **p<0.01; ***p<0.001. (C) Representative flow cytometry analysis and quantification of CD8+ T cells (CD45+CD3+CD8+) in MC38 tumours on day 28 with different treatments as indicated (n=3). (D) Representative flow cytometry analysis and quantification of cytolytic CD8+ T cells (CD45+CD3+CD8+IFN-γ+) in MC38 tumours on day 28 with the indicated treatment (n=3). (E) Interferon (IFN)-γ production in CD8+ T cells. MC38 tumours in wild type (WT) mice given the indicated treatment were removed on day 28. Tumours were digested to purify the CD8+ T cells. Purified CD8+ T cells were restimulated with 1 µg/ mL SIINFEKEL for 48 hours and then analysed by IFN-γ ELISPOT assay. (F) Representative flow cytometry analysis and quantification of CD8+ T cells (CD45+CD3+CD8+) and cytolytic CD8+ T cells (CD45+CD3+CD8+IFN-γ+) in tumour draining lymph nodes of the mice treated with LGG (n=4–6). (G) The levels of IFN-γ and tumour necrosis factor (TNF)-α in MC38 tumours after different treatments. MC38 tumours in WT mice given the indicated treatment were removed on day 28. The levels of IFN-γ and TNF-α in tumours were measured using the LEGENDplex cytokine kit. (C, D, E, F and G) Data are means±SEM; unpaired two-tailed Student’s t-tests; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. (H) Body weight change after Salmonella challenge. The WT mice were orally administered LGG (2×109 CFU) prior to oral infection with Salmonella (5×106 CFU). The body weight of mice was measured every 2 days. The percent weight change over the course of Salmonella infection is shown. Data are expressed as mean±SEM and were analysed by using two-way ANOVA; **p<0.01; ns p>0.05. DLN, draining lymph nodes.
To further determine the underlying immunological mechanism of LGG-mediated T cell immunity, we measured the abundance of CD8+ T cell populations by flow cytometry (fluorescence-activated cell sorting (FACS)). Compared with anti-PD-1 monotherapy, the combination treatment significantly increased the numbers of tumour infiltrating CD8+ T cells (figure 3C, p=0.0006). Moreover, LGG alone also markedly increased the CD8+ T cells compared with the isotype control antibody treatment (figure 3C, p=0.0027). We then examined the effector function of CD8+ T cells by intracellular IFN-γ staining and observed an increased percentage of IFNγ+CD8+ T cells in MC38 tumours treated with the combination treatment (figure 3D). ELISPOT assays for the IFN-γ secreting capacity of CD8+ T cells derived from the draining lymph nodes (DLN) of MC38 tumour-bearing mice showed that both LGG monotherapy and the combination treatment significantly increased IFN-γ production (figure 3E, p=0.0207 and p=0.0011, respectively). Consistently, the percentages of CD8+ and IFN-γ+ CD8+ T cells in DLN were also increased after LGG treatment (figure 3F). Similarly, cytokine analysis of MC38 tumours revealed increased levels of IFN-γ and tumour necrosis factor (TNF)-α (figure 3G), consistent with an enhanced cytotoxic function of CD8+ T cells.30 31 We also observed increased levels of CCL4 and CCL5, mainly produced by effector CD8+ T cells,32 in LGG or LGG+anti-PD-1 treated tumours (online supplemental figure S3A,B).
Because IFN-γ+ CD8+ T cells are known to promote clearance of intracellular pathogens,33 34 we sought to examine whether high systemic levels of IFN-γ+ CD8+ T cells, induced by oral administration of LGG, could augment host protective immunity against infection of Salmonella typhimurium, which can colonise the gut, invade intestinal tissues and cause enterocolitis.35 Feeding mice with live LGG prior to oral administration of S. typhimurium enhanced S. typhimurium clearance, as evidenced by markedly reduced weight and intestinal tract length losses (figure 3H and online supplemental figure S3C). Collectively, these findings indicate that oral administration of live LGG activates cytotoxic function of CD8+ T cells systemically.
Given that Th1 cells (CD4+ IFN-γ+) play a key role in activating CD8+ T cells,36 we also detected the tumour infiltrating CD4+ T cells in MC38 tumours. FACS results revealed a significant increase in the infiltration of Th1 cells in mice treated with either LGG or combination treatment (figure S3D). In order to further confirm the activation of effector CD4+ T cells, we measured the Th1-associated cytokines in MC38 tumours and observed an increase in levels of interleukin (IL)-6 and IFN-γ (online supplemental figure S3E and figure 3G). To determine whether LGG directly stimulates CD8+ T cells in vitro, we cocultured purified CD8+ T cells from OT-I mice with LGG. The IFN-γ production in CD8+ T cells treated with LGG is similar to that without treatment (online supplemental figure S3F, p>0.05), suggesting that LGG alone was insufficient to stimulate CD8+ T cells. We therefore hypothesised that the activation of T cells by LGG requires antigen cross-presentation via professional antigen presenting cells (APCs).
