Article Text

Abstract
Objective Spontaneous bacterial peritonitis (SBP) is a life-threatening complication of liver cirrhosis with a 1-year mortality of 66%. Bacterial translocation (BT) from the intestine to the mesenteric lymph nodes is crucial for the pathogenesis of SBP.
Design Since BT presupposes a leaky intestinal epithelium, the integrity of mucus and epithelial cell junctions (E-cadherin and occludin) was examined in colonic biopsies from patients with liver cirrhosis and controls. SBP-inducing Escherichia coli (E. coli) and Proteus mirabilis (P. mirabilis) were isolated from ascites of patients with liver cirrhosis and co-cultured with Caco-2 cells to characterise bacteria-to-cell effects.
Results SBP-derived E. coli and P. mirabilis led to a marked reduction of cell-to-cell junctions in a dose-dependent and time-dependent manner. This effect was enhanced by a direct interaction of live bacteria with epithelial cells. Degradation of occludin is mediated via increased ubiquitination by the proteasome. Remarkably, a novel bacterial protease activity is of pivotal importance for the cleavage of E-cadherin.
Conclusion Patients with liver cirrhosis show a reduced thickness of colonic mucus, which allows bacteria-to-epithelial cell contact. Intestinal bacteria induce degradation of occludin by exploiting the proteasome of epithelial cells. We identified a novel bacterial protease activity of patient-derived SBP-inducing bacteria, which is responsible for the cleavage of E-cadherin structures. Inhibition of this protease activity leads to stabilisation of cell junctions. Thus, targeting these mechanisms by blocking the ubiquitin-proteasome system and/or the bacterial protease activity might interfere with BT and constitute a novel innovative therapeutic strategy to prevent SBP in patients with liver cirrhosis.
- peritonitis
- bacterial translocation
- liver cirrhosis
- E. coli
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of the study
What is already known on this subject?
Spontaneous bacterial peritonitis (SBP) is a life-threatening complication of liver cirrhosis with a 1-year mortality of up to 66%.
New treatment options of SBP are urgently needed; medication is limited to antibiotics with high recurrence rates and increasing antibiotic resistance.
Bacterial translocation (BT) due to impaired intestinal epithelial barrier function is crucial for the pathogenesis of SBP.
What are the new findings?
Patients with liver cirrhosis display changes in colonic mucus and cell junction proteins as entry sites for SBP-inducing bacteria.
Escherichia coli (E. coli) and Proteus mirabilis (P. mirabilis) isolated from ascites from patients with SBP trigger degradation of occludin and cleavage of E-cadherin, which represent two essential cell junction components.
Bacteria-induced reduction of occludin is mediated via endogenous proteasomal degradation.
Of note, we identified a novel bacterial protease activity of E. coli and P. mirabilis, which is responsible for the cleavage of E-cadherin and thus constitutes a druggable therapeutic target.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
These findings lead to a better understanding of the molecular pathomechanisms of SBP and enable the identification of novel druggable targets to protect the mucoepithelial barrier function and prevent BT.
Inhibition of the ubiquitin-proteasome system is a therapeutic option to preserve cell–cell contacts by stabilisation of occludin.
Inhibition of the newly identified bacterial protease activity represents a direct therapeutic approach which targets specifically pathogenic E. coli and P. mirabilis inhibiting BT by preventing E-cadherin cleavage.
Introduction
Spontaneous bacterial peritonitis (SBP) is one of the most harmful complications of liver cirrhosis defined as bacterial infection of ascites without intra-abdominal inflammatory sources. With a 1-year mortality of up to 66%, SBP remains a serious challenge in healthcare.1 2 Currently, SBP is diagnosed by >250 polymorphonuclear granulocytes/µL ascitic fluid. Despite antibiotic therapies with cephalosporins or piperacillin-tazobactam,3 4 SBP reoccurs in 70% of cases and worsens chances of survival.1 5 6 Hence, there is an urgent need for novel treatment options of SBP. We know that bacterial translocation (BT)—with Escherichia coli (E. coli), Klebsiella pneumoniae and streptococci being the most common microbes—from the gut to mesenteric lymph nodes is crucial for the development of SBP.7–9 BT is driven by a suppressed intestinal immune system with small intestinal bacterial overgrowth and reduced microbial diversity.8 10–14 In addition, increased permeability of the intestinal epithelial barrier is of major importance. Thus, cell-to-cell junctions play an important role in the development of BT.15–17
To improve diagnosis and develop new treatment options for SBP, molecular mechanisms of BT need to be further characterised to identify druggable targets. We investigated changes in mucus and expression of cell-to-cell junction proteins in colonic biopsies from patients with liver cirrhosis and SBP. Moreover, we established an in vitro model and studied the effects of patient-derived SBP-inducing E. coli and Proteus mirabilis (P. mirabilis) on tight and adherens junctions in intestinal epithelial cells. We identified a novel role of bacterial proteases in destabilisation of the intestinal epithelium, adding a novel piece to the understanding of SBP pathogenesis and highlighting the significance of the gut-liver axis. Of clinical relevance, inhibition of this newly identified bacterial protease activity by matrix metalloproteinase (MMP) inhibitors as proof of concept represents a novel therapeutic option that targets specifically pathogenic E. coli and P. mirabilis inhibiting BT by preventing E-cadherin cleavage.
Materials and methods
Patients
Human biopsies from left-sided colon were obtained from patients undergoing colonoscopy (ethical vote 16-101-0382, University of Regensburg, Regensburg, Germany). Written informed consent was obtained from all patients. Colonoscopy was performed using a standard high-definition endoscope (Olympus CF HQ 190L) under conscious sedation using midazolam and/or propofol. Diagnosis of liver cirrhosis was established by clinical, laboratory or imaging tests. Exclusion criteria were pregnancy, GI diseases, inflammatory conditions, HIV and hepatitis C virus infection. Characteristics of patients are given in online supplemental tables 1–5.
Supplemental material
Cell culture and bacteria
Human Caco-2 cells (DSMZ, Braunschweig, Germany) were cultured using minimal essential medium (Sigma-Aldrich, Taufkirchen, Germany) supplemented with 10% fetal bovine serum (Bio&Sell, Nürnberg, Germany) without supplementation of antibiotics. For culture of HCT-116 cells (DSMZ, Braunschweig, Germany), McCoy’s 5A modified medium (Thermo Fisher Scientific, Waltham, Massachusetts, USA) was supplemented with 10% fetal bovine serum without antibiotics. Ten different E. coli strains were isolated from ascitic fluid of patients with SBP and serotyping was performed by Robert Koch Institute (Berlin, Germany). For assay development, verification and validation, E. coli ATCC25922 (O6:Hnt) (ATCC, Manassas, Virginia, USA) was used. In addition, a P. mirabilis strain was extracted from ascites of a patient with SBP (for antibiograms, see online supplemental table 6). Bacteria were grown in Luria-Bertani broth at 37°C under agitation. Co-culture experiments were performed with live bacteria at multiplicities of infection (MOI) 0–10, supernatant of bacterial overnight culture (SN) or heat-inactivated (HI) bacteria (65°C, 5 min). For transwell experiments, see the Supplementary material and methods section.
Immunoprecipitation and detection of ubiquitination of occludin and E-cadherin
Cell lysates were incubated with anti-occludin and anti-E-cadherin antibodies coupled to protein A agarose and analysed for ubiquitination by Western blot. For further details, refer to the Supplementary material and methods section.
Frozen sections of colonic biopsies
Colonic biopsies were transferred into Hanks’ balanced salt solution (Sigma-Aldrich, Taufkirchen, Germany), embedded in Tissue-Tek (Sakura Finetek Europe, Alphen aan den Rijn, Netherlands) and snap frozen in liquid nitrogen. Tissue blocks were cut in 8 µm sections using a cryomicrotome (Leica CM3050 S Cryostat, Leica Biosystems, Wetzlar, Germany).
