Article Text

Abstract
Objective Idiopathic Parkinson’s disease (PD) is characterised by alpha-synuclein (aSyn) aggregation and death of dopaminergic neurons in the midbrain. Recent evidence posits that PD may initiate in the gut by microbes or their toxins that promote chronic gut inflammation that will ultimately impact the brain. In this work, we sought to demonstrate that the effects of the microbial toxin β-N-methylamino-L-alanine (BMAA) in the gut may trigger some PD cases, which is especially worrying as this toxin is present in certain foods but not routinely monitored by public health authorities.
Design To test the hypothesis, we treated wild-type mice, primary neuronal cultures, cell lines and isolated mitochondria with BMAA, and analysed its impact on gut microbiota composition, barrier permeability, inflammation and aSyn aggregation as well as in brain inflammation, dopaminergic neuronal loss and motor behaviour. To further examine the key role of mitochondria, we also determined the specific effects of BMAA on mitochondrial function and on inflammasome activation.
Results BMAA induced extensive depletion of segmented filamentous bacteria (SFB) that regulate gut immunity, thus triggering gut dysbiosis, immune cell migration, increased intestinal inflammation, loss of barrier integrity and caudo-rostral progression of aSyn. Additionally, BMAA induced in vitro and in vivo mitochondrial dysfunction with cardiolipin exposure and consequent activation of neuronal innate immunity. These events primed neuroinflammation, dopaminergic neuronal loss and motor deficits.
Conclusion Taken together, our results demonstrate that chronic exposure to dietary BMAA can trigger a chain of events that recapitulate the evolution of the PD pathology from the gut to the brain, which is consistent with ‘gut-first’ PD.
- intestinal barrier function
- gut inflammation
- intestinal bacteria
- neurobiology
Data availability statement
Data are available upon reasonable request. The data that support the findings of this study are available from the corresponding author upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Parkinson’s disease (PD) is a multifactorial disease characterised by a long prodromal phase that in at least some cases includes gastrointestinal symptoms.
A direct correlation between gut dysbiosis and disease progression was found in PD patients.
Chronic dietary exposure to BMAA is believed to be the cause of amyotrophic lateral sclerosis/parkinsonism-dementia complex in specific populations.
BMAA targets the cysteine-glutamate antiporter encoded by SLC7A11 (Solute Carrier Family 7 member 11), a gene whose promoter hypermethylation in PD was associated to downregulation of antiporter expression consistent with an environmental exposure linked to PD risk.
What are the new findings?
In vivo BMAA administration depleted ileum levels of ‘Candidatus Arthromitus’, a group of bacteria that regulate gut mucosal immune homeostasis in mice leading to an increase in gut inflammation, disruption of gut barrier integrity and gut aSyn aggregation.
BMAA specifically targeted mesencephalic mitochondria inducing their fragmentation and cardiolipin exposure, which in turn activated innate immune responses such as NOD-like receptor 3 activation and aSyn aggregation.
Gut inflammation potentiated blood-brain barrier permeability and neuroinflammation, which culminated in nigrostriatal neuronal damage and PD-like motor dysfunction.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
This work generated cutting edge knowledge by establishing a direct link between chronic dietary consumption of an environmental microbial toxin and PD features. The specific antibiotic effect of foodborne BMAA identified here and the confirmed role of the dysbiotic gut as a trigger for the cascade of events that culminate in the brain, will have decisive public health implications regarding the revision of food safety guidelines to monitor and control this food toxin. This gives rise to an unprecedented opportunity for new bidirectional synergies between gastroenterology and neurology disciplines, which opens up new innovative possibilities for prevention or early diagnosis of PD.
Introduction
Idiopathic Parkinson’s disease (PD) is an age-related neurodegenerative disorder characterised by motor symptoms such as tremor, postural imbalance, bradykinesia and rigidity.1 2 Preceding non-motor features also include loss of smell and gastrointestinal (GI) dysfunction.3 Constipation, appearing during the prodromal phase, is perceived as a risk factor for developing PD.3 The histopathological postmortem hallmarks of PD are the presence of α-synuclein (aSyn)-containing insoluble fibrous aggregates, termed Lewy bodies and Lewy neurites as well as the loss of dopaminergic neurons in the substantia nigra pars compacta.1 Aggregated aSyn can also be found in the GI tract and in organs innervated by the vagus nerve4 5 years to decades before detectable involvement of the central nervous system (CNS). Those observations allowed the formulation of a hypothesis arguing that microbes or their products could travel from the gut to the brain via vagal retrograde axonal transport.6 This hypothesis found indirect support in data from humans who underwent complete truncal vagotomies and who appear to have a lower risk of developing PD.7 Additionally, certain microbial products occurring in a dysbiotic gut have been proposed to trigger aSyn overexpression and aggregation.8 ,9 Faecal and mucosa-associated gut microbiota differ between patients with PD and healthy controls,10 but functional interpretation is still controversial.11 Gut dysbiosis in patients with PD is correlated with intestinal inflammation, inferred from the upregulation of proinflammatory cytokines.12 13 In mice, intestinal homeostasis is maintained by tissue-resident T helper 17 (Th17) cells that differentiate in response to symbiont mucosa-associated segmented filamentous bacteria (SFB) ‘Candidatus Arthromitus’, which fulfil important roles in barrier integrity protection.14 15 SFB adherence to epithelial cells signals the release of serum amyloid A that acts on CD11c+ cells in the lamina propria to stimulate the production and release of interleukin (IL)-6 and IL-23 that stimulate differentiation and activation of Th17 cells. IL-17 released from tissue-resident homeostatic intestinal Th17 cells signals epithelial cells to produce antimicrobial peptides and tight-junction proteins.15 However, gut dysbiosis may also induce non-resident Th17 cells, either by erosion of protective microbes (mice SFB) or by the proliferation of pathobionts, driving the production of pro-inflammatory cytokines with peripherical effects.14 Recent data showed that inflammatory bowel disease patients may be at significantly higher risk of developing PD.16 Although PD has often been associated with a leaky intestinal barrier,17 18 the mechanisms through which gut dysbiosis disrupts barrier integrity are still incompletely understood. Accumulating evidence suggests that toxins produced by some gut microbes could impact host physiology.19 β-N-methylamino-L-alanine (BMAA), a natural non-proteinogenic diamino acid produced by cyanobacteria or other microbes and most often detected in aquatic food products, may be involved in neurodegeneration.19 BMAA was initially discovered in the seeds of cycad plant and its consumption has been proposed to cause amyotrophic lateral sclerosis/parkinsonism-dementia complex (ALS-PDC).20 Evidence suggests that BMAA can be misincorporated in proteins in place of serine, or bind electrostatically to protein nascent chains causing misfolding, aggregation, endoplasmic reticulum (ER) stress and apoptosis.19 Moreover, BMAA can induce mitochondrial dysfunction interfering with oxidative phosphorylation while also deregulating calcium homoeostasis and leading to reactive oxygen species (ROS) overproduction.21 Although there is still no evidence that members of the gut microbiota can produce BMAA, hypermethylation of cg06690548 on chromosome 4 in the promoter of SLC7A11 gene in PD patients is associated with downregulation of this cysteine–glutamate antiporter, a known target of BMAA,22 which is argued to be consistent with a PD-related environmental exposure. In fact, chronic exposure to environmental toxins that may lead to mitochondrial dysfunction has been advanced as a possible cause for PD in genetically susceptible individuals.23 Building on the mitochondrial cascade hypothesis for PD,23 we theorised that specific microbial molecules could travel from the gut to the brain and target neuronal mitochondria,24 thus generating a self-amplified harmful cycle primed by neuronal innate immune activation. Accordingly, in vivo BMAA administration-induced strong expression of proinflammatory cytokines.25 Our work further revealed that chronic treatment of mice with BMAA significantly eroded the levels of SFB, microbiota abundant in the ileum mucosa, with consequent alteration of local immune cell responses that led to increasing gut inflammation and disruption of gut barrier integrity. Surprisingly, we also observed that mesencephalic mitochondria were dysfunctional and accumulated aSyn aggregates which in turn led to further activation of neuronal innate immunity and neuroinflammation. Finally, we demonstrated a caudo-rostral progression of the disease from the gut to the brain most likely through the vagus nerve, as evidenced by the aSyn trail that culminated in substantia nigra (SN) degeneration and motor impairment, all archetypal features of PD.
Methods
Animal model and experimental design
C57BL/6 male mice obtained from Charles River (Barcelona, Spain) were housed under a 12 hours light/dark cycle with free access to water and food. At 26 weeks mice were randomly divided in two groups (untreated (Unt) and BMAA-treated) and daily orally administered with BMAA (0.1g/kg bw) for 12 weeks in gelatin pellets, until they were sacrificed. This BMAA dose was selected according to previous studies.26 Untreated animals received vehicle. Behavioural analyses were performed (30–40 weeks), mice were sacrificed, and samples were collected for analysis, as described in detail in online supplemental material. All reagents used are listed in online supplemental table S1.
Supplemental material
Microbiome profiling
Faecal, ileum and cecum mucosa-associated material were collected to assess microbiome profiles using 16S rRNA gene sequencing as previously described27 28 and using the mothur package V.1.44.129 and Silva reference files (release 138)30 for treatment of raw data, clustering and taxonomic annotation. Data analyses were performed with the online tool MicrobiomeAnalyst31 and its R package DESeq232 software as described in online supplemental material.