LGG increases tumour infiltrating DCs and activates the type I IFN signaling
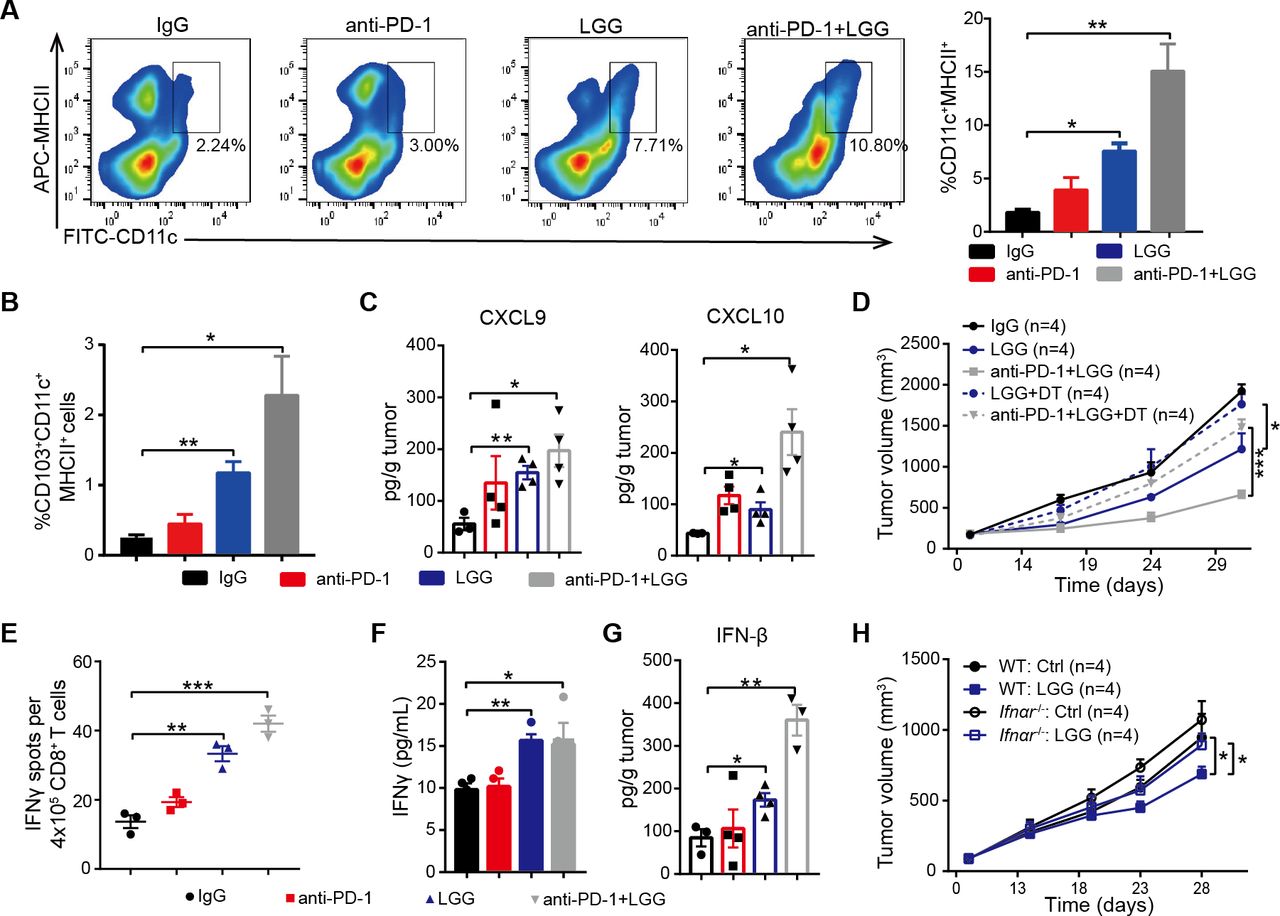
Considering DCs are potent APCs, we isolated the DCs from MC38 tumours treated with LGG or combination treatment and analysed by FACS. Both the combination treatment and LGG monotherapy significantly expanded the population of tumour infiltrating CD11c+ cells (figure S4A). Next, we analysed the population of DCs (CD11c+MHC II+) and observed a significant increase in mice treated with LGG or combination treatment (figure 4A). These findings are consistent with our hypothesis that LGG improves the antigen cross-presentation function of DCs. Elevated CCL4 is also known to mediate CD103+ DC tumour trafficking,37 we observed markedly increased CD103+ DCs in MC38 tumours on LGG or combination treatment (figure 4B). We also consistently observed increased levels of CXCL9 and CXCL10 (figure 4C), two chemokines produced by DCs that strongly mediate T cell tumour infiltration.38 To further investigate whether the LGG-induced antitumour immunity specifically relies on DCs, we inoculated MC38 cells into CD11c-DTR mice. Notably, treatment with DT significantly abrogated the antitumour response of both LGG and combination treatment (figure 4D), suggesting that the antitumour activity is dependent on DCs.