Mucus staining
Frozen sections of colonic biopsies were air dried for 30 min. Slides were fixed with 4% formaldehyde for 30 min, washed in phosphate buffered saline (PBS) and stained with alcian blue according to the manufacturer’s protocol (alcian blue stain kit, Abcam, Berlin, Germany).
Immunofluorescence and microscopy
To analyse the expression of occludin and E-cadherin, cells and colonic biopsies were fixed with formaldehyde and permeabilised with saponin. Thereafter, occludin and E-cadherin were stained using mouse anti-occludin-594 (for colonic biopsies), mouse anti-occludin-488 (for Caco-2 cells) or anti-rat E-cadherin-594 (for colonic biopsies and Caco-2 cells) antibodies. For further details, see the Supplementary material and methods section.
Protease activity assays
Cellular and bacterial protease activities were tested using fluorescence-labelled MMP substrate and a gelatin in-gel assay.18
The broad-spectrum MMP inhibitor BB-9419 was used to block human proteases (MMP inhibitor profiling kit; BML-AK016, Enzo Life Sciences, Lörrach, Germany). Different purified human MMPs were preincubated with 10 µM BB-94 for 1 hour at 37°C. Following addition of the substrate, the fluorescent signal was recorded after 240 s using a Tecan microplate reader Infinite pro 200 (Extinction/Emission spectra: 328/393). In addition, protease activities of Caco-2 cells and (HI) SBP-derived bacteria (E. coli, P. mirabilis) were analysed with or without inhibitor BB-94 using a protease fluorescent detection kit according to the manufacturer’s protocol (PF0100, Sigma-Aldrich, Taufkirchen, Germany).
Statistical analysis
Results are depicted as means±SD. Data were analysed using Welch’s t-test or Mann-Whitney rank-sum test. Normal distribution and homogeneity of variance were tested using the Shapiro-Wilk test and Brown-Forsythe test. Sigma Plot V.14.0 software (Systat, Erkrath, Germany) was used for graphics and statistical analysis.
Supplementary material and methods
Extended online supplemental information on material and methods is available at the journal website. This includes primer sequences applied for quantitative PCR (qPCR) (online supplemental table 7).
Results
Liver cirrhosis is associated with changes in the colonic mucus layer and destabilisation of cell junctions
We characterised mucus thickness and tight and adherens junction proteins as essential elements of the epithelial barrier in colonic biopsies of patients with (n=14) and without (n=19) liver cirrhosis. Both groups were matched regarding age and sex. Characteristics of patients, that is, Child-Pugh score, medication and comorbidities, are summarised in online supplemental tables 1–5.
Interestingly, the thickness of the colonic mucus—as first line of defence—from patients with liver cirrhosis was significantly reduced (figure 1A, p=0.036). Proton pump inhibitors (PPIs) are discussed to induce dysbiosis, small intestinal bacteria overgrowth and SBP.20–23 To exclude an influence of PPI on mucus thickness, we split our patient cohort into a PPI-untreated group and a PPI-treated group. PPI treatment had no effect on mucus thickness, neither in control nor in cirrhotic patients (figure 1B). Reduced thickness of mucus may contribute to direct/closer contact of bacteria and epithelial cells and therefore bacteria-driven destabilisation of epithelial integrity.
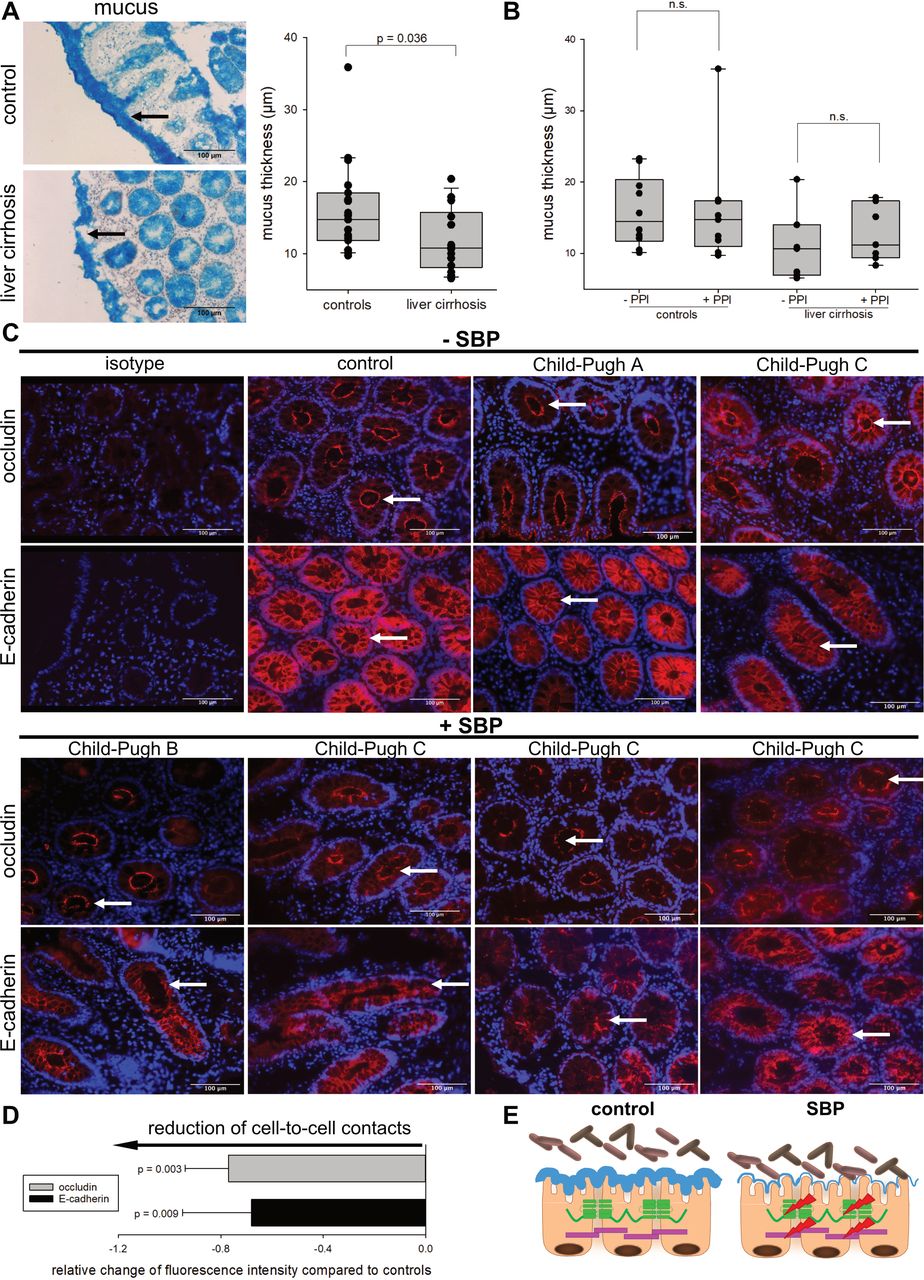
Reduced thickness of colonic mucus in patients with liver cirrhosis and destabilisation of cell junctions in patients with spontaneous bacterial peritonitis (SBP). (A, left panel) Analysis of colonic mucus via alcian blue staining in biopsies of patients with liver cirrhosis (n=14) and healthy controls (n=19). Images are displayed at 20xmagnification. (A, right panel) Quantification of mucus thickness. One dot represents one patient. A significant reduction of mucus thickness was observed in patients with liver cirrhosis (p=0.036, Mann-Whitney rank-sum test). (B) Impact of proton pump inhibitor (PPI) therapy on mucus thickness in controls with (n=9, online supplemental table 3) or without PPI (n=10, online supplemental table 2) and patients with liver cirrhosis with (n=7, online supplemental table 5) or without PPI treatment (n=7, online supplemental table 4). One dot represents one patient. Treatment with PPIs did not affect colonic mucus layer thickness. (C) Immunohistochemistry of occludin and E-cadherin in colonic biopsies. One control, one Child-Pugh class A patient, one Child-Pugh class B patient with SBP and four Child-Pugh class C patients with or without SBP are shown (compare online supplemental tables 3–5, blue). The patient cohort was composed as follows: upper panel (from left to right): control patient 19, patients with liver cirrhosis 10, 6; lower panel (from left to right): patients with liver cirrhosis 5, 12, 14, 13 (compare online supplemental tables 3–5, blue). From the ascites of Child-Pugh C patient 5 and 14, Escherichia coli were isolated as the SBP-inducing bacteria (compare online supplemental tables 4–6). Arrows point to specific morphological changes regarding occludin and E-cadherin. All images are presented at 20x magnification. (D) Quantification of fluorescence intensity of occludin and E-cadherin in colonic biopsies of patients with SBP. Fluorescence intensity of patients with SBP was normalised to median fluorescence intensity of controls. Controls are shown as baseline. Cell junction proteins occludin (p=0.003) and E-cadherin (p=0.009) were significantly reduced (mean±SD, Welch’s t-test). (E) Schematic illustration of the epithelial barrier in controls and patients with SBP. Reduction of mucus thickness in patients with liver cirrhosis may facilitate bacteria-to-cell interaction. In SBP the essential cell junction proteins occludin and E-cadherin are downregulated. Thus, the mucoepithelial barrier is impaired (red arrow). Occludin is depicted in green and E-cadherin in purple.