Behavioural analyses
Mice behavioural analyses were assessed as described before for beam walking,33 hindlimb clasping,34 35 inverted grid,9 36 open field33 and T-maze37 as described in online supplemental material.
Immunohistochemistry, immunofluorescence and microscopy analyses
At the end of the experiment, animals were transcardially perfused. Brain and ileum samples were collected, cryoprotected and stored at −80°C. Immunohistochemistry and immunofluorescence staining for brain and gut samples were performed as published38 and as described in detail in online supplemental material. In gut samples we assessed (a) the integrity of intestinal barrier by immunofluorescence, using a score system scale18 39; (b) The number of CD11b divided per the total counting area (mm2) and by immunohistochemistry (c) the number of CD4 cells divided per the total counting area (mm2); and (d) the optical density (OD) of aSyn aggregates and the presence of phosphorylated-aSyn (p-aSyn) inclusions. In brain samples, we used immunofluorescence to determine (a) the number of in Iba1+ cells in and Trem2 expression in SN and (b) the number of TH + and ChAT + cells in dorsal motor nucleus of the vagus (DMV) divided per the total counting area (mm2). We used immunohistochemistry to assess (c) the OD of aSyn aggregates in striatum (STR), SN and DMV, the presence of p-aSyn in DMV and SN and the OD of tyrosine hydroxylase (TH) in STR, (d) the stereological quantification of TH + cells in SN and (e) the number of IgG-immunopositive staining in the perivascular area per total area (mm2) in the cortex, STR and SN to assess the blood-brain barrier (BBB) integrity. All image acquisitions were performed under well-established blinded protocols and randomised processes, and with different technical and biological replicates, as comprehensively described in online supplemental material.
Flow cytometry
Blood was collected by cardiac puncture, transferred to Histopaque 1083 solution (Sigma) and centrifuged. The peripheral blood mononuclear cell (PBMC) fraction was isolated, washed and incubated with Anti-Mouse CD45 PerCP (Clone 30F11), Anti-Mouse CD3 FITC (Clone REA641), Anti-Mouse CD4 APC (Clone REA604) and Anti-Mouse CD8 PE (Clone REA601) (1/50) (Miltenyi Biotec). Cell suspension acquisition was performed in BD FACSCalibur cytometer (BD Bioscience) and analysed in FlowJo software (BD Bioscience) (see online supplemental material for comprehensive description).
Western blotting, spectrophotometry and ELISA determinations in brain, intestine homogenates and blood
After completing the behavioural tests, Unt and BMAA-treated mice were euthanised and blood, mesencephalon, striatum, ileum and cecum samples were isolated. Tissues were snap frozen, stored at −80°C and homogenised as described in online supplemental material. Plasma interferon (IFN)γ and IL-6 levels were determined. In mesencephalic homogenates we determined synaptic markers and innate immunity markers by western blot, caspase-1 activity by spectrophotometry and innate immunity markers and aSyn oligomers with ELISA kits and western blot. In striatal homogenates we determined dopamine levels with ELISA Kit. In ileum and cecum homogenates we determined caspase-1 activity by spectrophotometry, and innate immunity markers and aSyn oligomers with ELISA kits and by western blot (see online supplemental material).
Western blotting, spectrophotometry and ELISA determinations in cellular extracts
NT2 Rho+, Rho0 cells40 and primary mesencephalic neurons obtained from mesencephala of C57Bl/6 mice embryos brains at gestation day 14/15 were cultured as described previously with some modifications41 42 and treated with 3 mM BMAA for 48 hours with 1 µM CCCP (cells) or 5 µM (neurons) for 2 hours before cell harvesting or with 20 mM NH4Cl and/or 20 µM Leupeptin for 4 hours to the culture medium wherever indicated. Cells were washed in ice-cold phosphate-buffered saline (PBS 1×), scraped and homogenised as described in online supplemental material. In cellular extracts we determined aSyn oligomers, LC3II and innate immunity markers by western blot. In neuronal mitochondrial fractions we determined phospho-Drp1 levels and aSyn oligomers by western blot. In neuronal cytosolic fractions we analysed innate immunity markers with ELISA kits and Caspase-1 activation by spectrophotometry (see online supplemental material).
Immunocytochemistry and confocal microscopy analysis
Primary mesencephalic neurons, NT2 Rho+ and Rho0 cells were grown on glass coverslips (16 mm diameter) in 12-well plates or in ibidi μ-Slide 8-well plates. Following treatments, we determined mitochondrial movements with MitoTracker Green in neurons, mitochondrial membrane potential with TMRM Probe in both cells and neurons and cardiolipin distribution and fluorescence using the 10-N-Nonyl acridine orange in neurons. Immunocytochemistry and confocal microscopy procedures are described in online supplemental material.
Mitochondria isolation, oxygen consumption rate, Ca2+ handling capacity and glycolytic fluxes (ECAR) analyses
Mouse mesencephalic and cortical mitochondria were isolated by Percoll gradient (see online supplemental material). Oxygen consumption rate (OCR) was measured in fresh mesencephalic or cortical mitochondria with a Seahorse XF24 Extracellular Flux analyser (Seahorse Bioscience, Billerica, Massachusetts, USA). For the respiratory coupling experiments, the following determinations were calculated according to the ensuing ‘rate measurement equation’ (see online supplemental material).43 Mitochondrial Ca2+ uptake was measured fluorometrically in the presence of the Ca2+-sensitive fluorescent dye Calcium Green 5N (150 nM), using excitation and emission wavelengths of 506 nm and 532 nm, respectively.44 In primary mesencephalic neurons the OCR and ECAR were determined using a Seahorse XF24 Extracellular Flux analyser (Seahorse Bioscience). The experimental set-up is described in online supplemental material.
Statistical analyses
Microbiome population statistics are described in the detailed online supplemental materials and methods. Statistical analyses of data sets were performed using GraphPad Prism V.8 (GraphPad Software) software as summarised in online supplemental table 2. All data are represented as the mean±SEM. Normality distribution analysis (Shapiro-Wilk test) was applied to determine the subsequent parametric or non-parametric tests. Pair-wise comparisons were performed by unpaired Student’s t test or Mann-Whitney test. Comparisons of multiple groups were performed with one-way analysis of variance followed by Dunnett’s post-hoc test or Kruskal-Wallis test followed by Dunn’s post-hoc test. Correlation analysis between two variables was performed by Pearson’s correlation test. All statistical tests were two-tailed and the annotation for significance values was: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. P and N values are indicated at each figure legend.
Results
Gut microbiota alterations in mice treated orally with BMAA
BMAA is a microbial neurotoxin that can be ingested through the diet due to its accumulation in the food chain, especially from aquatic ecosystems.19 We have orally administered BMAA for 12 weeks to wild-type (WT) C57BL/6 male mice and observed no alterations in their weight or glycaemia (online supplemental figure S1A-C) although a significant increase in stool water content was observed (data not shown). After treatment, faecal material was collected, and mice were sacrificed to isolate the ileum and the cecum. A comprehensive analysis of the ileum-associated (figure 1), faecal (figure 2) and cecum-associated bacteria (online supplemental figure S2) was performed using Illumina MiSeq technology at our Next Generation Sequencing Unit Genoinseq (https://www.cnc.uc.pt/en/services). We obtained on average 1876±1650 reads per ileum sample, and detected 102 genera, 59 families, 40 orders, 14 classes and 10 bacterial phyla. From the stools we obtained 52 952±12 996 reads per sample and identified 165 genera, 82 families, 56 orders, 18 classes and 11 phyla. From the cecum samples we obtained on average 31 001±7140 reads per sample and detected 121 genera, 62 families, 39 orders, 15 classes and 11 phyla. We observed an increase in the diversity of ileum-associated bacteria in BMAA-treated mice (alpha-diversity, which measures within-sample taxonomic diversity; figure 1A) most likely due to the loss of the most representative SFB members, although the overall community diversity was similar between environments (beta-diversity, which evaluates the similarity or dissimilarity between two communities; figure 1B). Sequences of five genera were significantly altered in the ileum-associated mucosa of BMAA-treated mice with the most significant decreases of ‘Candidatus Arthromitus’ (approximately 17-fold decrease) and Turicibacter (approximately 13-fold decrease) and increases of Colidextribacter (approximately 6-fold increase), Bacteroides (approximately 7-fold increase) and Lachnospiraceae_NK4A136 (approximately 11-fold increase) (figure 1C–I). Most microbiome studies have been focused on the transient stools’ microbiota, which may not translate the real impact of different disease states. In the faecal material, no significant changes were detected in the richness of microbiota between BMAA-treated and untreated mice at any taxonomic level (alpha diversity; figure 2A) and some overlap in the microbial composition of both communities was observed (beta diversity; figure 2B). We found that 11 genera were significantly altered in faecal samples of BMAA-treated mice: Olsenella, Mucispirillum, Eggerthellaceae_unclassified and Roseburia were enriched while RF39_ge, Monoglobus, Family_XIII_AD3011_group, Akkermansia, Tyzzerella, Turicibacter and Clostridia_UCG_014_ge were depleted (figure 2C–I). Although Roseburia (figure 2G) have been associated with short chain fatty acids (SCFAs) production and were found to be depleted in patients with PD,45 they were enriched in the stools of BMAA-treated mice. On the other hand, the abundance of Akkermansia known to improve gut barrier function by modulating the mucus layer with beneficial impact in the immune response, was lower in BMAA-treated mice stools (figure 2H).45 Interestingly, BMAA treatment elicited significant depletion of members of the Turicibacter and of members of the cryptic Clostridia_UCG_014_ge (figure 2F,I). Our results link the erosion of the ileum-associated Turicibacter with that observed in the transient faecal microbiota and in the cecum microbiota, the latter showing overall decreased diversity and a distinct microbial community composition (online supplemental figure S2A,B). As observed in the faecal material, the abundances of Turicibacter and Clostridia_UCG_014_ge were also depleted in the cecum after BMAA treatment (online supplemental figure S2C-F, I). Bacteroides were enriched in both the ileum and cecum of BMAA-treated mice (figure 1H, online supplemental figure S2C-E, G). Of notice was the near disappearance of cecum Bifidobacterium levels (online supplemental figure S2H), whose members are thought to promote health and restore mucus growth via SCFAs production.