Lactobacillus rhamnosus GG (LGG) increases tumour infiltrating dendritic cells and activates type I interferon (IFN) signalling. (A) Representative flow cytometry analysis and quantification of dendritic cells (DCs) (CD45+CD11c+MHC II+) in MC38 tumours on day 28 given the indicated treatment (n=3). (B) Representative flow cytometry analysis and quantification of CD103+ DCs (CD45+CD11c+ MHC II+ CD103+) in MC38 tumours on day 28 given the indicated treatment (n=3). (C) The levels of CXCL9 and CXCL10 in MC38 tumours. MC38 tumours in wild type (WT) mice given the indicated treatment were removed on day 28. The levels of CXCL9 and CXCL10 in tumours were measured using a LEGENDplex chemokine kit. (D) CD11c-DTR were subcutaneously inoculated with MC38 cells and treated with IgG control antibody, oral administration of LGG (2×109 CFU) or a combination of LGG and anti-programmedcell death 1 (PD-1) antibody (n=4). Diphtheria toxin was administered 1 day before LGG or anti-PD-1 treatment (+DT). (E) IFN-γ production in CD8+ T cells after coculturing with DCs. MC38-OVA tumours in WT mice were given the indicated treatment. Tumour draining lymph nodes were removed and digested to purify the CD11c+ cells. The CD11c+ cells were cocultured with CD8+ T cells from OT-I mice for 3 days and the cross-priming activity of DCs was analysed by IFN-γ ELISPOT assay. (F) IFN-γ levels in the cell supernatant from DCs and CD8+ T cell coculture as described in (E). (G) IFN-β levels in MC38 tumours in WT mice. MC38 tumours in WT mice given the indicated treatment were removed on day 28. The level of IFN-β in tumours were measured using LEGENDplex cytokines kit. (A, B, C, E, F and G) Data are means±SEM; unpaired two-tailed Student’s t-tests; *p<0.05; **p<0.01; ***p<0.001. (H) WT or Ifnαr −/− mice were inoculated with 1×106 MC38 cells. Mice (n=4) were treated with oral administration of LGG (2×109 CFU). Oral administration of phosphate-buffered saline (PBS) served as the negative control. Tumour growth was measured twice per week. (D and H) Data are expressed as mean±SEM, tumour volume results were analysed by using two-way analysisof variance; *p<0.05; ***p<0.001; ns p>0.05.
To determine whether LGG enhances the cross presentation of tumour antigens on DCs, we performed cross-priming assay by an IFN-γ ELISPOT assay with purified CD11c+ cells from the DLN of MC38-OVA tumour-bearing mice. DCs sorted from the mice treated with either LGG or combination treatment showed increased priming functions as assessed in terms of both IFN-γ spots and IFN-γ production (figure 4E,F). DCs in MLN were purified and their antigen-specific cross-priming ability was examined. We observed an increased abundance of IFN-γ+ T cells in wells containing DCs with LGG treatment (online supplemental figure S4B). In addition, LGG treatment significantly increased the numbers of CD11c+ cells in MLN (online supplemental figure S4C, p<0.0001), suggesting that oral administration of live LGG might activate DCs in MLN. Taken together, these results indicate that LGG can systemically enhance the antigen processing and presenting capacities of DCs.
In order to further confirm that the combination treatment activated DCs, we performed RNA sequencing (RNA-seq) of CD11c+ cells purified from MC38 tumour bearing mice treated with either anti-PD-1 antibody or LGG plus anti-PD-1 antibody. Combination treatment increased the expression of IFN-related and cytokine genes (online supplemental figure S4D). Gene ontology (GO) analysis revealed stimulation of the T cell activation pathway, positive regulation of cytokine production pathway and activation of the IFN induction related pathway (online supplemental figure S4D), consistent with the aforementioned findings. We also found similarly activated immune response-related pathways in DCs from mice treated with LGG compared with the control IgG antibody.