Focusing on the epithelial barrier as second line of defence, patients with SBP showed a disruption of occludin rings and loss of E-cadherin structures compared with controls (figure 1C). In addition, we observed a significant reduction of occludin (p=0.003) and E-cadherin (p=0.009) protein levels (figure 1D). Thus, these data provide evidence for a destabilised colonic epithelial barrier in patients with SBP (figure 1E).
E. coli strains disrupt the epithelial junction barrier via reduction of occludin and E-cadherin
To analyse the molecular mechanisms of bacteria-to-cell interactions, an in vitro model was established. Therefore, different colon cell lines, for example, Caco-2 and HCT-116, were tested. Caco-2 cells displayed proliferation in a monolayer and showed cellular polarisation and formation of cell-to-cell junctions. In contrast, HCT-116 cells grew in a multilayer without polarisation (figure 2A). Thus, Caco-2 cells which have been established as an optimised in vitro model of the intestinal mucosa24 represent an ideal tool to analyse bacteria-to-cell interactions under standard conditions. At day 6 of culture, Caco-2 cells were differentiated as shown by expression of differentiation marker alkaline phosphatase (ALPI). Protein levels of E-cadherin and occludin were stable up to day 6 and day 8, respectively, and slightly decreased with longer incubation time (figure 2B). After 6 days, Caco-2 cells were 100% confluent. With a longer incubation time, a cell multilayer developed with concomitant induction of cell death (figure 2C and online supplemental figure 1). In summary, the Caco-2 cell model at day 6 provided the optimal in vitro conditions with respect to (1) polarisation, (2) formation of cell-to-cell junctions, (3) differentiation and (4) cell viability.
Supplemental material

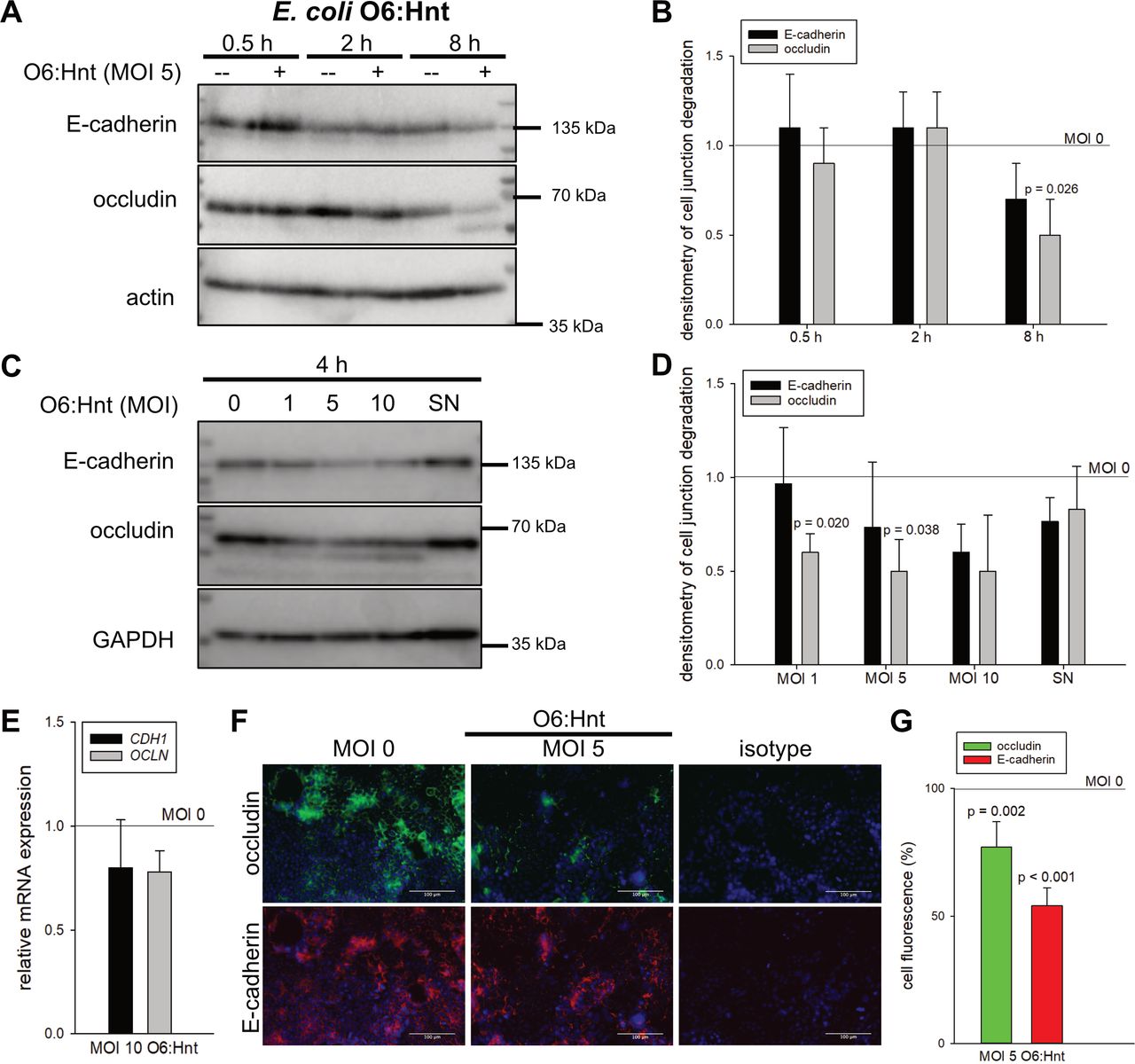
Establishment of an in vitro model of the intestinal mucosa to study bacteria-to-cell interactions. (A) Electron microscopy analysis of polarised Caco-2 and unpolarised HCT-116 cells precultured for 6 days. Microvilli and cell-to-cell contacts in Caco-2 cells are indicated by arrows. (B) Western blot analyses of differentiation marker ALPI, E-cadherin and occludin. Caco-2 cells were cultured for 1–14 days. One exemplary blot with densitometric analysis out of three replicates is shown. (C) Caco-2 cells were cultured for 6, 8 and 14 days and labelled with 1 µg/mL DAPI. Quantification of DAPI-positive/dead Caco-2 cells is depicted (n=3, mean+SD, Welch’s t-test). (D) Electron microscopy imaging of Caco-2 cells with or without Escherichia coli O6:Hnt (multiplicity of infection (MOI) 5, 4 hours). Co-culture with E. coli O6:Hnt did not affect essential cellular structures, for example, brush borders and mitochondria (indicated by arrows). (E) Caco-2 cells were cultivated up to 8 days and cell junction regulation was examined by Western blot in the presence and absence of E. coli O6:Hnt for 4 hours. Densitometric analysis is given below each exemplary blot. (F) E. coli O6:Hnt-stimulated Caco-2 cells were stained with crystal violet and cell death was quantified (n=3, mean±SD). (G) Cell cycle analysis on E. coli O6:Hnt stimulation was performed by flow cytometry (n=3, mean±SD, Welch’s t-test).
