Article Text

Statistics from Altmetric.com
Recently in Gut, several publications reported an increased prevalence of hypomorphic (defective) sucrase-isomaltase (SI) gene variants in patients with irritable bowel syndrome (IBS),1 2 and the association with impaired cell-surface expression and reduced digestive function of the corresponding enzyme.3 In addition, hypomorphic SI carriers have shown reduced response compared with non-carriers in a low-FODMAP (fermentable oligo- di- mono-saccharides and polyols) trial of IBS patients with diarrhoea.4 These and other studies,5 6 including recent large population-based surveys,7 raise the question whether genetic defects in the SI gene may be sought to explain abdominal symptoms in some patients with IBS.
A 23-year-old patient was referred with a diagnosis of IBS due to his long-standing postprandial diarrhoea associated with bloating, abdominal pain and nausea. He also had marked fatigue, headaches and mouth ulcers. He had symptoms since infancy without signs of malnutrition or failure-to-thrive. Extensive investigations excluded alternate causes of diarrhoea, including coeliac and inflammatory bowel disease, pancreatic exocrine insufficiency and bile acid diarrhoea. A low FODMAP diet worsened symptoms and psychological interventions were not helpful.
We hypothesised the involvement of SI defects. Resequencing of the gene identified three coding variants of interest (H1684Q, G1760V and V15F), which were assigned to a heterozygous compound combination as [H1684Q, G1760V] + [V15F] (figure 1A) based on sequencing data from additional family members (not shown). While [V15F] is known to have 35% reduced disaccharidase activity, an in vitro system was developed in order to study the [H1684Q, G1760V] variant by testing its disaccharidase activity in transfected COS cells, as previously described.1 3 This variant showed no expression at the cell surface (figure 1B), corresponding to only residual disaccharidase activity (25%) when coexpressed with [V15F], also defective, mimicking the patient’s heterozygous state (figure 1C).
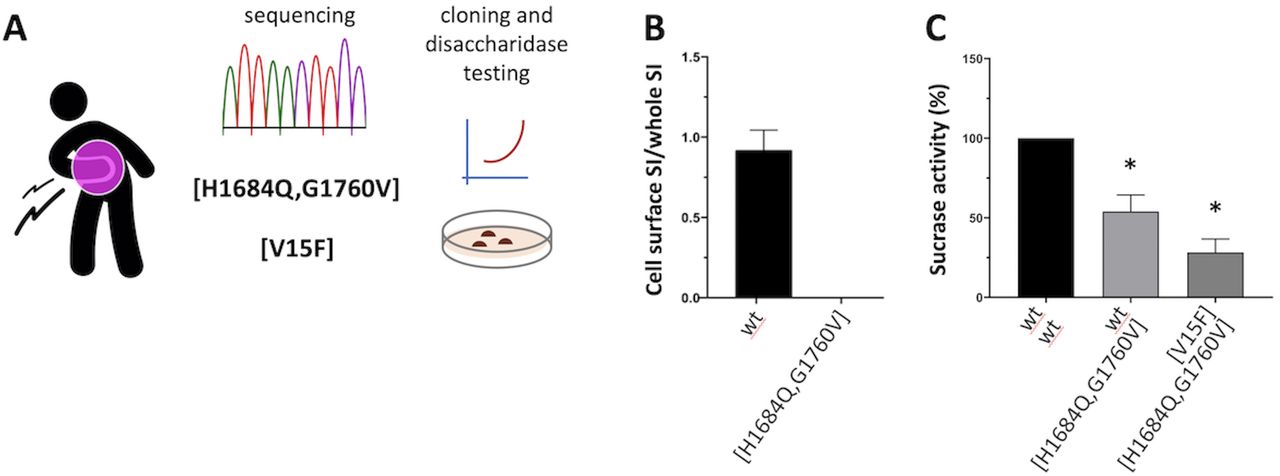
Patient’s SI genetic profiling and in vitro characterisation of the enzymatic activity of corresponding DNA variants. (A) The patient’s DNA was sequenced via clinical exome sequencing (Illumina) service at the Australian Genomic Research Facility (www.agrf.org.au), and confirmed via ad hoc resequencing of the SI gene (Illumina targeted assay) at IKMB in Kiel Germany; (B/C) COS-1 cells were transfected or cotransfected with cDNAs encoding SI variants of interest (wt, [H1684Q, G1760V] and [V15F]), and cell surface expression (B) and sucrase activity (C) determined relative to wt (set as reference 100%), upon immunoprecipitation and quantification/normalisation with appropriate antibodies as previously described.3 *Student t-test p<0.05. SI, sucrase-isomaltase.