Given that bacteria are able to trigger local and systemic antitumour immunity via type I IFNs,39 we tested whether LGG modulates the antitumour function through regulating type I IFN expression. We measured IFN-β concentrations in MC38 tumours and observed a greater than threefold increase in IFN-β following combination treatment compared with anti-PD-1 alone (figure 4G). We found that expression of CXCL10, a type I IFN-stimulated chemokine, was also increased (figure 4C). To further assess the potential role of type I IFN in LGG-mediated antitumour immunity, MC38 tumours were inoculated into wild type (WT) or Ifnαr −/− mice and treated with LGG. The antitumour effects disappeared in Ifnαr −/− mice (figure 4H). We also treated MC38 tumours in WT mice with an anti-IFN alpha receptor (IFNAR) blocking antibody and found that treatment with anti-IFNAR significantly impaired the antitumour activity of LGG (online supplemental figure S4E, p=0.0427). Thus, these findings demonstrate that LGG mediated activation of DCs and type I IFN signalling is required for antitumour efficacy of LGG treatment.
LGG induces type I IFN response in DCs via an MyD88-independent manner
It has been reported that the adaptor protein, myeloid differentiation primary-response protein 88 (MyD88), plays a vital role in the induction of type I IFN production.40 To investigate whether MyD88 was required to mediate the response to LGG therapy, we implanted MC38 tumour cells on flanks of Myd88-deficient mice. Surprisingly, absence of host MyD88 did not affect the antitumour activity of LGG or combination treatment (figure 5A). To further verify this, we performed a FACS experiment to analyse the abundance of both infiltrated T cells and DCs in MC38 tumour bearing-Myd88 −/− mice. CD8+ T cells and CD4+ Th1 cells were significantly increased not only in the combination treated MC38 tumours, but also in LGG treated MC38 tumours (online supplemental figure S5A,B). Populations of DCs and CD103+ DCs were also increased (figure 5B). RNA-seq analysis of CD11c+ cells isolated from MC38 tumour-bearing Myd88 −/− mice given combination treatment exhibited increased the expression of genes associated with T cell activation, cytokine-related and response to IFN pathways, similar to those of WT mice (figure 5C). These findings demonstrate that host MyD88 is not essential for LGG-induced DCs and T cells activation.

Lactobacillus rhamnosus GG (LGG) induces a type I interferon (IFN) response via a non-MyD88 dependent manner in dendriticcells (DCs). (A)Myd88 −/− mice were subcutaneously inoculated with MC38 cells and treated with IgG control antibody, anti-PD-1 antibody, oral administration of live LGG (2×109 CFU) or combination of LGG and anti-programmedcell death 1 (PD-1) antibody (n=5). (B) Representative flow cytometry analysis and quantification of DCs and CD103+ DCs in MC38 tumours from Myd88 −/− mice on day 21 given the indicated treatment as described in (A) (n=3). (C) Heatmap illustrating the relative expression of genes and the partitioning of the clusters of genes (gene ontology-GO analysis) based on the RNA-seq analysis of CD11c+ cells in mesenteric lymph nodes (MLN) of MC38 tumour-bearing Myd88 −/− mice given anti-PD-1 antibody or combination treatment with anti-PD-1 antibody and LGG. (D) MC38 tumours in Myd88 −/− mice given the indicated treatment were removed on day 21. The level of IFN-β in tumours was measured using a LEGENDplex cytokine kit. (E)Myd88 −/− mice were subcutaneously inoculated with MC38 cells and treated with IgG control antibody, anti-PD-1 antibody, oral administration of live LGG (2×109 CFU) or a combination of LGG and anti-PD-1 antibody (n=5). Two hundred micrograms anti-IFNAR1 antibody was administered intraperitoneally (i.p.) 24 hours before other treatments. (A and E) Data are expressed as mean±SEM, tumour volume results were analysed by using two-way analysisof variance; *p<0.05; **p<0.01; ***p<0.001. (F) IFN-γ levels in CD8+ T cells from Myd88 −/− mice. The Myd88 −/− mice were gavaged with LGG two times for 1 week. CD11c+ cells were purified from the MLN in and then cocultured with CD8+ T cells and the cross-priming activity of DCs was analysed by IFN-γ ELISPOT assay. (B, D and F) Data are means±SEM; unpaired two-tailed Student’s t-tests; *p<0.05; **p<0.01.