To mimic bacterial infection, E. coli ATCC25922 (O6:Hnt) was used in different MOI for up to 8 hours. Luria-Bertani broth was used as control (MOI 0). Relevant cellular structures, for example brush borders, mitochondria or other cell organelles, were unchanged by co-culture with E. coli O6:Hnt and Caco-2 cells (figure 2D, MOI 5). Of note, stimulation with E. coli O6:Hnt (MOI 10) downregulated E-cadherin and occludin by up to 60%. This effect was most prominent in Caco-2 cells that had been precultured for 6 days prior to bacterial treatment. Longer preculture resulted in less pronounced downregulation of occludin and E-cadherin presumably due to cell death (figure 2C,E). In addition, crystal violet staining revealed no decrease in cell confluence after E. coli stimulation (figure 2F, MOI 10). A G1 arrest with 17.7% more cells in the G0G1 fraction (p=0.043) was detected, whereas the subG1 fraction was not increased in the presence of E. coli O6:Hnt (figure 2G).
In conclusion, interference with essential tight and adherens junction proteins is a key pathophysiological feature of SBP-relevant bacteria like E. coli.
Whereas short-time incubation of 30 min to 2 hours did not influence protein levels of E-cadherin and occludin, bacterial treatment for 4–8 hours resulted in a dose-dependent reduction of both cell-to-cell contact proteins by up to 50% (figure 3A–D). These destabilising effects of E. coli O6:Hnt at MOI 5 were additionally confirmed by immunohistochemistry with a significant decrease by 23% for occludin (p=0.002) and by 46% for E-cadherin (p<0.001) (figure 3F,G). Interestingly, mRNA levels were not affected (figure 3E). Furthermore, and of clinical relevance, minor or no effects were seen on protein levels of cell-to-cell junction proteins when E. coli SN was used (figure 3C,D) instead of live bacteria.
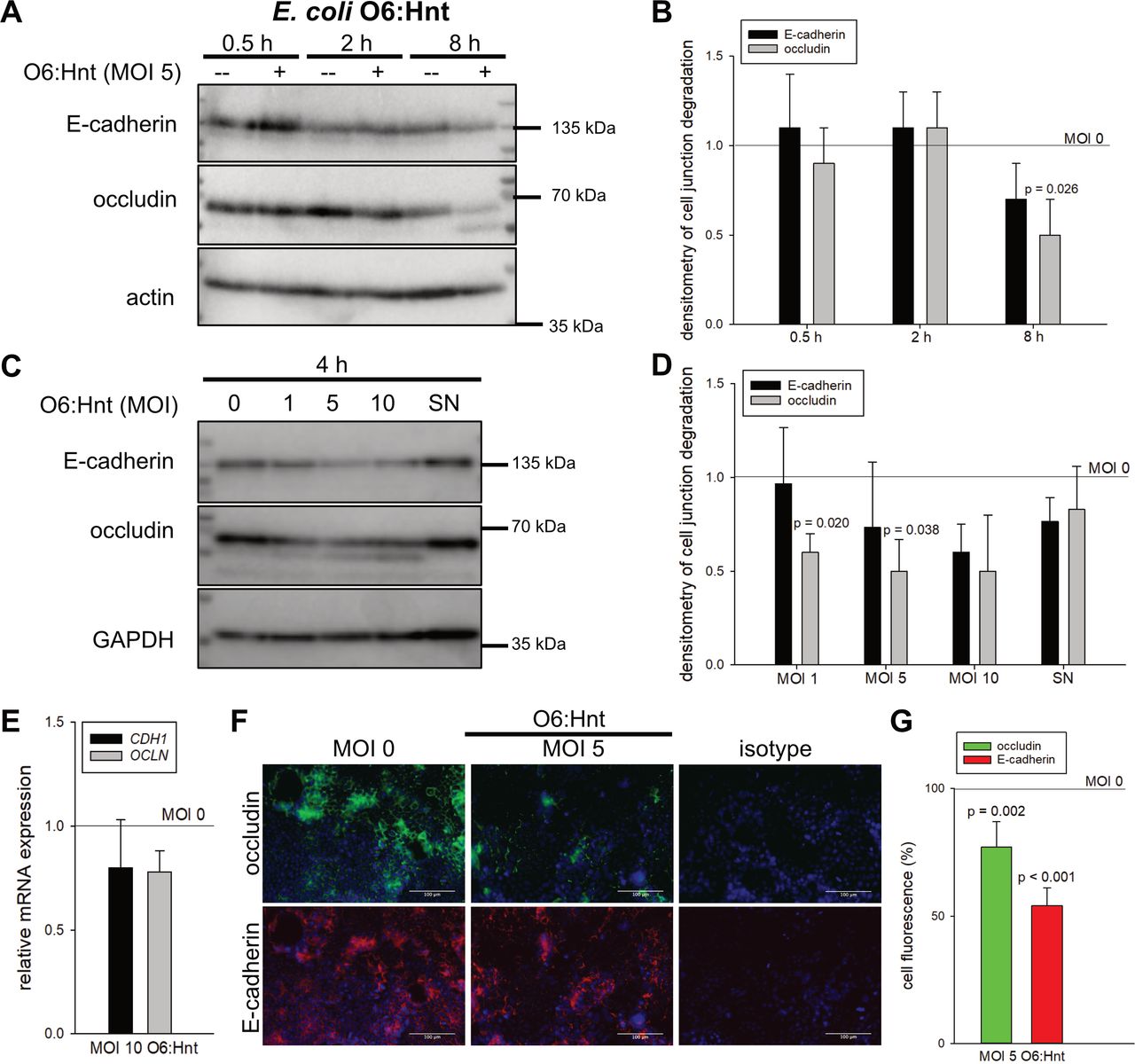
Escherichia coli induced dose-dependent and time-dependent destabilisation of the intestinal epithelial barrier. Caco-2 cells were cultivated for 6 days. E-cadherin and occludin were analysed by Western blot following (A) a time kinetic or (C) incubation with increasing multiplicities of infection (MOI) of E. coli O6:Hnt. Densitometry of three experiments is given in (B) and (D) (mean±SD, Welch’s t-test). (C, D) In addition to live bacteria, supernatant of bacterial overnight culture (SN) was used for a 4 hour stimulation of Caco-2 cells (mean±SD, Welch’s t-test). (E) Quantitative PCR analysis of Caco-2 cells stimulated with E. coli O6:Hnt at MOI 10 for 4 hours (n=3, mean±SD). (F) Immunohistochemistry and (G) corresponding quantification of cell fluorescence of occludin and E-cadherin following E. coli O6:Hnt stimulation (n=3, mean±SD, Welch’s t-test).
These data indicate that bacterial co-culture of Caco-2 cells induces a time-dependent and dose-dependent epithelial destabilisation and an arrest of the cell cycle in G1.
Patient-derived pathogenic E. coli strains induce destabilisation of epithelial cell-to-cell junctions
To investigate the clinical relevance of the bacteria-induced epithelial destabilisation, 10 E. coli strains were isolated from ascites of patients with SBP (patient-derived E. coli=PDEC 1–10). Five out of 10 (50%) of these E. coli strains caused a reduction of both E-cadherin and occludin (online supplemental figure 2). For further analyses, E. coli O16:H5 (PDEC 1—with minor effects) and E. coli Ont:H7 (PDEC 2—with major effects) were used. Treatment of Caco-2 cells with E. coli O16:H5 downregulated E-cadherin by 14%. E. coli Ont:H7 reduced E-cadherin (p=0.029) and occludin (p=0.007) protein levels by up to 73% and concurrently led to induction of cell death (figure 4A–D). Of importance, HI bacteria did not affect the levels of cell junction proteins (figure 4A). To analyse the role of bacterial membrane components on epithelial destabilisation, Caco-2 cells were treated with 0.1 and 1 µg/mL E. coli lipopolysaccharide (LPS) (O55:H5). In the presence of LPS, protein levels of occludin and E-cadherin did not change (figure 4E). In accordance, TLR2 and TLR4 were not detected on the surface of Caco-2 cells, neither on naive cells nor after stimulation with phorbol 12-myristate 13-acetate (PMA) or E. coli LPS (O55:H5) that can trigger TLR2 or TLR4 expression (online supplemental figure 3). These data underline that live bacteria are required to induce the reduction of occludin and E-cadherin.