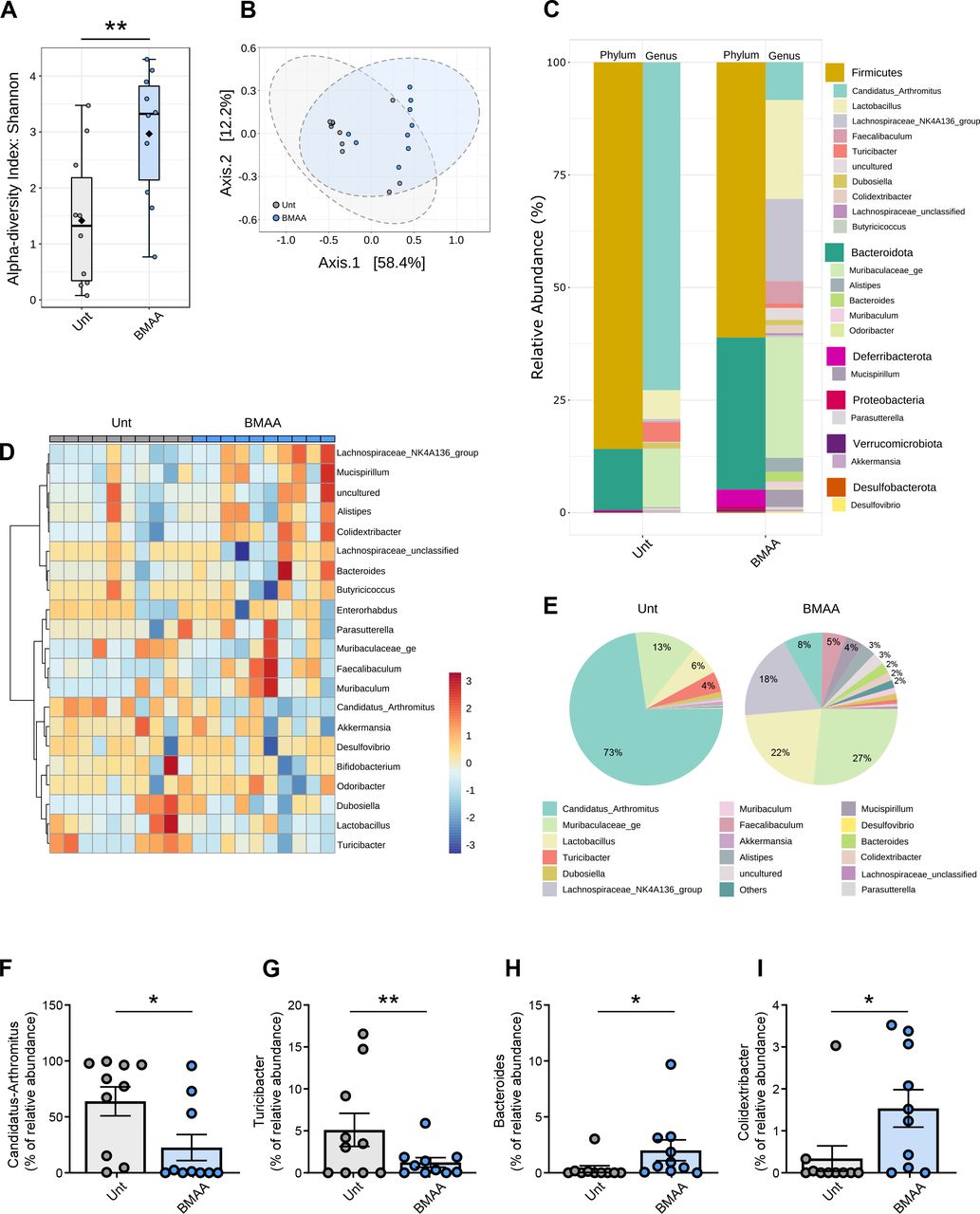
Ileum mucosa-associated microbiome diversity in β-N-methylamino-L-alanine (BMAA)-treated mice. (A) Alpha diversity measured using the Shannon index at operational taxonomic unit (OTU) level derived from 16S rDNA sequencing of ileum intestinal samples from untreated (Unt) or BMAA-treated mice (n values for Unt=10 and BMAA=10, Unt vs BMAA, Mann-Whitney test, **p=0.0089). (B) Beta diversity evaluated by principal coordinate analysis (PCoA) based on Bray-Curtis index of OTUs derived from 16S rDNA sequencing of ileum intestinal samples from Unt or BMAA-treated mice (n values for Unt=10 and BMAA=10; PERMANOVA: r2=0.205, *p<0.015; PERMDISP: F=0.773, p=0.391). (C) Taxonomic diversity of ileum intestinal samples from Unt or BMAA-treated mice at the phylum and genus levels. (D) Heatmap of the relative abundances of genera detected in ileum intestinal samples from Unt or BMAA-treated mice using Pearson’s correlation coefficient as a distance metric, with clustering based on Ward’s algorithm. (E) Pie-charts showing the proportional taxonomic composition at the genus level of ileum intestinal microbiota samples from Unt or BMAA-treated mice. (F–I) Differential abundance of selected genera in ileum intestinal samples from Unt or BMAA-treated mice (n values for Unt=10 and BMAA=10, Unt vs BMAA, DESeq2 statistical analysis). (F) ‘Candidatus Arthromitus’ (*padj=0.0296). (G) Turicibacter (**padj=0.0027). (H) Bacteroides (*padj=0.0150). (I) Colidextribacter (*padj=0.0103).

Microbiome diversity in the stools of β-N-methylamino-L-alanine (BMAA)-treated mice. (A) Alpha diversity measured using the Shannon index at operational taxonomic unit (OTU) level derived from 16S rDNA sequences obtained from faecal samples from untreated (Unt) or BMAA-treated mice (n values for Unt=17 and BMAA=16, Unt versus BMAA, Mann-Whitney test, p=0.136). (B) Beta diversity evaluated by principal coordinate analysis (PCoA) based on Bray-Curtis index of OTUs derived from 16S rDNA sequencing of faecal samples from Unt or BMAA-treated mice (n values for Unt=16 and BMAA=16; PERMANOVA: r2=0.108, ***p<0.001; PERMDISP: F=2.378, p=0.134). (C) Taxonomic diversity of faecal microbiota from Unt or BMAA-treated mice at the phylum and genus levels. (D) Heatmap of relative abundances of the genera detected in the faeces from Unt or BMAA-treated mice using Pearson’s correlation coefficient as a distance metric, with clustering based on Ward’s algorithm. (E) Pie-charts showing the proportional taxonomic composition at the genus level of faecal microbiota samples from Unt or BMAA-treated mice for two selected class level taxa affected by BMAA treatment, Bacilli and Coriobacteria. (F–I) Differential abundance of selected genera in faecal samples from Unt or BMAA-treated mice (n values for Unt=17 and BMAA=16, Unt vs BMAA, DESeq2 statistical analysis). (F) Turicibacter (*padj=0.0169). (G) Roseburia (*padj=0.0242). (H) Akkermansia (**padj=0.0078). (I) Clostridia_UCG_014_ge (*padj=0.0190).