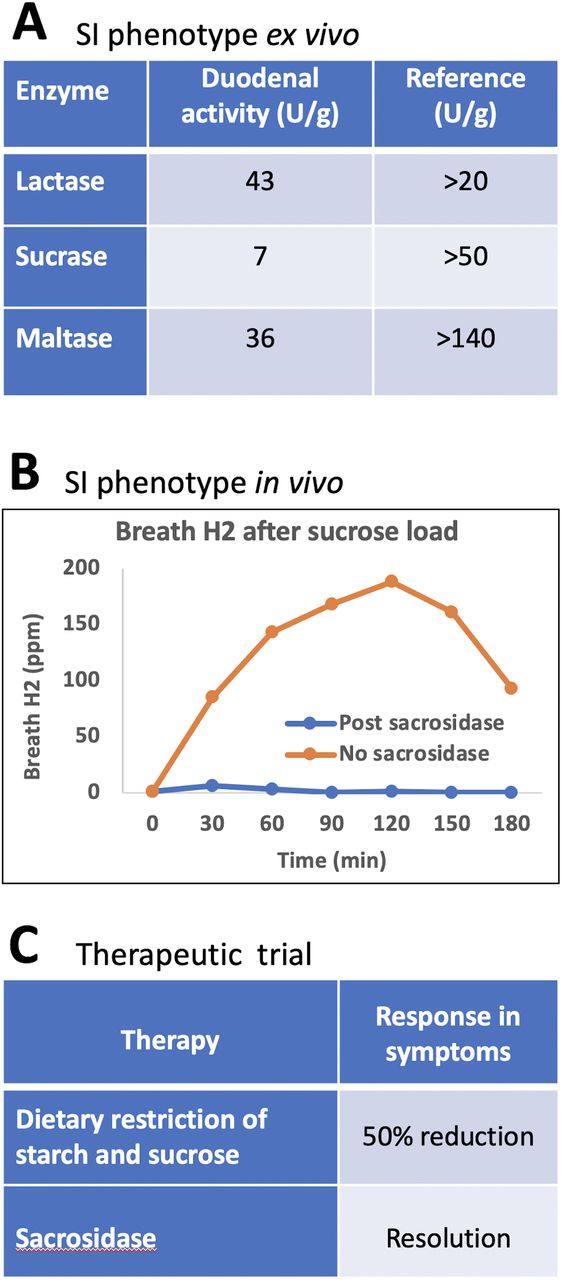
Follow-up clinical investigations of SI function showed marked impairment of sucrase and maltase activities in duodenal biopsies and, on breath hydrogen tests, a sustained hydrogen response to sucrose, which was abrogated in the presence of sacrosidase (figure 2). Formal dietary reduction of starches and sucrose was associated with a 50% symptom response, and a therapeutic trial of sacrosidase (Sucraid, QOL Medical, FL) led to complete resolution of symptoms (figure 2). Withholding then reinstituting sacrosidase verified response to it.
{kind=link}
{kind=link}
Characterisationof SI deficiency in the studied patient. (A) Enzymatic activity ex vivo in duodenal biopsies; (B) Sucrose breath hydrogen testing with and without sucrosidase; (C) Therapeutic trials with sucrose-reducing and starch-reducing diet, and sucrosidase. SI, sucrase-isomaltase.
These results indicate that our patient has congenital SI deficiency (CSID), considered a rare autosomal recessive condition most commonly identified in infants with symptoms of diarrhoea, malabsorption and failure-to-thrive. While a few CSID cases have also been described in adults,8 there is accumulating evidence that partial SI deficiency (possibly as in hypomorphic carriers) is associated with increased risk of IBS in the general population. Hence, a clinical continuum across sucrose and starch malabsorption may be envisaged, which spans a spectrum of functionally diverse DNA variations in the SI gene. These include homozygous and heterozygous combinations of variously defective SI variants, resulting in a genotype-mediated gradient of disease risk ranging from mild(er) IBS to severe CSID.
Thus, in a small fraction of patients with IBS, symptoms might be wholly or partly due to SI dysfunction associated with hypomorphic variants of the SI gene. Symptoms that raise the possibility of a defective SI gene may include onset in childhood, predominantly postprandial timing and a poor response to FODMAP restriction, which does not reduce sucrose and only partially reduces starch content. If a role for SI deficiency is suspected, it should be pursued and, while the clinical interpretation of tissue hydrolase activities and hydrogen breath tests requires further elucidation (especially in milder SI defects), SI genotyping and/or sequencing represents a valuable contribution to clinical profiling. Such findings can lead to gratifying amelioration of symptoms, although specific enzyme therapy is currently limited by cost and availability.
Ethics statements
Patient consent for publication
Ethics approval
Case report, formal ethics approval not needed in accordance with local ethics department guidelines.
Footnotes
PRG and MD are joint senior authors.
Twitter @damato_mauro
Correction notice This article has been corrected since it published Online First. The author's name, Hassan Y Naim, has been updated.
Contributors AFo, EPH, SRF and PRG patient characterisation; DMH and HN in vitro experimentation and disaccharidase assays; B-SL, AFr and MD'A genome sequence analysis and interpretation; MD'A and PRG: study conception and supervision, project planning, data interpretation, manuscript drafting; AFo, EPH, DMH, SRF, B-SL, AFr, MD'A and PRG critical revision of the manuscript.
Funding The study was supported by grants from the Spanish Ministry of Science and Innovation (MICINN PID2020-113625RB) to MD'A, the German Research Foundation (DFG 331/13–1) to HN, and QOL Medical to MD'A and HN.
Competing interests MD'A and HN have received financial support from QOL Medical, in the form of unrestricted research grants; PRG, EPH: Monash University financially benefits from the sales of a digital application, booklets and online courses on the FODMAP diet; PRG has published two educational/recipe books on diet; other authors have nothing to declare.
Provenance and peer review Not commissioned; internally peer reviewed.