Supplemental material
Supplemental material
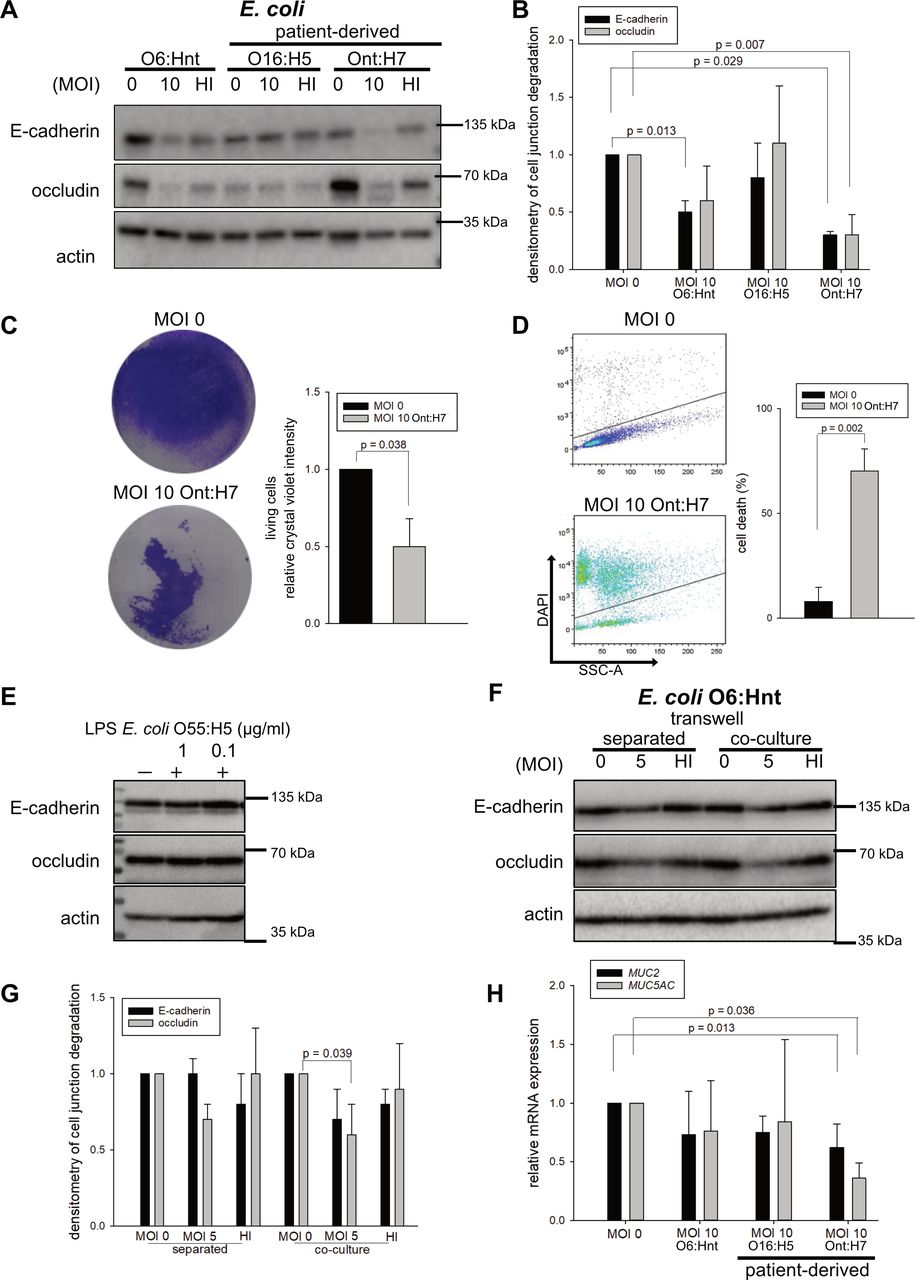
Patient-derived spontaneous bacterial peritonitis (SBP)-inducing isolates with direct contact to epithelial cells trigger epithelial destabilisation. (A) Caco-2 cells were stimulated with different Escherichia coli at multiplicity of infection (MOI) 10 for 4 hours. In addition to E. coli O6:Hnt, E. coli SBP isolates (O16:H5, Ont:H7, compare online supplemental table 6) from ascites of two patients were used for stimulation. E-cadherin and occludin were analysed on protein level by Western blot with densitometric analysis (B) of three replicates (mean±SD, Welch’s t-test and Mann-Whitney rank-sum test). (C) Caco-2 cells were infected with E. coli Ont:H7 (MOI 10, 4 hours), fixed and stained with crystal violet (n=3, mean±SD, Welch’s t-test). (D) DAPI exclusion analysis of Caco-2 cells stimulated with patient-derived E. coli Ont:H7 (MOI 10, 4 hours) (n=3, mean±SD, Welch’s t-test). (E) Caco-2 cells were stimulated with 0.1 and 1 µg/mL E. coli O55:H5 LPS for 4 hours. E-cadherin and occludin protein levels were analysed by Western blot (n=3). One exemplary blot is shown. (F) Caco-2 cells were grown in 6-well plates and heat-inactivated (HI) E. coli O6:Hnt bacteria were added onto transwell inserts on top of Caco-2 cells (separated). As control, Caco-2 cells were stimulated with E. coli O6:Hnt in the same well (co-culture). Protein levels of E-cadherin and occludin were assayed by Western blot with densitometric analysis (G) of three replicates (mean±SD, Welch’s t-test). (H) Quantitative PCR analysis of MUC2 and MUC5AC52 expression of Caco-2 cells stimulated with different (patient-derived) E. coli (O6:Hnt, O16:H5 and Ont:H7, MOI 10) for 4 hours (n=3, mean±SD, Welch’s t-test and Mann-Whitney rank-sum test).
To elucidate whether destabilisation of the epithelial barrier requires direct bacteria-to-cell contact, transwell experiments were performed. With direct contact between E. coli and Caco-2 cells, occludin and E-cadherin were downregulated by 30%–40% (occludin, p=0.039). Without direct contact, occludin levels were reduced by 30%, while E-cadherin levels remained unchanged. HI bacteria affected the protein levels of neither E-cadherin nor occludin (figure 4F,G). Since a reduced thickness of mucus was observed in patients with liver cirrhosis (figure 1A,B), we hypothesised that SBP-inducing bacteria might regulate mucus production. Therefore, we measured mucin expression of MUC2 and MUC5AC representative for colon and stomach in Caco-2 cells following bacterial treatment. Especially, patient-derived E. coli Ont:H7 reduced MUC2 (p=0.013) and MUC5AC (p=0.036) expression (figure 4H). These data indicate that degradation of the epithelial barrier requires direct interaction of live bacteria and epithelial cells and that bacteria interfere with MUC2 expression, the essential component of the colonic mucus.
Bacteria induce downregulation of occludin via enhanced proteasomal degradation
Since effects on occludin and E-cadherin were detected on protein but not on mRNA level, we assumed regulation via degradation. Therefore, Lys-48 ubiquitination of occludin and E-cadherin was studied by immunoprecipitation. Caco-2 cells were lysed and occludin and E-cadherin were precipitated and analysed by Western blot. E-cadherin showed no alteration in ubiquitination (figure 5A). In contrast, precipitated occludin displayed a 33% increase in ubiquitination on bacterial co-incubation (figure 5B). Thus, occludin is tagged for proteasomal degradation on co-cultivation with bacteria. To verify proteasomal degradation, proteasome inhibitor clasto-lactacystin β-lactone (LA) (2.5 and 5 µM) was administered 4 hours prior to stimulation with E. coli O6:Hnt. Proteasome inhibition did not prevent E-cadherin reduction. Of note, occludin degradation was reduced up to 80% in the presence of LA (5 µM) (figure 5C,D). This result is in accordance with Lys-48 ubiquitination and confirms that occludin is subjected to E. coli-induced proteasomal degradation.