BMAA-induced gut dysbiosis impairs GI function and exacerbates inflammation
BMAA-induced erosion of ‘Candidatus Arthromitus’ from the ileum mucosa (figure 1E–F) was accompanied by an increase of taxa that may signal intestinal epithelial cells and CD11b+ positive macrophages or dendritic cells to promote tissue inflammation, which we also found to be increased in the ileum of BMAA-treated mice (figure 3A–B). Activation of resident immune cells or amplified diffusion of noxious molecules by pathobionts proliferating in the ileum mucosa indeed induced inflammation, as inferred from the increased levels of proinflammatory mediators such as tumour necrosis factor (TNF)-α (figure 3C), IL-8 (figure 3D) and IL-17 (figure 3E) and from the lower levels of IL-10, an anti-inflammatory cytokine (figure 3F). Ileum SFB are known to actively stimulate gut immunity and induce homeostatic Th17 cell response and promote intestinal barrier integrity.15 Our results indicate that the increase in IL-17 levels were not due to SFB-induced tissue-resident homeostatic Th17 cells, but instead to the stimulation of inflammatory Th17 cells.14 The loss of SFB in BMAA-treated mice may have hypothetically allowed overgrowth of pathobionts able to stimulate inflammatory Th17 cells. Under these inflammatory conditions NOD-like receptor (NLRP) 3 inflammasome is activated due to the translocation of nuclear factor kappa-B (NF-kB) p65 into the nucleus (figure 3G) allowing the activation of caspase 1 and the production of IL-1β (figure 3H–I). This seems to be a specific effect of ‘Candidatus Arthromitus’ depletion in the ileum since BMAA effects in the cecum are not consistent with a major inflammatory profile (online supplemental figure S3A–D). Increased levels of TNF-α and IL-1β are known to enhance intestinal permeability due to the internalisation of tight junction proteins, namely ZO-1 and occludin.46 We observed a significant decrease in ZO-1 (figure 3J,K) and occludin (figure 3J,L) levels in the ileum of BMAA-treated mice, which indicates disruption of intestinal barrier integrity. Since infiltrated T cells are mostly CD4-expressing ‘helper’ T cells, we tackled the levels of CD4+ cells in ileum lamina propria of BMAA-treated mice but did not detect relevant differences (figure 3M,N). Nevertheless, we observed an alteration in CD4/CD8 ratios in the blood of BMAA-treated mice (figure 3O,P), due to a decrease in CD4 cells (online supplemental figure S3E,F), which might indicate systemic inflammation and concomitantly increased permeabilisation of the BBB. Indeed, we observed an increase in IFNγ and IL-6 levels in the plasma of BMAA-treated mice (figure 3Q), similarly to what is observed in patients with PD47 and that may impact the BBB integrity.48 We observed a positive microvascular leak in SN in BMAA-treated mice (figure 3R,S), but no alterations in the striatum or cortex (online supplemental figure S3G–J). Additionally, we did not observe CD4+ cellular infiltration in SN at this time point, despite the increased BBB permeability (online supplemental figure S3K). Noteworthy, the cortex and striatum have increased permeability to blood-borne components compared with midbrain areas,49 leaving the question of why we see BBB permeability alteration in the SN, but not in the cortex or striatum unanswered.
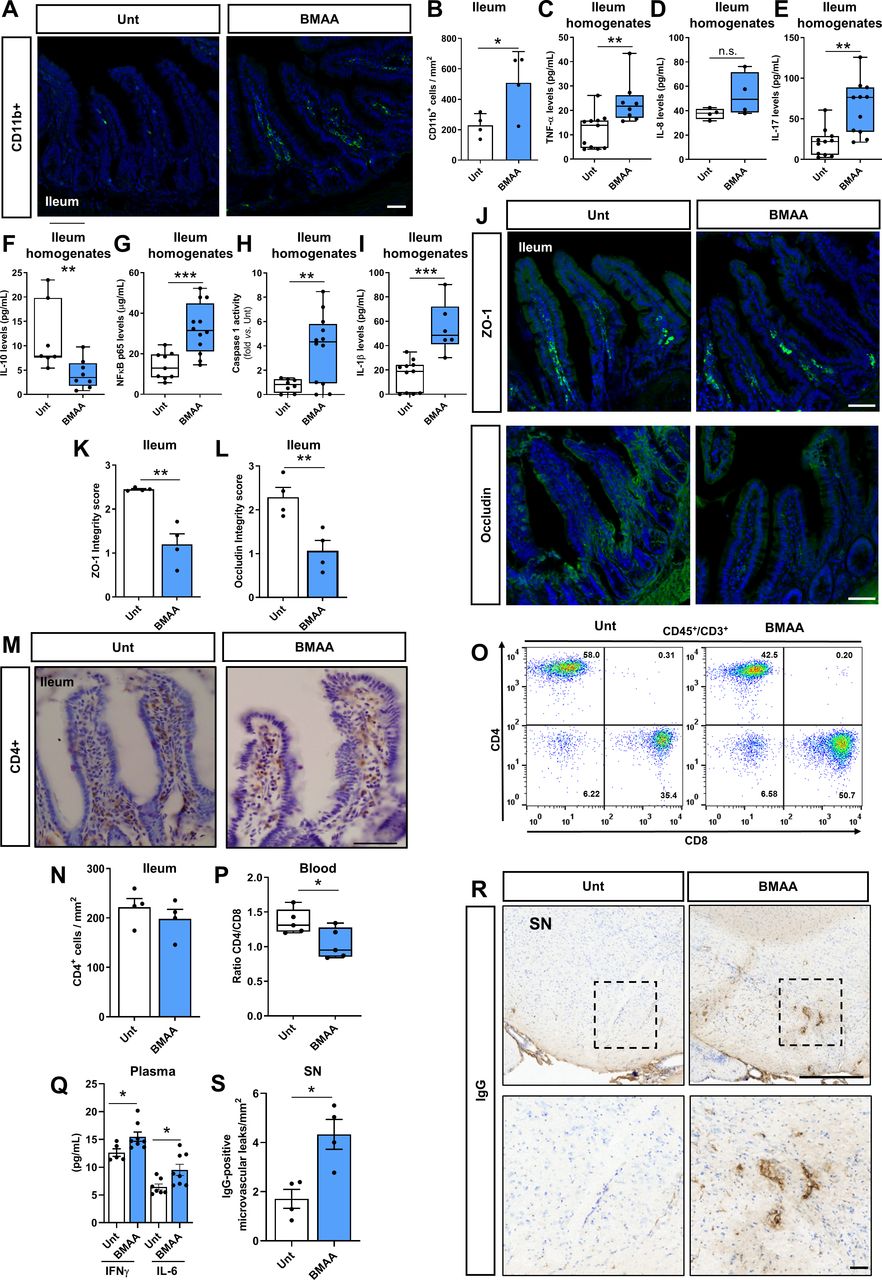
β-N-methylamino-L-alanine (BMAA) induces ileum inflammation and substantia nigra (SN) microvascular leaks. (A) Representative images of transversal ileum sections stained with anti-CD11b in untreated (Unt) and BMAA-treated mice. (B) Quantification of CD11b+ cells per mm2 in the ileum (n values for all conditions=4, Unt vs BMAA, *p=0.0435). (C–I) Measurement of specific mediators involved in the inflammatory response in ileum intestinal samples from Unt or BMAA-treated mice. (C) Tumour necrosis factor measured by ELISA (n values for Unt=11 and BMAA=8, Unt vs BMAA, **p=0.0036), (D) Interleukin (IL)-8 measured by ELISA (n values for Unt=4 and BMAA=4, Unt vs BMAA p=0.135), (E) IL-17 measured by ELISA (n values for Unt=11 and BMAA=11, Unt vs BMAA, **p=0.0011), (F) IL-10 measured by ELISA (n values for Unt=7 and BMAA=8, Unt vs BMAA, **p=0.009), (G) NF-κB measured by ELISA (n values for Unt=9 and BMAA=12, Unt vs BMAA, ***p=0.0008), (H) Caspase-1 activity. Values were subtracted to Unt (n values for Unt=8 and BMAA=12, Unt vs BMAA, **p=0.0068) and (I) IL-1β measured by ELISA (n values for Unt=7 and BMAA=6, Unt vs BMAA ***p=0.0002). (J–L) Assessment of intestinal barrier integrity. (J) Immunofluorescence staining for zonula occludens-1 (ZO-1) and occludin. (K) ZO-1 integrity score (n values for all conditions=4, Unt vs BMAA, **p=0.0019). (L) Occludin integrity score (n values for all conditions=4, Unt vs BMAA, **p=0.0095). (M) Representative images of transversal ileum sections stained with anti-CD4 in Unt and BMAA-treated mice. (N) Quantification of CD4+ cells per mm2 in ileum (n values for all conditions=4, Unt vs BMAA, p=0.411). (O–P), Determination of lymphocyte populations in the blood by flow cytometry. (O) Representative plots of lymphocytes (CD45+CD3+) CD4 and CD8 populations in Unt and BMAA-treated mice. (P) Quantification of the CD4/CD8 ratio (n values for all conditions=5, Unt vs BMAA, *p=0.036). (Q) Interferon (IFN)γ and IL-6 levels in the plasma were measured by ELISA (n values for Unt=5–7 and BMAA=8, Unt vs BMAA *p=0.0346 for IFNγ and *p=0.0238 for IL-6). (R–S) Assessment of IgG-positive microvascular leaks in the SN of Unt and BMAA-treated mice. (R) Representative images of SN coronal sections stained with IgG. (S) Quantification of IgG-positive microvascular leaks per mm2 in SN (n values for all conditions=4, Unt vs BMAA, *p=0.0108). Scale bars are 50 µm except in R (upper box)=500 µm. Data represent mean+SEM. Statistical analysis: Unpaired Student’s t-test was performed in B, D–E, G–I, K–L, N, P–Q and S. Mann-Whitney test was performed in C and F.