In order to identify whether LGG modulates the antitumour functions of DCs and CD8+ T cells by regulating type I IFN expression in Myd88 −/− mice, we measured the concentration of IFN-β protein in MC38 tumours. The absence of MyD88 did not affect IFN-β production, and IFN-β was significantly increased in tumours treated with either LGG or in combination (figure 5D). We next examined whether type I IFNs were required for the increased antitumour effect, we blocked type I IFN signalling following LGG therapy using anti-IFNAR. As observed previously in WT mice, the antitumour effect of LGG in Myd88 −/− mice was abrogated by administration of IFNAR antibody (figure 5E). We also evaluated the cross-presentation ability of Myd88 −/− DCs in MLN by IFN-γ ELISPOT. Myd88 −/− mice were gavaged with live LGG twice weekly. We observed a significant increase in IFN-γ spot-forming cells in wells containing CD11c+ cells isolated from LGG-treated mice (figure 5F). Collectively, these findings indicate that LGG-mediated enhancement of DC cross-presentation is not dependent on MyD88 signalling.
LGG induces IFN-β production via the cGAS/STING/TBK1/IRF7 axis in DCs
Since LGG induced production of type I IFNs in an MyD88-independent manner, it was conceivable that LGG might trigger IFN-β induction through a cGAS/STING-dependent pathway. Thus, we reanalysed the RNA-seq results of DCs sorted from MLN of both WT and Myd88 −/− mice. Gene set enrichment analysis identified that the gene signature ‘Response to Interferon Beta’ was strongly skewed toward either the LGG treatment (figure 6A,B) or the combination treatment (online supplemental figure S6A,B). We subsequently analysed the expression of type I IFN-related genes and found that both Irf7 and Ifnb1 were each markedly upregulated (online supplemental figure S6C,D). We also observed that the cytosolic DNA-sensing pathway was activated. To confirm this, we treated bone marrow-derived DCs (BMDCs) with genomic DNA of LGG and noted that LGG-DNA also significantly increased IFN-β production compared with the control (figure 6C). Collectively, these results suggest that LGG may induce IFN-β production in a cGAS/STING-dependent manner.

Lactobacillus rhamnosus GG (LGG) induces interferon (IFN)-β production via cyclic GMP-AMP synthase (cGAS)/stimulator of IFN genes (STING)/TANK binding kinase 1 (TBK1)/interferon regulatory factor 7 (IRF7) axis in dendriticcells (DCs). (A–B) Gene set enrichment analysis plots of enplot-response to IFN-β were positively correlated with LGG treatment based on the RNA-seq data of CD11c+ cells derived from mesenteric lymph nodes (MLN) in wild type (WT) (A) and Myd88 −/− (B) mice. (C) Bone marrow-derived DCs (BMDCs) were derived from WT mice and then treated with LGG or LGG chromosomal DNA (50 µg/mL) overnight. The supernatants were collected to measure IFN-β by ELISA assay. (D) The mRNA levels of Ifnβ in BMDCs from WT, Sting−/− or Cgas−/− mice. BMDCs were derived from WT, Sting−/− or Cgas−/− mice and then treated with LGG or LGG chromosomal DNA (50 µg/mL) overnight. BMDCs were collected and RT-qPCR was performed to measure the mRNA levels of Ifnβ. (E) BMDCs were derived from WT, Sting−/− or Cgas−/− mice and then treated with LGG or LGG chromosomal DNA (50 µg/mL) overnight. The supernatants were collected to measure IFN-β by ELISA assay. (F) Western blot analysis of phosphorylated p-TBK1, total TBK1, p-IRF3, total IRF3, p-IRF7 and total IRF7 in WT and Cgas −/− BMDCs. (G–H) Ifnβ mRNA level in BMDCs and IFN-β levels in supernatant after siRNA treatments. BMDCs were treated with siRNAs targeting Tbk1, Irf3 and Irf7, respectively. Twenty-four hours later, cells were harvested and cocultured with live LGG. After overnight incubation, the cells were collected for qPCR analysis of Ifnβ mRNA level (G) and the supernatant was collected for IFN-β ELISA (H). (C–E and G–H) Data are expressed as means±SEM; unpaired two-tailed Student’s t-tests; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
To confirm that type I IFN production pathway activated in DCs by LGG is cGAS/STING dependent, we conducted a series of qPCR experiments with BMDCs derived from WT, Sting−/− (Tmem173−/− ) and Cgas−/− mice. Consistently, treatment of either LGG or LGG-DNA strongly upregulated Ifnb mRNA expression in WT BMDCs, while STING-deficient or cGAS-deficient BMDCs had great reduction of Ifnb expression (figure 6D). It is worth noting that Ifnb mRNA expression was close to be undetectable in cGAS-deficient BMDCs. Furthermore, we examined the expression of an array of immune activation-related genes in these BMDCs. As expected, expression of Tnfα, Il12, Cxcl9, Cxcl10, Cxcl11 and Cx3cr1 were all upregulated in WT BMDCs following LGG or LGG-DNA treatment (online supplemental figure S7A). Interestingly, cGAS deficiency drastically reduced the mRNA levels of these genes. Similar results were also observed in Sting−/− BMDCs, with the exception of Tnfα and Cxcl9 (online supplemental figure S7A). Others have previously demonstrated that Cgas mRNA is increased in activated DCs during bacterial infection.41 Consistently, we observed upregulation of Cgas mRNA in both WT and Sting−/− BMDCs on treatment with LGG (online supplemental figure S7B). These results support the notion that cGAS is essential for type I IFN pathway activation in DCs.