Occludin reduction via enhanced proteasomal degradation. (A) Immunoprecipitation to analyse ubiquitination of E-cadherin and occludin. Cell lysates were incubated with bead-coupled antibodies (IP). As control, pellets of preclearing (clear) and 20 µg cell lysates (input) were loaded. (B) Densitometry of ubiquitin normalised to precipitated occludin (n=3, mean±SD). (C) Co-cultivation of Caco-2 cells with proteasome inhibitor clasto-lactacystin β-lactone (2.5 and 5 µM) for 4 hours followed by stimulation with Escherichia coli O6:Hnt at multiplicity of infection (MOI) 5 for additional 4 hours. Protein regulation of E-cadherin and occludin was analysed by Western blot. (D) Occludin densitometric analysis of three replicates (mean±SD, Welch’s t-test).
E-cadherin is cleaved by a novel bacterial protease activity
Since degradation of E-cadherin is independent of the proteasome, we hypothesised that different proteases may be responsible for bacteria-induced downregulation of this protein. E-cadherin is a known cellular substrate of MMPs.25 26 To analyse whether E-cadherin is a target of MMPs, we used the broad-spectrum MMP inhibitor batimastat (BB-94) as a potential blocker of E-cadherin degradation.
First, we tested the effect of BB-94 on endogenous MMPs. Activated MMPs are secreted into the extracellular space. Therefore, we treated supernatant of Caco-2 cells with BB-94 and detected a reduction of total protease activity (figure 6A). In addition, the effect of BB-94 on specific recombinant human MMPs was analysed. All MMPs tested were significantly inhibited up to 90% (figure 6B). Bacterial proliferation was not affected by the MMP inhibitor (figure 6C). Therefore, relevant pleiotropic effects of BB-94 on bacterial protein synthesis can be excluded. Thus, we concluded that this inhibitor could effectively be applied in our bacteria-Caco-2-model to further investigate and potentially restore the cell-to-cell junction protein E-cadherin.
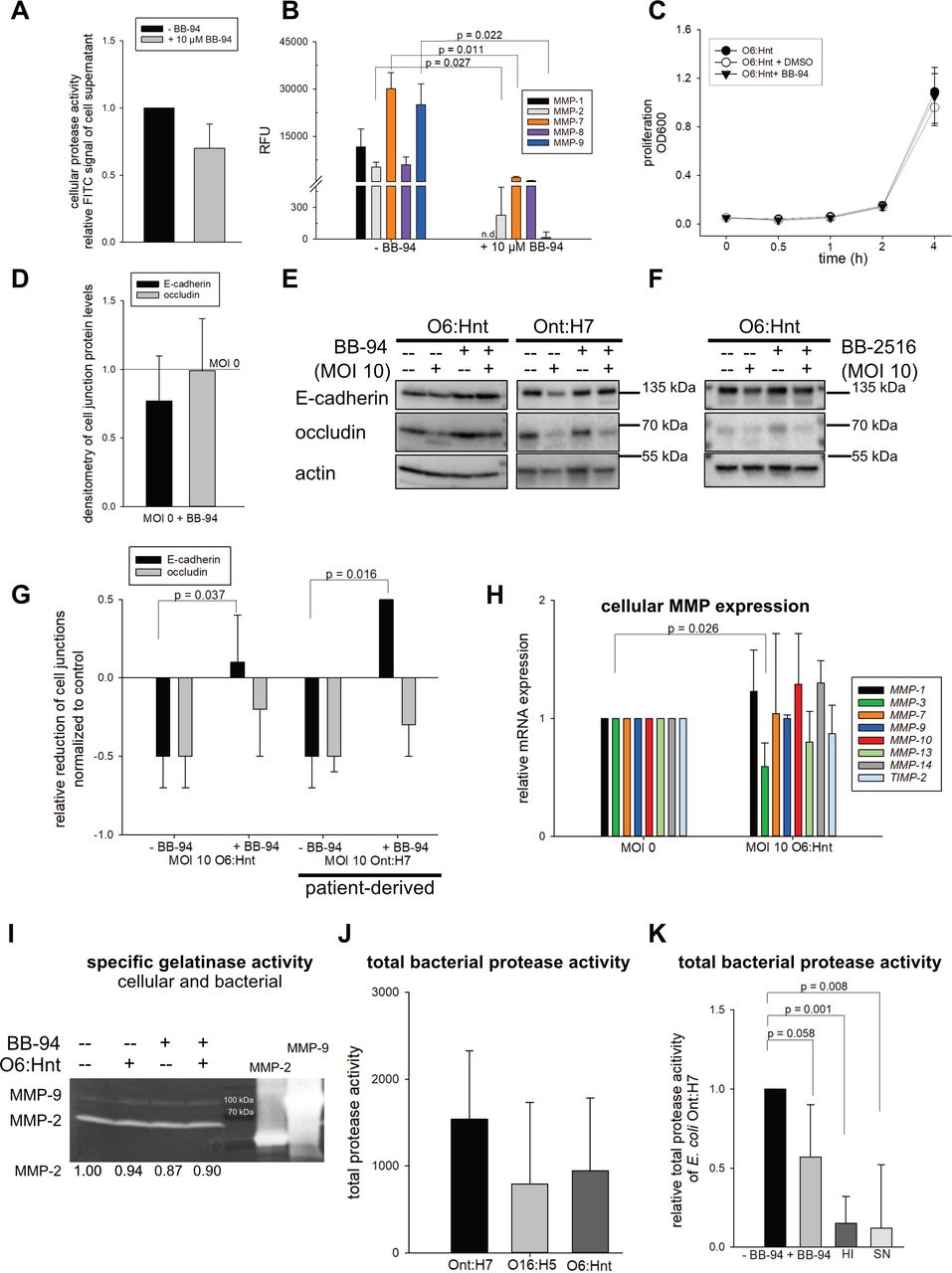
E-cadherin is a target of a bacterial protease activity. (A) Cellular protease activity±matrix metalloproteinase (MMP) inhibitor BB-94 was measured by a fluorescein isothiocyanate (FITC)-coupled protease substrate. Relative FITC signal of three experiments is shown (mean±SD). (B) Inhibition of MMP activity by BB-94. Recombinant MMPs (MMP-1 (0.5 µM), MMP-2 (0.3 µM), MMP-7 (0.3 µM), MMP-8 (0.1 µM) and MMP-9 (0.3 µM)) were preincubated with BB-94 (10 µM) for 1 hour at 37°C. Fluorescent MMP substrate was added and MMP cleavage was recorded as relative fluorescence units (RFU) (n=3, mean±SD, Welch’s t-test). (C) Proliferation of Escherichia coli O6:Hnt ± BB-94 or DMSO was unchanged (n=3, mean±SD). (D) Densitometric analysis of cell junction proteins E-cadherin and occludin in the presence of 10 µM BB-94 (n=4, mean±SD). (E, F) Following treatment with BB-94 or BB-2516 (10 µM) for 20 min, Caco-2 cells were stimulated with E. coli O6:Hnt or patient-derived E. coli Ont:H7 at multiplicity of infection (MOI) 10 for 4 hours. E-cadherin and occludin were analysed by Western blot and (G) densitometric analysis (n=3, mean±SD, Welch’s t-test). (H) Qualitative PCR analysis of different MMPs and tissue inhibitor 2 of MMPs (TIMP-2) of E. coli O6:Hnt-stimulated Caco-2 cells (n=2–6, mean±SD, Welch’s t-test). (I) Cellular MMP activity was analysed by gelatin zymography. A 10% acrylamide gelatin gel (1 mg/mL) was loaded with supernatant of (un)stimulated Caco-2 cells. Recombinant MMP-2 and MMP-9 served as positive controls. One representative out of three gelatin zymography assays is shown. (J) Total protease activities of E. coli O6:Hnt and patient-derived E. coli strains (Ont:H7 and O16:H5) were quantified by a fluorescent (FITC) protease substrate (n=5, mean±SD). (K) Total protease activity of patient-derived E. coli Ont:H7±10 µM BB-94, heat-inactivated (HI) bacteria and bacterial supernatant of bacterial overnight culture (SN) was quantified by a fluorescent (FITC) protease substrate (n=5, mean±SD, Welch’s t-test and Mann-Whitney rank-sum test).