BMAA targets mesencephalic mitochondria
Our findings support the notion that BMAA, a non-proteinogenic amino acid of microbial origin, may target their ancient relatives, the mitochondria. After BMAA treatment we isolated mice mesencephalic mitochondria and observed a reduction in mitochondrial function, namely a decrease in basal and maximal respiration and a decrease in ATP synthesis (figure 4A–D), which reduced mitochondrial pool calcium-buffering capacity (figure 4E–F). However, cortical mitochondria isolated from BMAA-treated mice were not affected (figure 4G–L). To investigate if BMAA contributes to neurodegeneration by specifically targeting neuronal mitochondria, we performed in vitro experiments using pure isolated mitochondria, to find that acute BMAA administration decreased oxidative phosphorylation both in isolated mesencephalic mitochondria (online supplemental figure S4A–G) and in cortical mitochondria of WT mice.21 Our results indeed confirm that both brain areas are differentially affected, not due to differences in the mitochondrial pools between mesencephalon and cortex, but as a consequence of blood-borne translocation or vagal rostral trajectory of BMAA or of other unknown effectors that could not reach cortical areas at the time point defined in this work. Additionally, we also determined OCR in primary mesencephalic neurons exposed to BMAA and observed a decrease in basal mitochondrial respiration and ATP synthesis, similar to those elicited by CCCP (online supplemental figure S4H–L). Moreover, we observed a BMAA-induced decrease in glycolysis, glycolytic capacity rate and spare glycolytic capacity in primary mesencephalic neurons, effects comparable to those caused by CCCP (online supplemental figure S4M–P). Accordingly, and due to the observed mitochondrial dysfunction in BMAA-treated primary mesencephalic neurons, we detected a decrease in the mitochondrial membrane potential (online supplemental figure S5A). Interestingly, BMAA did not further decrease the mitochondrial membrane potential in Rho0 cells (mitochondrial-deficient cells that lack mitochondrial DNA) (online supplemental figure S5B). While a dysfunctional mitochondrial pool needs fragmentation to be degraded by mitophagy and avoid mitochondrial-mediated death,50 we found that BMAA increased the levels of the fission protein phosphoDrp1 in the mitochondria of primary mesencephalic neurons (figure 4M,N) thus leading to a fragmented mitochondrial network (figure 4O–S). Remarkably, the toxin was unable to further fragment the mitochondrial pool in cells devoid of functional mitochondria (online supplemental figure S5C–E). A decreased mitochondrial function with increased fragmentation leads to the exposure of cardiolipin, a lipid occurring both in the mitochondrial inner membrane and in bacteria.51 Indeed, we confirmed that treatment of primary mesencephalic neurons with BMAA triggered cardiolipin exposure (figure 4T,U). To overcome the exposure of this danger-associated molecular pattern (DAMP), dysfunctional and fragmented mitochondria are expected to be removed by mitophagy.52 We examined mitochondrial movement and macroautophagy in primary mesencephalic neurons and observed a reduction in mitochondrial average velocity induced by BMAA (online supplemental figure S6A,B). Moreover, we also observed a decrease in the autophagic flux in primary mesencephalic neurons treated with BMAA (online supplemental figure S6C–E), which indicates a decreased turnover of dysfunctional mitochondria. We further showed that mitochondria were located in autophagosomes (online supplemental figure S6F–H) but not co-localised with autolysosomes (online supplemental figure S6I–K), which points to an accumulation of dysfunctional mitochondria in BMAA-treated primary mesencephalic neurons with the undesirable overexposure of cardiolipin.

β-N-methylamino-L-alanine (BMAA) induces neuronal mitochondrial dysfunction, fragmentation and cardiolipin exposure. Isolated mesencephalic mitochondria from untreated (Unt) and BMAA-treated mice were examined. (A) Representative graph showing oxygen consumption rate (OCR); (B) basal respiration; (C) ATP synthesis; (D) maximal respiration. Values are pmol O2/min/μg protein. (B–D, n values for all conditions=7, Unt vs BMAA, B, *p=0.0125, C *p=0.0286 and D, *p=0.020). (E–F) Mesencephalic mitochondria’s ability to uptake calcium was evaluated with the fluorescent probe Calcium-green (n values for all conditions=4, Unt vs BMAA, ****p<0.0001). Isolated cortical mitochondria from Unt and BMAA-treated mice were examined. (G) Representative graph showing OCR in the cortex; (H) basal respiration; (I) ATP synthesis; (J) maximal respiration. Values are pmol O2/min/μg protein. (H–J) n values for all conditions=4, Unt vs BMAA; H, p=0.4456; I, p=0.1246 and J, p=0.9689). (K–L) Cortical mitochondria’s ability to uptake calcium was evaluated with the fluorescent probe calcium-green (n values for all conditions=4, Unt vs BMAA, p=0.1870). (M–U) Primary mesencephalic neuronal cultures were treated with 3 mM BMAA and 1 µM CCCP for 2 hours. (M) Representative immunoblot for phospho-Drp1 levels and (N) respective densitometric analysis. The blots were re-probed for TOM20 to confirm equal protein loading and mitochondrial fraction purity (n values for Unt and CCCP=4, BMAA=3. Unt vs CCCP, p=0.0711; Unt vs BMAA, *p=0.0375). (O) Primary mesencephalic neurons were immunostained with Tom20. (P–S) Alterations in mitochondrial network were calculated with an ImageJ Macro tool (n values for all conditions=6). (P) Number of mitochondrial networks (Unt vs CCCp, p=0.2674; Unt vs BMAA, p=0.3169). (Q) Number of mitochondrial branches (Unt vs CCCP, ****p<0.0001; Unt vs BMAA, ***p=0.0002). (R) Mitochondrial footprints (Unt vs CCCP, *p=0.0247; Unt vs BMAA, *p=0.0241). (S) Number of mitochondrial individuals (Unt vs CCCP, **p=0.026; Unt vs BMAA, p=0.0968). (T) Representative images of cardiolipin exposure which was visualised with the fluorescent dye 10-N-nonyl acridine orange. (U) Cardiolipin fluorescence was calculated with ImageJ. Data is reported as absolute values (n values for all conditions=6, Unt vs CCCP, p=0.676; Unt vs BMAA, *p=0.0255). Scale bars in O=50 µm and T=33 µm. Data represents mean+SEM. Statistical analysis: Unpaired Student’s t-test was performed in B–E, H–J and L. One-way analysis of variance followed by Dunnett’s test was performed in N, Q–S and U. Kruskal-Wallis test followed by Dunn’s test was performed in P.
Mitochondrial exposure of cardiolipin drives innate immunity activation and neuroinflammation
Dysfunctional mitochondria exposing cardiolipin, a recognised DAMP,52 activate innate immune responses in a self-amplified loop that culminates in progressive neurodegeneration. To tackle neuronal contribution to innate immunity activation we confirmed a low level of glial cell contamination in primary mesencephalic neuronal cultures (less than 1% of Iba1+, Trem2+, CD11b+ cells and less than 20% of glial fibrillary acidic protein, GFAP-positive cells) (data not shown), which led us to propose that neurons can mount an innate immune response mediated by BMAA-induced mitochondrial dysfunction. We observed that BMAA induced an increased expression of toll-like receptor (TLR) 4 in primary mesencephalic neurons (figure 5A–B). TLR signalling leads to NF-κB activation and translocation into the nucleus, where it binds to the promotor region of IL-1β gene inducing its transcription.53 We also observed NF-κB activation after BMAA treatment (figure 5C) and the expected increase in pro-IL-1β levels (figure 5A,D). Caspase 1 that cleaves pro-IL-1β was also found to be activated in neurons after BMAA treatment (figure 5E), leading to an increase in mature IL-1β levels (figure 5F).

β-N-methylamino-L-alanine (BMAA) activates brain neuronal innate immunity in vitro and in vivo. Primary mesencephalic neuronal cultures from naive mice were treated with 1 µM CCCP for 2 hours and 3 mM BMAA for 48 hours. (A) Representative immunoblot for toll-like receptor (TLR)4 and pro-interleukin (IL)-1β levels. Blots were re-probed for βIII-Tubulin to confirm equal protein loading. (B) Densitometric analysis of the levels of TLR4 was normalised with βIII-Tubulin (n values for all conditions=4, untreated (Unt) vs CCCP, p=0.076; Unt vs BMAA, **p=0.0023). (C) Nuclear factor kappa-B (NF-κB) levels were calculated using NF-κB p65 ELISA kit. Values are μg/mL (n values for all conditions=7, Unt vs CCCP, *p=0.0039, Unt vs BMAA, *p=0.013). (D) Densitometric analyses of the levels of pro-IL-1β were normalised with βIII-Tubulin (n values for all conditions=5, except BMAA=4, Unt vs CCCP, p=0.1213; Unt vs BMAA, *p=0.044). (E) Caspase-1 activation (n values for all conditions=4, Unt vs CCCP, *p=0.047, Unt vs BMAA, **p=0.002). (F) IL-1β levels in the isolated cytosolic fraction was determined using an IL-1β ELISA kit. Values are pg/mL (n values for all conditions=5, Unt vs CCCP, p=0.5928; Unt vs BMAA, *p=0.018). Homogenates from the mesencephalon of mice treated with or without BMAA were examined. (G) Representative immunoblot for TLR7, TLR4 and pro-IL-1β levels. The blots were re-probed for βIII-tubulin to confirm equal protein loading. (H) Densitometric analyses of TLR7 levels normalised against βIII-tubulin (n values for Unt=7 and BMAA=6, Unt vs BMAA, *p=0.027). (I) Densitometric analyses of TLR4 levels normalised against βIII-tubulin (n values for Unt=6 and BMAA=4, Unt vs BMAA, **p=0.004). (J) NF-κB levels were calculated using NF-κB p65 ELISA kit. Values are μg/mL (n values for Unt=6 and BMAA=4, Unt vs BMAA, **p=0.002). (K) Densitometric analyses of pro-IL-1β levels normalised against βIII-tubulin (n values for Unt=6 and BMAA=3, Unt vs BMAA, **p=0.007). (L) Caspase-1 activation (n values for Unt=10 and BMAA=8, Unt vs BMAA, **p=0.003). (M) IL-1β levels were determined using an IL-1β ELISA kit. Values are pg/mL (n values for Unt=6 and BMAA=4, Unt vs BMAA, **p=0.003). (N) IL-17 levels were determined using an IL-17 ELISA kit. Values are pg/mL (n values for Unt=5 and BMAA=6, Unt vs BMAA, p=0.8029). (O) IL-10 levels were determined using an IL-10 ELISA kit. Values are pg/mL (n values for Unt=5 and BMAA=8, Unt vs BMAA, p=0.483). (P–R), Iba1 and Trem2 expression in SN from Unt and BMAA-treated mice by immunofluorescence. (P) Representative images of brain coronal sections stained with Iba1 (microglial and macrophage-specific calcium-binding protein), Trem2 (triggering receptor expressed on myeloid cells 2) and Hoechst 33 342 as nuclei marker in SN. Enlarged boxes show the area of Trem2 (white pixels) contained in Iba1 signal. (Q) Quantification of the number of Iba1+ cells per mm2 (n values for all conditions=4, Unt vs BMAA, *p=0.028). (R) Percentage of Trem2 area contained in Iba1 expression (n values for all conditions=4, Unt vs BMAA, ***p=0.0002). Scale bars are 50 µm. Data represent mean+SEM. Statistical analysis: One-way analysis of variance followed by Dunnett’s test was performed in B, D–E. Kruskal-Wallis test followed by Dunn’s test was performed in C and F. Unpaired Student’s t-test was performed in H–N and Q–R and Mann-Whitney test in O.