Next, to investigate whether the downstream signalling of cGAS/STING is activated in BMDCs with LGG treatment, we first detected the IFN-β production using ELISA assay. Both cGAS and STING deficiency significantly reduced the induction of IFN-β on LGG or LGG-DNA treatment (figure 6E). Of note, LGG-DNA cannot stimulate IFN-β production in Cgas−/− BMDCs (figure 6E). To validate the cGAS/STING signalling is responsible for the IFN-β induction on LGG treatment, we collected WT and Cgas−/− BMDCs stimulated with LGG to detect TBK1 phosphorylation (p-TBK1), IRF3 phosphorylation (p-IRF3) and IRF7 phosphorylation (p-IRF7). The protein levels of p-TBK1 and p-IRF7 were mildly increased in WT BMDCs on LGG stimulation, while the level of p-IRF3 stayed unchanged (figure 6F). In comparison, the increase of p-TBK1 or p-IRF7 was compromised when cGAS was deficient (figure 6F and online supplemental figure S7C). These findings indicate that TBK1 and IRF7 are two critical components of the type I IFN pathways activated by LGG.
To further confirm the role of IRF3 and IRF7 in IFN-β production pathway induced by LGG, we knocked down TBK1, IRF3 or IRF7 in WT BMDCs using the respective siRNA (online supplemental figure S7D). We then treated these BMDCs with LGG and detected the IFN-β expression by qPCR and ELISA. On LGG treatment, either TBK1 knockdown or IRF7 knockdown significantly decreased IFN-β expression not only at the mRNA level but also at the protein level (figure 6G,H). Similar results, but to a lesser extent, were observed in IRF3 knockdown BMDCs (figure 6G,H). The IFN-β protein level in IRF3 knockdown BMDCs was much higher than that in IRF7 knockdown BMDCs (figure 6H). Thus, even though we cannot definitively exclude the possibility that IRF3 contributes to LGG-induced IFN-β production, the data suggest that IRF7 play a major role in cGAS/STING/TBK1 pathway activation during LGG treatment.