Second, BB-94 alone (without bacteria) did not affect cell junctions (figure 6D). Of note, BB-94 led to restoration of E-cadherin on co-culture of Caco-2 cells with both E. coli strains, O6:Hnt (p=0.037) and SBP-derived E. coli Ont:H7 (p=0.016) (figure 6E,G). As proof of concept, a second broad-spectrum MMP inhibitor marimastat (BB-2516) reconstituted E-cadherin protein levels downregulated by E. coli O6:Hnt (figure 6F). Thus, MMP inhibitors like BB-94 and BB-2516 protect the epithelial barrier and, therefore, may prevent BT and development of SBP. Given the protective effect of BB-94 and BB-2516, we investigated whether cellular MMPs are responsible for destabilisation of cell-to-cell junctions. Surprisingly, gene expression (figure 6H) and gelatinase activity of endogenous MMPs (figure 6I) were not induced on co-cultivation of Caco-2 cells with E. coli. This implies that cellular MMPs are not the mediators of bacteria-induced cleavage of E-cadherin. We hypothesised that if not cellular proteases, then bacterial proteases instead were responsible for E-cadherin degradation.
Third, we assessed total protease activity of the different E. coli strains. Of clinical interest, patient-derived E. coli Ont:H7 with the most prominent effect on downregulation of cell contact proteins exhibited a high protease activity. E. coli O6:Hnt and O16:H5 displayed lower protease activities (figure 6J), in line with their ability to degrade E-cadherin (figure 4A). BB-94 treatment resulted in a reduction of 43% (p=0.058) of total bacterial protease activity. HI bacteria and bacterial SN showed a significantly 85% and 88% lower (p=0.001 and p=0.008, respectively) total bacterial protease activity. This suggests that the bacterial protease activity is only to a minor extent released into the SN (figure 6K). In conclusion, these data demonstrate that the cell junction protein E-cadherin is degraded by a bacterial protease of live bacteria.
SBP-derived P. mirabilis displays the novel protease activity
To generalise our observation that a novel bacterial protease activity is responsible for the cleavage of E-cadherin, we tested additional bacterial strains involved in the pathogenesis of SBP. We isolated a P. mirabilis strain from ascites of a patient who suffered from acute-on-chronic liver failure. This P. mirabilis reduced E-cadherin and occludin protein levels by up to 63% and concurrently led to induction of cell death (figure 7A,B). As shown above, BB-94 had no influence on bacterial proliferation (figure 7C). Of note, treatment with BB-94 (figure 7D,E) and BB-2516 (figure 7J,K) restored E-cadherin levels when added to a Caco-2–P. mirabilis co-culture. These data were in accordance with the effects of the SBP-derived E. coli strain Ont:H7. Thus, patient-derived bacteria strains—E. coli and P. mirabilis—exhibited high BB-94-sensitive and BB-2516-sensitive protease activities resulting in cleavage of E-cadherin (figures 6E–G,K and 7D–F,J,K). To exclude that P. mirabilis-induced cell death is the reason for the observed reduction of E-cadherin and occludin protein levels, we analysed Caco-2 cells co-cultured with P. mirabilis (1) in total (attached+detached), (2) attached and (3) detached. Attached cells with almost no signs of cell death in flow cytometry exhibited a reduction of E-cadherin and occludin protein levels. Again, E-cadherin was reconstituted by BB-94 (figure 7G–I and online supplemental figure 4). Furthermore, the mode of cell death observed on bacteria-Caco-2 co-culture was caspase-independent and consequently is not detachment-induced anoikis (online supplemental figure 4). Thus, we show that bacteria-induced cell death and not E-cadherin and/or occludin degradation is responsible for the detachment of Caco-2 cells.
Supplemental material

The novel bacterial protease activity cleaves cell junction protein E-cadherin and is a feature of different spontaneous bacterial peritonitis (SBP)-inducing bacteria. (A) Caco-2 cells were treated with SBP-derived Proteus mirabilis (compare online supplemental table 6) at multiplicity of infection (MOI) 10, heat-inactivated (HI) P. mirabilis and bacterial supernatant of bacterial overnight culture (SN) for 4 hours. E-cadherin and occludin were analysed on protein level by Western blot. (B) Caco-2 cells were co-cultured with P. mirabilis (MOI 10), fixed and stained with crystal violet. Cell survival was quantified (right panel, n=3, mean±SD). (C) Proliferation of patient-derived P. mirabilis±BB-94 or DMSO was photometrically measured (n=3, mean±SD). (D) Following treatment with BB-94 (10 µM) for 20 min, Caco-2 cells were co-cultured with P. mirabilis at MOI 10 for 4 hours. Protein levels of occludin and E-cadherin were analysed by Western blot and (E) quantified by densitometry (n=3, mean±SD, Welch’s t-test). (F) Total protease activity of P. mirabilis±BB-94 and HI bacteria was measured by a fluorescein isothiocyanate (FITC)-coupled protease substrate (n=3, mean±SD, Welch’s t-test). (G) Following treatment with BB-94 (10 µM, 20 min), Caco-2 cells were treated with patient-derived P. mirabilis at MOI 10 for 4 hours. Attached versus detached cells were analysed on protein level for E-cadherin and occludin or (H, I) induction of cell death via DAPI exclusion by flow cytometry. Total cells represent the combination of attached and detached cells (n=3, mean±SD, Welch’s t-test). (H, I) Blue lines and bars represent MOI 0, while red lines and bars represent MOI 10 P. mirabilis. (J, K) Analysis of E-cadherin and occludin protein levels by Western blot with densitometric analysis. Prior to P. mirabilis co-culture (MOI 10, 4 hours), Caco-2 cells were stimulated with 10 µM BB-2516 for 20 min (n=3, mean±SD).
In summary, in patients with liver cirrhosis and SBP, the thickness of the colonic mucus layer is reduced and tight and adherens junction proteins E-cadherin and occludin are downregulated. Our Caco-2 model highlights a direct bacteria-to-cell interaction to be essential for reduction of cell-to-cell contact proteins. On E. coli and P. mirabilis stimulation, cells downregulate occludin via endogenous proteasomal degradation, whereas bacterial proteases are responsible for the cleavage of E-cadherin.
Discussion
With this study, we provide novel insights on both host (human) and bacterial factors as mediators for BT and consequent infection of ascites (SBP) in patients with liver cirrhosis. We identified the following novel pathomechanisms and potential therapeutic targets in SBP. Patients with liver cirrhosis display (1) a reduced thickness of colonic mucus, which allows bacteria-to-epithelial cell contact. Intestinal bacteria induce (2) proteasomal degradation of occludin in epithelial cells and (3) cleavage of E-cadherin by a novel bacterial protease activity. These mechanisms (1–3) allow intestinal bacteria to reach the epithelium, destabilise the protective epithelial barrier and thus promote BT and SBP (figure 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Novel pathomechanisms for spontaneous peritonitis (SBP). Patients with liver cirrhosis display (1) a reduced thickness of colonic mucus, which allows bacteria-to-epithelial cell contact. Intestinal bacteria induce (2) proteasomal degradation of occludin in epithelial cells and (3) cleavage of E-cadherin by a novel bacterial protease activity. These mechanisms (1–3) allow intestinal bacteria to reach the epithelium, destabilise the protective epithelial barrier and thus promote BT and SBP.