To demonstrate that BMAA treatment selectively induces neuroinflammation through mitochondria-dependent neuronal innate immunity activation, we isolated mice mesencephalon to tackle NLRP3 inflammasome activation and observed that BMAA induced an increased expression of TLR7 and TLR4 receptors in vivo (figure 5G–I). Both TLR7, located in endosomes, and TLR4 in the plasma membrane, are able to trigger innate immune responses in neurons.54 These receptors signal NF-κB (figure 5J), which will activate NLRP3 inflammasome. Indeed, we observed an increase in pro-IL-1β levels (figure 5G and K) and the activation of caspase 1 (figure 5L), which allowed the release of the pro-inflammatory IL-1β (figure 5M). Taken together, our results strongly suggest that orally-administered BMAA activated mice brain innate immunity probably as a result of mitochondrial exposure of DAMPs. While recent studies link the production of IL-17 in the brain with BBB leakage and inflammatory disease progression,55 our mouse model fails to reveal significant increase in mesencephalic levels of IL-17 (figure 5N) or statistically significant alterations in the levels of anti-inflammatory IL-10 (figure 5O). We observed the activation of microglia in the SN with enlarged cell bodies and processes visualised with Iba1 (figure 5P–Q), and detected increased localisation of Trem2-positive disease-associated microglia (figure 5P–R). Activated microglia also releases pro-inflammatory cytokines, namely TNF-α and IL-1β that may favour BBB permeabilisation and subsequent infiltration of peripheral leucocytes into the CNS.55 Nevertheless, at the selected time point we did not observe CD4+ cell infiltration in the SN of BMAA-treated mice (online supplemental figure S3K).
BMAA treatment elicits propagation of aSyn aggregates from the gut to the nigrostriatal regions of the brain likely via the vagus nerve
aSyn aggregates are major histopathological hallmarks of PD. Recent data from humans and PD mouse models suggest that the upregulation of aSyn expression and the subsequent oligomerisation and aggregation occurs initially in gut enteric neurons and later in SN dopaminergic neurons.3 5 Indeed, we observed aSyn aggregates and p-aSyn (S129P) in ileum samples of BMAA-treated mice (figure 6A–E). Ileum samples from BMAA-treated mice revealed increased levels of aSyn aggregates and/or oligomers by immunohistochemistry (figure 6A,B), ELISA (figure 6C) and western blot (figure 6D–E). These differences were significant in the ileum, but not in the cecum (online supplemental figure S7A). Furthermore, and in agreement with Braak’s hypothesis proposing that a pathogen or a toxin may travel from the gut to the brain via the vagus nerve,5 we could detect aSyn aggregates and p-aSyn (S129P) forms in the DMV (figure 6F–G), showing significant high levels of aSyn aggregates in BMAA-treated mice (figure 6G). Finally, we detected aSyn aggregates in the mesencephalon of BMAA-treated mice, but not p-aSyn (S129P) forms (figure 6H–L). Indeed, it was proposed that only after the accumulation of degradation-resistant aSyn aggregates we would see extensive phosphorylation in the aggregates.56 In agreement with Braak’s stages, it is possible that insufficient degradation-resistant aggregates are present to induce extensive phosphorylation in SN at the time point of our experiment. Accordingly, our data did not show an increase in aSyn aggregates immunoreactivity in the cortex of BMAA-treated mice (online supplemental figure S7B). Herein, we showed that BMAA promoted overexpression and oligomerisation of aSyn probably due to its effect on neuronal mitochondria, and essentially because BMAA promoted aSyn oligomerisation in primary mesencephalic neurons (figure 6M–O) and in Rho+ cells, but failed to increase aSyn aggregation in Rho0 cells (figure 6P–Q). Indeed, data from the literature indicate that different cellular and animal models of PD can be obtained by targeting mitochondrial function, as is the case of PD cybrids and of the MPTP-mice model,57 both developing PD-related neuropathological hallmarks including aSyn oligomerisation. In fact, we also observed aSyn oligomers’ accumulation in the mesencephalic mitochondrial fraction of BMAA-treated mice (online supplemental figure S7C,D).

β-N-methylamino-L-alanine (BMAA) induces caudo-rostral alpha-synuclein (aSyn) aggregation. (A) Photomicrographs represent histology for aSyn aggregates and phosphorylated-aSyn (p-aSyn) (S129P) immunoreactivity in the ileum from untreated (Unt) and BMAA-treated mice. (B) Quantitative analysis of optical density (OD) for aSyn aggregates immunoreactivity in myenteric plexuses. Data was normalised to Unt group (n values for all conditions=4, Unt vs BMAA, ****p<0.0001). (C) aSyn oligomers were assessed in ileum homogenates with an ELISA kit. Values are pg/mL (n values for all conditions=8, Unt vs BMAA, *p=0.0425). (D) Representative immunoblot showing aSyn monomer and oligomers in ileum homogenates. The blots were re-probed for βIII-Tubulin to confirm equal protein loading. (E) Densitometric analyses of the levels of aSyn normalised against βIII-Tubulin. Data are expressed relatively to Unt group (n values for all conditions=4, Unt vs BMAA *p=0.0286). (F) aSyn aggregates and p-aSyn (S129P) histological immunoreactivity in the dorsal motor nucleus of the vagus (DMV). (G) Quantitative analysis of OD for aSyn aggregates immunoreactivity in DMV. Data normalised to Unt group (n values for all conditions=4, Unt vs BMAA, *p=0.0109). (H) aSyn aggregates and p-aSyn (S129P) histological immunoreactivity in substantia nigra (SN). (I) Quantitative analysis of OD for aSyn aggregates immunoreactivity in the SN. Data normalised to Unt group (n values for all conditions=4, Unt vs BMAA, **p<0.0047). (J) aSyn oligomeric levels were calculated using an ELISA kit in mesencephalic homogenates of Unt and BMAA-treated mice. Values are pg/mL (n values for Unt=8 and BMAA=4, Unt vs BMAA, **p=0.0042). (K) Representative immunoblot showing aSyn monomer and oligomers in mesencephalic homogenates. The blots were re-probed for βIII-Tubulin to confirm equal protein loading. (L) Densitometric analyses of the levels of aSyn normalised against βIII-Tubulin. Data are expressed relatively to Unt group (n values for all conditions=4, Unt vs BMAA *p=0.0286). (M) Cytosolic aSyn oligomeric levels from primary mesencephalic neuronal cultures treated with 1 µM CCCP for 2 hours and 3 mM BMAA for 48 hours were calculated using an ELISA kit. Values are pg/mL (n values for all conditions=3, Unt vs CCCP, p=0.2615; Unt vs BMAA, **p=0.0063). (N) Representative immunoblot showing aSyn monomer and oligomers in mesencephalic neuronal cultures treated with 3 mM BMAA for 48 hours. The blots were re-probed for β-Actin to confirm equal protein loading. (O) Densitometric analyses of the levels of aSyn normalised against β-Actin. Data are expressed relatively to Unt neurons (n values for all conditions=4, Unt vs BMAA *p=0.0286). (P) NT2-Rho+ and Rho0 cells treated with 5 µM CCCP for 2 hours and 3 mM BMAA for 48 hours were evaluated by western blot. Representative immunoblot showing aSyn oligomers. The blots were re-probed for α-βIII-Tubulin to confirm equal protein loading. (Q) Densitometric analyses of the levels of aSyn normalised against α-βIII-Tubulin in Rho+ cells (n values for all conditions=6, Unt vs CCCP, *p=0.0236, Unt vs BMAA, **p=0.0018) and Rho0 cells (n values for all conditions=4, p>0,5). Histology samples were counterstained with cresyl violet. Scale bars are 50 µm (enlarged inner square) and 1 mm. Data represent mean+SEM. Statistical analysis: Unpaired Student’s t-test was performed in B–J and Mann-Whitney test in L and O. One-way analysis of variance (ANOVA) followed by Dunnett’s test was performed in M. In Q one-way ANOVA followed by Dunnett’s test was performed to compare different treatments against Unt group, and unpaired Student’s t-test was performed to compare Rho+ vs Rho0 cells.