cGAS/STING is required to mediate the antitumour effects of LGG and combination treatment with anti-PD-1 antibody
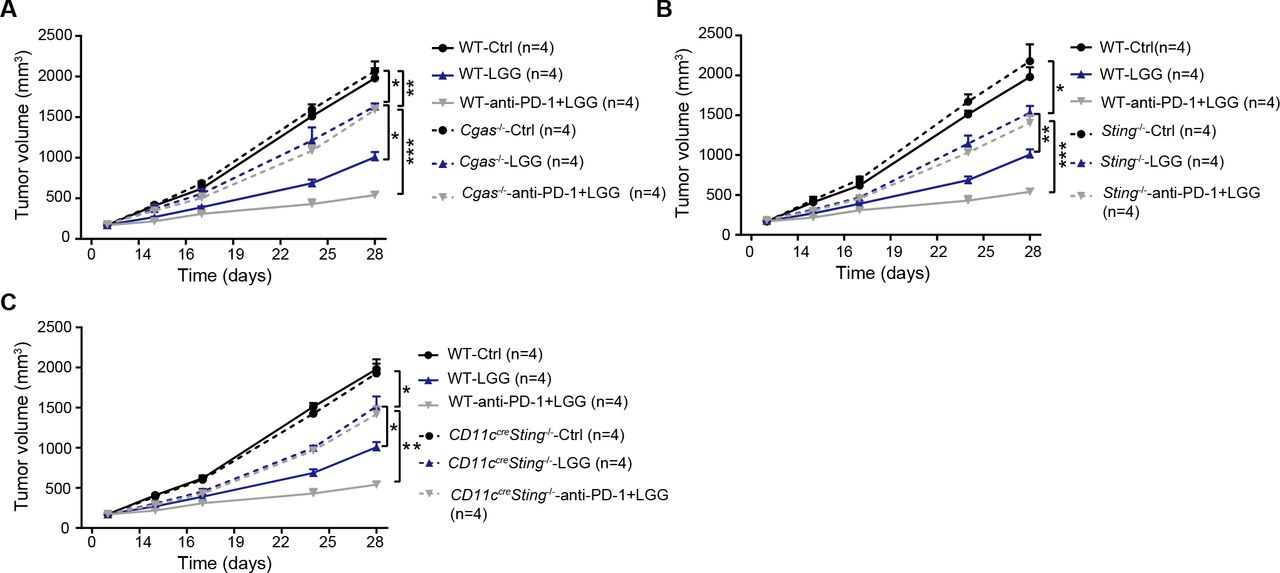
Having identified that live LGG was able to induce cGAS/STING-dependent IFN-β production in vitro, we next inquired whether cGAS/STING signalling is involved in the LGG-induced antitumour effect using a murine colon cancer model. MC38 cells were subcutaneously inoculated in WT and Cgas −/− mice and treated with LGG or the combination treatment with LGG and anti-PD-1 antibody. WT and Cgas −/− mice exhibited comparable tumour growth (figure 7A). Deficiency of cGAS significantly attenuated tumour volume reductions induced by LGG or the combination treatment (figure 7A). Similar results were also observed in Sting −/− mice (figure 7B). To evaluate the role of DC signalling, we employed the Cd11c cre Sting f/f mice and monitored tumour growth after LGG treatment. In Cd11c cre Sting f/f mouse, Sting undergoes deletion specifically in CD11c+ cells such as DCs. MC38 tumours grew more rapidly in Cd11c cre Sting f/f mice than control mice treated with LGG or the combination treatment (figure 7C). These results demonstrate that both cGAS and STING in DCs play an important role in the antitumour activity of live LGG and the combination treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
cGAS/STING is required to mediate the antitumour effects of Lactobacillus rhamnosus GG (LGG) and combination treatment with anti-programmedcell death 1 (PD-1) antibody. (A) Cgas −/− and wild type (WT) mice were subcutaneously inoculated with MC38 cells and treated with IgG control antibody, oral administration of LGG (2×109 CFU) or a combination of LGG and anti-PD-1 antibody. (B) Sting −/− and WT mice were subcutaneously inoculated with MC38 cells and treated with IgG control antibody, oral administration of LGG (2×109 CFU) or a combination of LGG and anti-PD-1 antibody (n=4). (C) Cd11ccreSting f/f and WT mice were subcutaneously inoculated with MC38 cells and treated with IgG control antibody, oral administration of LGG (2×109 CFU) or a combination of LGG and anti-PD-1 antibody (n=4). (A–C) Data are expressed as mean±SEM. Tumour volume results were analysed by using two-way analysisof variance; *p<0.05; **p<0.01; ***p<0.001.
Discussion
Our study establishes an empirical basis for developing oral administration of live LGG probiotics in combination with ICB for cancer treatment. We demonstrate that cGAS/STING axis is required for the antitumour effect of LGG and the induction of type I IFN. The cGAS-STING-TBK1-IRF7-IFN-β cascade mediates a robust adaptive immune response to LGG in DCs. Furthermore, LGG treatment is associated with gut microbiota that likely contribute to the antitumour efficacy (graphical abstract). Our findings offer valuable insight into the molecular mechanism of LGG-mediated antitumour immunity and highlight the potential to improve ICB immunotherapy through combination with LGG oral administration.
Supplemental material
We provided evidence that oral administration of LGG increases the population and the function of DCs in MLN and raises the possibility of recruiting activated DCs from the gut to tumour sites. Our observations are similar to the findings that administration of a toll-like receptor (TLR) agonist strongly activates MLN DCs and stimulates high levels of cytokines.42 It is well known that intestinal DCs are in direct contact with harmless and pathogenic luminal contents.43 It is possible that LGG is phagocytosed by a migratory DC population once it reaches the lamina propria. This DC population then processes LGG and traffics to the MLN, where it activates naïve T cells. Both the activated DCs and T cells migrate to the DLN and the tumour sites. Consistent with this supposition, we have found large amounts of tumour infiltrated CD103+DCs and CD8+ T cells expressing IFN-γ in mice treated with either LGG or the combination treatment. These findings provide new insights into the regulation of immune cell responses in distant tumour sites through the crosstalk between the gut and tumour.