Changes in mucus and cell junction proteins in patients with liver cirrhosis as entry sites for SBP-inducing bacteria
The intestinal epithelium separates the luminal microbiota from the systemic tissues. The first line of defence however are the colonic mucus layers.27 Intestinal mucus contains many different proteins, and the O-glycosylated mucin 2 (MUC2) is the core molecule.28 29 The colon handles a large bacterial load with a two-layered mucus system which consists of an inner layer formed of densely packed MUC2 sheets which is impenetrable to bacteria and an outer layer which serves as a niche for the microbiota.30 Mice deficient in MUC2 develop severe colitis and eventually colon cancer.29 31–33 Johansson et al showed that colon mucus defects of patients with active ulcerative colitis (UC) allow bacteria to penetrate and reach the epithelium. To our knowledge, our data represent the first analysis of colonic mucus in patients with liver cirrhosis. We extend the observation of Johansson et al in patients with UC34 and of Sorribas et al in murine models of liver cirrhosis35 to patients with liver cirrhosis.
We demonstrate decreased mucus thickness in patients with liver cirrhosis. Van der Post et al described that major structural mucus components including MUC2 were reduced in patients with active UC.36 This is in line with our observation that MUC2 mRNA is less abundant in our in vitro model when Caco-2 cells were co-cultured with patient-derived E. coli. There are different strategies by which commensal and pathogenic bacteria are able to alter the intestinal mucus layer, for example, a reduction in mucus synthesis, secretion, viscosity and thickness, an increase in mucus degradation and penetrability and an alteration of the mucus composition.37 Of note, mucin synthesis is also regulated at transcriptional and epigenetic levels. Numerous transcription factors have been shown to regulate MUC2. They can be linked to specific bacteria or microbial products and are mainly acting through the activation of nuclear factor (NF)-κB binding to a specific site in the promoter of MUC2. In addition, specific inflammatory markers including tumour necrosis factor and interleukins engage the NF-κB pathway and transactivate MUC2, whereas activation of the Janus kinase by tumour necrosis factor represses the MUC2 gene.38 39 In colon cancer, epigenetic mechanisms such as promotor methylation, histone modifications and additionally micro-RNAs contribute to the regulation of MUC2.
Furthermore, we provide novel data on structural changes associated with reduction of colonocyte expression of the junction proteins occludin and E-cadherin in patients with SBP. These combined results indicate that compositional alterations in the mucus of patients with liver cirrhosis lead to mucus barrier weakening, increased host–bacteria contact and reduction of cell junction proteins. These data provide a platform for further studies to therapeutically restore the mucoepithelial barrier. Some limits of our study merit consideration: The murine models allow to dissect the effects of portal hypertension using prehepatic portal hypertensive mice from those of cirrhosis (in comparison to bile duct ligation and CCl4-induced cirrhotic mice) and thus Sorribas et al concluded that cirrhosis, but not portal hypertension, impairs the mucoepithelial barrier and enables BT.35 In addition, murine models allow to study the gut-vascular barrier because translocation is not solely limited to the epithelial gut interface.17 The Caco-2 co-culture model bears the advantage of standardised analyses of specific bacterial strains and their interaction with epithelial junction proteins.
Intestinal bacteria induce proteasomal degradation of the cell junction protein occludin and destabilisation of the epithelial barrier
Another important finding of our study is that bacteria can enhance Lys-48 ubiquitination of occludin. Lys-48 polyubiquitination constitutes a signal targeting occludin for proteasomal degradation. Although the ubiquitin system is unique to eukaryotes, bacteria have developed a plethora of strategies that are capable of exploiting the host ubiquitination pathway.40 Protein ubiquitination is a cascade catalysed by an E1 (ubiquitin activating enzyme), an E2 (ubiquitin-conjugating enzyme) and an E3 (ubiquitin ligase). There are two major classes of E3s, HECT type and RING/U-box type E3s. Bacteria express molecular mimics of both of them. Furthermore, F-box domain containing proteins mediate ubiquitination and have also been described in bacterial effectors.41 42 Targeting host protein ubiquitination through a set of ubiquitin ligases is an important virulence strategy adopted by microbes to modify key host signalling pathways to establish conditions favourable for their survival and growth.43 44 We assume that similar mechanisms of ubiquitination may be responsible for degradation of occludin in our bacteria-Caco-2 model. Alternative mechanisms of action of these effectors are (1) catalysing E1-independent and E2-independent ubiquitination, (2) post-translational modification of the ubiquitination cascade and (3) coupling their catalytic activity to components of the ubiquitination machinery.41 In conclusion, host ubiquitination is not only essential for eukaryotic cell development and homeostasis but also plays a critical role for the outcome of many bacterial infections. Thus, targeting proteasomal degradation by pharmacological inhibition of bacterial ubiquitin ligase-like enzymes should be considered as a novel strategy to treat bacterial infections. Inhibition of the proteasome has been validated as an important strategy in anticancer therapy.45–49 We suggest that proteasome inhibition may offer a novel option to prevent degradation of occludin and to restore intestinal barrier function.
A novel bacterial protease activity cleaves E-cadherin, which leads to disruption of the integrity of the host epithelium
Furthermore, we highlight a novel bacterial protease activity, which is responsible for the cleavage of E-cadherin. Only live bacteria and to a lesser extent the SN, but not LPS, induced a decrease in cell junctional components. Thus, the respective protease activity must either be part of live bacteria or be secreted/released into the supernatant. Highest protease activity was detected in the patient-derived SBP isolates E. coli Ont:H7 and P. mirabilis. As the protease activity could be partially blocked by the MMP inhibitors BB-94 and BB-2516, we suggest that patient-derived E. coli strains express a protease targeting E-cadherin. We could verify a bacterial protease activity in different SBP-inducing E. coli strains and in one P. mirabilis strain. This may allow generalising our hypothesis that intestinal bacteria destabilise cellular junctions via induction of proteasomal degradation of occludin and bacterial protease-induced cleavage of E-cadherin.
Established for cancer therapy, protease inhibition has evolved from strategies targeting large spectrum proteases to strategies targeting specific proteases and to indications beyond cancer.47 Our data suggest that protease targets in SBP (analogous to the treatment options in inflammatory bowel disease) should include ubiquitin-proteasome system inhibitors.
Targeting bacterial proteases provides novel opportunities in anti-infective therapy. Resistance is of significant concern in patients with liver cirrhosis and SBP.50 There is an obvious need for more efficacious agents due to the failure of current antibiotics.51 We identified a novel protease activity, which constitutes a druggable target in SBP. We support the idea that next-generation anti-infective protease inhibitors will have wider clinical coverage compared with current ones that are mainly antiviral.
Acknowledgments
We thank our patients for kindly donating biopsies. We thank Peter Oefner (Institute of Functional Genomics, University Regensburg, Regensburg, Germany) and Franz Paul Armbruster (Immundiagnostik, Bensheim, Germany) for helpful scientific discussions. We thank Florian Zeman (Centre for Clinical Studies, University Hospital Regensburg, Regensburg, Germany) for his assistance with statistical analysis. We thank Susanne Modrow, Jonathan Jantsch, Michaela Simon and Patrick Neubert (Institute for Medical Microbiology and Hygiene, University of Regensburg, Regensburg, Germany) for constant support. We thank Angelika Fruth (Robert Koch Institute Berlin, Germany) for characterisation of patient-derived bacterial strains and constant support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MM initiated SBP project and designed the work; MM, MH, KG, LB and CK were responsible for experimental design; AK, VP and AM performed colonoscopies and collected human tissue samples; MH, PN, ER, AS and HG performed the majority of experiments; CB performed electron microscopy experiments; MH drafted manuscript, prepared figures; SS analysed the clinical data; MH, KG and MM wrote and edited the manuscript. All authors approved the final version of the manuscript.
Funding This research was supported by the Federal Ministry for Economic Affairs and Energy, Germany (Zentrales Innovationsprogramm Mittelstand, KF 2525606CS3).
Competing interests MM and CK report grants from Federal Ministry for Economic Affairs and Energy (Germany) during the conduct of the study.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.