β-N-methylamino-L-alanine (BMAA) induces dopaminergic neurodegeneration in vivo. (A) Representative immunofluorescence photomicrographs of the localisation of tyrosine hydroxylase (TH) and choline acetyltransferase (ChAT)-positive neurons in the dorsal motor nucleus of the vagus (DMV) from untreated (Unt) and BMAA-treated mice. (B) Quantification of the number of TH-positive neurons per mm2 in the DMV region (n values for all conditions=4, Unt vs BMAA, ***p=0.004). (C) Quantification of the number of ChAT-positive neurons per mm2 in the DMV region (n values for all conditions=4, Unt vs BMAA, p=0.41). (D) Representative immunoblot for TH, synaptophysin and PSD95 proteins. The blots were re-probed for βIII-tubulin to confirm equal protein loading. (E) Densitometric analysis of TH, (F) synaptophysin and (G) PSD95. Data were normalised with βIII-tubulin and expressed relatively to Unt mice (E–F, n values for Unt=10 and BMAA=8, G, Unt=9 and BMAA=7, Unt vs BMAA; E, **p=0.006, F, p=0.8286 and G, p=0.2799). (H) Representative photomicrographs of brain coronal sections immunostained with TH in striatum (STR) and substantia nigra (SN) from Unt and BMAA-treated mice. (I) Optical density analysis of the TH-positive fibres in the STR normalised to Unt group (n values for Unt=6 and BMAA=5, Unt vs BMAA, **p=0.0016). (J) Total number of nigral TH-positive neurons in SN assessed by stereological analysis (n values for Unt=6 and BMAA=5, Unt vs BMAA, *p=0.0135). (K) Dopamine levels were assessed in STR homogenates from Unt and BMAA-treated mice by Dopamine ELISA kit. Values are pg/mL (n values for Unt=6 and BMAA=7, Unt vs BMAA, *p=0.035). Scale bars are 50 µm (A) and 100 µm (H) (enlarged inner square) and 200 µm (A) and 1 mm (H). Data represents mean+SEM. Statistical analysis: Unpaired Student’s t-test was performed in B–C, E, G, I–J and Mann-Whitney test in F and K.
Mice treated orally with BMAA developed nigrostriatal degeneration and motor dysfunction
BMAA-induced erosion of ‘Candidatus Arthromitus’ from the ileum mucosa (figure 1E–F) led to GI inflammation and aSyn caudo-rostral aggregation, which may have contributed to the loss of TH-positive neurons. Our study revealed that the levels of TH-positive neurons decreased in BMAA-treated animals, but not of ChAT-positive neurons in the DMV (figure 7A–C). This reflects an increased susceptibility of dopaminergic neurons to BMAA that can be propagated to the SN through vagal catecholaminergic fibres located in the abdominal vagus nerve where roughly 70% of the neurons are TH-positive.58 To further examine whether BMAA-induced neuroinflammation and aSyn oligomerisation led to dopaminergic neuronal damage we analysed TH levels in mesencephalon and STR of BMAA-treated mice. Interestingly, we observed a loss of dopaminergic neurons in the mesencephalon as determined by the decrease in TH levels (figure 7D–E), without any detectable alteration of other synaptic markers, such as PSD95 and synaptophysin (figure 7D and F–G). We also observed depletion of TH-positive fibres in the STR (figure 7H–I) and a decrease of the total number of nigral TH-positive cells in the SN (figure 7H and J) of BMAA-treated mice, whose loss led to lower dopamine levels in the STR (figure 7K). Correlational analysis indicates that the levels of aggregated aSyn in the DMV and SN contributes to depletion of mesencephalic TH-positive neurons, although we observed that SN IgG leakage also contributes to the increase of aSyn aggregates levels in the SN, an effect that may not only be caudo-rostral, since SN IgG leakage does not correlate with aSyn aggregated levels in the DMV (online supplemental figure S7E–H).
To test if the BMAA-induced dopaminergic neuronal loss, potentiated by systemic low-grade inflammation (figure 3Q), could affect motor performance, we evaluated mice motor performance using different motor behavioural tests. We found that motor performance in the beam walking test (8 mm) was significantly impaired in BMAA-treated mice (figure 8A). We also observed that BMAA increased the hind clasping score (figure 8B) and decreased the latency in the inverted grip test (figure 8C). These results show that BMAA-induced motor impairment but had no apparent impact on the memory function as evaluated by the T-maze test at the selected time point (figure 8D–E). The locomotor activity in the open field test was also significantly decreased (figure 8F–I). Additionally, we also observed that BMAA treatment induced odour discrimination alterations, which agrees with PD symptomatology (data not shown).

β-N-methylamino-L-alanine (BMAA) in vivo administration induces motor behavioural changes. (A) Balance and motor coordination performance was assessed with the beam walking test (n values for untreated (Unt)=9 and BMAA=11, Unt vs BMAA, ****p<0.0001). (B) Hindlimb clasping reflex was monitored, as a quick phenotypic neurological scoring system for evaluating disease progression (n values for Unt=10 and BMAA=15, Unt vs BMAA, ***p=0.0008). (C) Inverted grip test was used to evaluate muscular strength of limb muscles (n values for all conditions=8, Unt vs BMAA, *p=0.0132). (D–E) Cognitive and memory ability was assessed using a T-maze. (D) Percentage of alternation between arms and (E) latency to respond (s) was assessed (n values for all conditions=8, Unt vs BMAA, D, p=0.2165; E, p=0.192). (F–I) Locomotor activity was evaluated in an open field arena. (F) Distance travelled (cm), (G) % time spent at the centre of the arena, (H) mean velocity (cm.s−1) and (I) % resting time (n values for all conditions=14, Unt vs BMAA, F, *p=0.0384; G, *p=0.0165; H, *p=0.0383 and I, *p=0.0495). Data represents mean+SEM. Statistical analysis: Unpaired Student’s t-test was performed in A, C and E–I, and Mann-Whitney test in B and D.
Discussion
Idiopathic PD represents approximately 90% of the disease cases but the aetiological factors remain elusive.1 It is currently accepted that motor symptoms are preceded by non-motor features such as GI dysfunction.2 Indeed, aSyn aggregates, a major PD pathological hallmark, were found to accumulate in some patients’ peripheral organs such as the gut, early in the prodromal phase.3–5 Accordingly, it was postulated that environmental factors, namely certain gut microbes or their products, could act as environmental triggers of PD.6 Understanding the impact of gut dysbiosis on the CNS has become critical for developing new diagnostic and therapeutic tools that in the future may arrest disease progression. This work shows that BMAA, an environmental microbial metabolite with known neurotoxic properties, depletes specific bacterial groups that essentially regulate ileum mucosal immunity and possibly gut motility. Such narrow-spectrum erosion was accompanied by exacerbated GI inflammation, barrier disruption and aSyn aggregation, and it was also further possible to track the progression of the pathology to the mesencephalon, where the mitochondria became dysfunctional. Consequently, mitochondrial network fragmentation and exposure of DAMPs activated neuronal innate immunity promoting aSyn aggregation, neuroinflammation, loss of dopaminergic neurons and motor alterations in WT mice. These observations support the idea that certain microbial metabolites can trigger ‘gut-first’ PD but they also reinforce the role of mitochondria as key players in disease progression.
Changes detected in the intestinal microbiota of patients with PD include increased proportions of bacterial groups with pro-inflammatory properties and concomitant decrease in the relative abundance of groups with anti-inflammatory action.45 Gut dysbiosis was also detected in individuals with rapid eye movement sleep behaviour disorder59 suggesting that these alterations occur early in the prodromal phase of ‘body-first’ PD cases.60 Here we show that BMAA exposure significantly alters the relative levels of key bacterial populations in the ileum and cecum mucosa, as well as in the faecal material, although the effects are more conspicuous in the ileum mucosa. Turicibacter are filamentous spore-forming bacteria that were significantly depleted after BMAA treatment. These bacteria were recently found to stimulate the release of serotonin from enterochromaffin cells, a neurotransmitter that regulates intestinal motility.61 Since constipation is a common PD symptom, we speculate that the extensive erosion of Turicibacter in the ileum and cecum mucosa and in the faecal material may contribute to impaired serotonin signalling which may hypothetically lead to impaired gut motility. Relevantly, BMAA treatment also significantly depleted ‘Candidatus Arthromitus’ the most prominent bacterial genus in the mice ileum mucosa. Since SFB have fundamental roles in mice gut homeostasis,14 15 their depletion could justify enhanced gut inflammation13 and the observed loss of intestinal barrier integrity. Indeed, depletion of SFB activated CD11b+ gut resident cells that released TNF-α and IL-1β, and may have also activated non-resident proinflammatory Th17 cells driving the production of IL-17 with effects in the periphery.14 Interestingly, circulating Th17 cells are increased in early-stage PD.62 Under inflammatory conditions, the intestinal epithelium is exposed to multiple cytokines that may synergistically impair intestinal barrier through reduction in occludin and ZO1 levels as well as cytoskeletal rearrangement.46 Additionally, we observed a decrease in CD4+ lymphocytes and an increase in the levels of IFNγ and IL-6 in the blood of BMAA-treated mice, resembling what has been described in patients with PD.48 63 The decline of CD4+ T cells and the increase of pro-inflammatory cytokines in PD patients’ blood reported in previous studies might suggest impairment of peripheral immunity with propensity to systemic inflammation.64 The increased mesencephalic IgG infiltration detected in our study suggests that BMAA promoted BBB permeability in the SN although no detectable effects were observed in the striatum or the cortex. BMAA has been shown to cross the BBB and has been detected in the brain of patients with ALS/PDC.20 Herein, we show that in vivo BMAA treatment impaired the OCR and calcium uptake by mesencephalic mitochondria but not in cortical mitochondria, despite the fact that direct acute in vitro treatment of isolated mesencephalic or cortical mitochondria with BMAA induced a decrease in OCR.21 BMAA treatment led to mitochondria dysfunction and fragmentation in primary mesencephalic neurons, which due to mitophagy impairment were not degraded and consequently exposed cardiolipin, a DAMP able to activate neuronal innate immunity.52 Interestingly, when the experiments were conducted in Rho0 cells that lack functional mitochondria, BMAA had no effect on the membrane potential or on the fragmentation of the mitochondrial net. We also observed that BMAA treatment activated the NLRP3 inflammasome accompanied by the release of IL-1β in enriched mesencephalic neuronal cultures. Although in the in vivo context we cannot affirm that NLRP3 activation and IL-1β release are restricted to neurons, the activation of the microglia in SN and the more intense co-localisation of Trem2-positive monocytes strongly indicate the involvement of systemic immunity.54 Since neurons express TLRs and major histocompatibility complex class I proteins and are able to release pro-inflammatory mediators,65 we propose that in BMAA-treated mice, neurons are the ones to be initially affected and only later signal microglia.