Based on our data, it was noted that LGG treatment eliminated the population of CD11c−MHC II+ cells in MC38 tumours. It was reported that myeloid-derived suppressor cells have high MHC II levels especially in tumours44 and can profoundly diminish CD8+ T cell activity. Moreover, it has been reported that high MHC II expression in monocytes was positive correlated with B16 tumour growth.45 Hence, we speculated that the antitumour effects of LGG might attribute to the decrease of CD11c−MHC II+. Further studies are needed to establish which subtype of the CD11c−MHC II+ suppressor-like cells could be inhibited by LGG. Furthermore, STING-mediated type I IFNs has been proved to induce natural killer (NK) cells activation and thus contribute to eliminating tumours, especially in B16 melanoma.46 47 We cannot rule out the important role of NK cells activation in controlling tumours. Considering LGG indeed mediated a STING-dependent type I IFN activation in DCs, it is reasonable to postulate that LGG could enhance NKs in tumours and improve the response to immunotherapy. Additional investigation will be needed to resolve this issue.
LGG can modulate the inflammatory immune response during cancer development and transformation, reducing acute and chronic diarrhoea associated with chemotherapy and radiotherapy in patients with cancer.48 It has also been reported to induce adaptive immune responses to support tumour regression.14 The induction of type I IFNs by bacteria is essential for the function of CD8+ T cells. We identify that LGG induces IFN-β production in DCs via the cGAS/STING/TBK1/IRF7 axis, with the limitation that we only determined the pathway in GM-CSF or FLT3L derived BMDCs, which may represent immature DCs or cDC2 and this needs to be verified using primary DCs in the future. To the best of our knowledge, this is the first report on the involvement of the cGAS/STING/TBK1/IRF7 axis in LGG-mediated type I IFN induction in cancer treatment. In this report, we are not able to definitively exclude the possibility that other mediators, which also trigger type I IFN induction, may contribute to the efficacy of oral administration of live LGG. For example, TIR-domain-containing adaptor protein inducing IFN-β or cathelicidin-related antimicrobial peptide (in mice and LL37 in human) may also be involved in induction of type I IFN production.49 50 To discern the LGG-mediated mechanism and further develop potential therapeutics targeting this pathway, it is essential to understand which mediator is required for type I IFN induction by LGG.
Furthermore, we provide evidence that combination treatment with LGG and anti-PD-1 antibody can modulate the gut microbiota community, probably be beneficial for promoting immune cell activation. We identify a potential signature of two specific enriched species (L. murinus and B. uniformis) present LGG treatment. It would be interesting to explore their potentials as predictive markers for ICB immunotherapy responses in humans. Regarding the mechanism for activating immune responses, L. murinus can enhance IL-12 expression and decrease OX40 expression in DCs and therefore promote activation.28 Upcoming studies will aim at exploring the molecular mechanisms by which these species influence innate and adaptive immunity.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors WS and LW designed research studies. WS, LW, QX and XD conducted experiments, and analysed data. WS, LW, XZ and HL wrote the manuscript. RRW, JB, KY, QX and YF edited the manuscript. LW, XZ and RRW supervised the project.
Funding This work was supported by the Chicago Tumour Institute, an endowment from the Ludwig Cancer Research Foundation (to RRW). This work was also partially supported by a discovery grant from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016–04715, to XZ). JB was supported by a Clinical Therapeutics Training Grant (T32GM007019).
Competing interests RRW has stock and other ownership interests with Boost Therapeutics, Immvira, Reflexion Pharmaceuticals, Coordination Pharmaceuticals, Magi Therapeutics, Oncosenescence. He has served in a consulting or advisory role for Aettis, Astrazeneca, Coordination Pharmaceuticals, Genus, Merck Serono S.A., Nano proteagen, NKMax America, Shuttle Pharmaceuticals. He has a patent pending entitled 'Methods and Kits for Diagnosis and Triage of Patients With Colorectal Liver Metastases' (PCT/US2019/028071). He has received research grant funding from Varian and Regeneron. He has received compensation including travel, accommodations or expense reimbursement from Astrazeneca, Boehringer Ingelheim and Merck Serono S.A.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.