One well described consequence of innate immunity activation is the production of antimicrobial peptides.66 Recent data suggest that, in the enteric nervous system (ENS), aSyn plays a role in innate immune defences of the GI tract. Indeed, increased expression of aSyn in the enteric neurites of the upper GI tract of paediatric patients positively correlated with the degree of acute and chronic inflammation induced during norovirus infection in the intestinal wall.67 It was also demonstrated that aSyn exhibits antibacterial activity against Escherichia coli and Staphylococcus aureus, which suggests that, in addition to a role in neurotransmitter release, aSyn may also function as a natural bacteriostatic protein.68 In BMAA-treated mice, we observed an evident increase in aSyn aggregation in the ileum and a caudo-rostral progression through the DMV nucleus into the SN. Our findings are in line with studies conducted in germ-free mice colonised with PD gut microbiota or with curli-producing E. coli, which showed increased aggregation of aSyn in the gut and in the brain,9 69 and that suggest an aSyn-dependent mechanism in the aetiology of some PD ‘gut-first’ cases.70 71 The proposal that a bacterial toxin such as BMAA can stimulate the expression and spread of aSyn through enteroendocrine cells that synapse with enteric nerves in the GI tract3 is innovative and may reveal important new targets to address prodromal, peripheral synucleinopathy. Regarding the ENS, a very interesting study performed in patients with PD’ duodenal biopsies failed to demonstrate alterations in the number of submucosal neurons as well as alterations in mitochondrial membrane potential and aSyn levels.72 Nevertheless, this data agrees with our study, since we did not observe an increase in duodenal aSyn aggregates in BMAA-treated mice (data not shown). Additionally, aSyn has been proposed to behave as prion-like protein spreading through a caudo-rostral trajectory from the gut to the brain via the vagus nerve.70 73–76 We also observed that aSyn accumulates within brain mitochondria, which may indicate a positive feedback mechanism further contributing to their dysfunction. Several in vitro studies showed that aSyn targets the mitochondria inducing morphological and functional alterations.77 Interestingly, rotenone, a complex I inhibitor that leads to mitochondrial dysfunction and fragmentation, also induces aSyn accumulation.78 We therefore propose that the PD neurodegenerative process is intimately associated to the role of mitochondria in the activation of innate immunity. This work further reinforces the crucial role of mitochondria in the aetiology of PD, essentially because BMAA treatment did not increase aSyn aggregation in Rho0 cells. The observation that BBB permeabilisation and mitochondrial dysfunction occur in the SN but not in the cortex led us to hypothesise the vital impact of the caudo-rostral progression of aSyn pathology through the vagus nerve. Indeed, and despite previous observations showing increased permeability of cortical and STR regions to blood-borne components,49 we observed a positive correlation between BBB permeabilisation and aSyn aggregates levels in the midbrain, which indicates that both the blood and the vagal routes are likely involved in the full deleterious effect of BMAA on the mitochondria. This will in turn fully activate innate immune responses and neuroinflammation, which will synergistically induce dopaminergic neurodegeneration and impair motor function. In summary, our findings indicate that in genetically susceptible PD patients, the pathology may start in the gut when triggered by an environmental toxin such as BMAA, a microbial product commonly found in seafood, shellfish and fish. From the observed effect of BMAA on specific members of the mice gut microbiome namely on ‘Candidatus Arthromitus’, we propose that this toxin may have initially evolved as an antimicrobial compound, possibly to provide some competitive advantage to its producers in a shared ecosystem, and accidentally targeted the mitochondria, acknowledged relatives of an ancient endosymbiotic proteobacteria. In support of the gut-to-brain paradigm, we demonstrate that gut inflammation is correlated with aSyn aggregation and highlight the key role of mitochondria in the downstream activation of neuronal innate immunity, aSyn aggregation and loss of DMV and SN TH-positive neurons. Although we did not determine ENS neuronal mitochondrial function, we share the view that the gut and the ENS act as a gateway through which BMAA and aSyn reach the DMV and the SN, target the mitochondria and gradually inflict damage to the most vulnerable TH-positive neurons.79 However, we cannot discard completely the possibility of peripheral inflammation and consequent BBB disruption being involved in the access of BMAA to the brain parenchyma, an important question that must be addressed in a future study.
We theorise that chronic exposure to microbial BMAA via dietary sources impacts the host and may elicit phenotypic alterations that might trigger PD. Our results suggest that BMAA possesses narrow-spectrum antibiotic activity against some bacterial ‘sentinels’ responsible for gut mucosal immune homeostasis, the SFB. Notwithstanding the role of host genetic susceptibility and of other contributors, namely those driven by gut dysbiosis as probable drivers of pathology progression, this work is important to alert health authorities to the pressing need to implement routine protocols to monitor the levels of this (and other) toxin in foods and food supplements. In conclusion, our work sheds additional light into a long-lasting discussion80 by providing the first proof-of-concept for the possible involvement of a microbial toxin in the aetiology of PD (figure 9), which stands as an important step towards development of innovative therapies that may successfully thwart the onset and chronic course of this neurodegenerative disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram of ‘Gut-first’ PD. Environmental microbial toxins lead to the erosion of segmented filamentous bacteria (SFB, green) in the ileum, which potentiates a Th17 proinflammatory response and the loss of intestinal barrier integrity. These events in the gut allow the progression of the disease into the brain either through the blood or the vagus nerve. Microbial toxins target mesencephalic mitochondria and activate neuronal innate immunity followed by aSyn expression, microglial activation and ultimately PD. aSyn, alpha-synuclein; BMAA, β-N-methylamino-L-alanine; IL, interleukin; PD, Parkinson’s disease; Th17, T helper 17. (This image was created at BioRender.com).
Data availability statement
Data are available upon reasonable request. The data that support the findings of this study are available from the corresponding author upon request.
Ethics statements
Patient consent for publication
Ethics approval
Animal procedures were approved by the Animal Welfare Committee of the Center for Neuroscience and Cell Biology and Faculty of Medicine, University of Coimbra, and certified by the Portuguese authorities (Direção Geral de Veterinária).
Acknowledgments
SMC dedicates this work to her mentor and PhD supervisor, Professor Catarina Resende de Oliveira. The authors acknowledge Inês Melo Marques for assistance on the blind quantification and scoring of IF/IHC images, Ana Cristina Rego for providing key Seahorse methods and Conceição Egas (Genoinseq, Next Generation Sequencing Unit, Center for Neuroscience and Cell Biology) for microbiome sequencing support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ARE and MFM-P contributed equally.
Correction notice This article has been corrected since it published Online First. The legend for figure 6 has been corrected.
Contributors NE and SMC conceptualised and designed the study and the research plan. NE supervised the microbiome work and SMC supervised the rest of the work. SA, IT and DN-C generated the microbiome data. DFS and ARE performed all the in vitro experiments. EC and MFM-P performed animal surgeries, tissue clearing. MFM-P and JDM performed histology analyses. MFM-P performed FACS analyses. EC, ARE and DFS performed cytokine analyses. EC, ARE, JDM, MFM-P and ARP-S performed behaviour experiments. ILF, ARE and DFS performed Seahorse technique. ARE and DFS performed protein analysis. ARE, DFS and MFM-P performed confocal imaging. MFM-P and DN-C performed data analysis. NE and SMC wrote the manuscript. All authors contributed to the material and methods section, results section and figure legends. SMC and NE are guarantors of this work.
Funding This work was funded by Santa Casa da Misericórdia de Lisboa, Portugal, through Mantero Belard Neurosciences Prize 2016 (MB-40-2016); by FMUC-PEPITA (2018); by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Programme under project CENTRO-01-0145-FEDER-000012 (HealthyAging 2020) and through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalization and by Portuguese national funds via FCT—Fundação para a Ciência e a Tecnologia under projects PTDC/MED-NEU/3644/2020, PINFRA/22184/2016/POCI-01-0145-FEDER-022184 and UIDB/04539/2020. EC was supported by fellowship MB-40-2016. IT was supported by IF/01061/2014 Investigator contract. JDM is supported by PhD fellowship PD/BD/146409/2019, DN-C is supported by PhD fellowship SFRH/BD/117777/2016 and ARP-S is supported by PhD fellowship PD/BD/2020.06543.BD.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Commentary