Article Text

Abstract
Objective Stromal barriers, such as the abundant desmoplastic stroma that is characteristic of pancreatic ductal adenocarcinoma (PDAC), can block the delivery and decrease the tumour-penetrating ability of therapeutics such as tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), which can selectively induce cancer cell apoptosis. This study aimed to develop a TRAIL-based nanotherapy that not only eliminated the extracellular matrix barrier to increase TRAIL delivery into tumours but also blocked antiapoptotic mechanisms to overcome TRAIL resistance in PDAC.
Design Nitric oxide (NO) plays a role in preventing tissue desmoplasia and could thus be delivered to disrupt the stromal barrier and improve TRAIL delivery in PDAC. We applied an in vitro–in vivo combinatorial phage display technique to identify novel peptide ligands to target the desmoplastic stroma in both murine and human orthotopic PDAC. We then constructed a stroma-targeted nanogel modified with phage display-identified tumour stroma-targeting peptides to co-deliver NO and TRAIL to PDAC and examined the anticancer effect in three-dimensional spheroid cultures in vitro and in orthotopic PDAC models in vivo.
Results The delivery of NO to the PDAC tumour stroma resulted in reprogramming of activated pancreatic stellate cells, alleviation of tumour desmoplasia and downregulation of antiapoptotic BCL-2 protein expression, thereby facilitating tumour penetration by TRAIL and substantially enhancing the antitumour efficacy of TRAIL therapy.
Conclusion The co-delivery of TRAIL and NO by a stroma-targeted nanogel that remodels the fibrotic tumour microenvironment and suppresses tumour growth has the potential to be translated into a safe and promising treatment for PDAC.
- pancreatic cancer
- drug resistance
- fibrosis
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All relevant data supporting the key findings of this study are available within the article and the supplementary files.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Pancreatic ductal adenocarcinoma (PDAC) frequently exhibits an abundant desmoplastic stroma, which substantially blocks the penetration of therapeutics and decreases the response to therapy.
Clinical trials for recombinant tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) or TRAIL receptor agonists have shown only a moderate therapeutic benefit for patients with advanced PDAC due to the short half-life of recombinant TRAIL and its low bioavailability in tumour tissues.
The carcinoma-associated fibroblast (CAF) diversity—most notably inflammatory CAFs expressing proinflammatory cytokines and myofibroblastic CAFs mainly producing extracellular matrix components plays a crucial role in tumour progression and resistance to chemotherapy and immunotherapy in various cancer types, including PDAC.
What are the new findings?
We adopted an in vitro–in vivo combinatorial phage display technology to identify novel tumour stroma-targeting peptide ligands that target tumour-associated pancreatic stellate cells in an orthotopic PDAC model.
Nitric oxide (NO) could normalise the inflammatory and desmoplasia phenotype of pancreatic stellate cells and triggers apoptosis sensitisation of PDAC.
A tumour stroma-targeted nanogel (modified with phage display-identified tumour stroma-targeting peptides) containing the NO donor and TRAIL could reprogramme the desmoplastic stroma and decrease the activity of the antiapoptotic pathway thereby facilitating tumour penetration by TRAIL and substantially enhancing the antitumour efficacy of TRAIL therapy.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
The tumour stroma-targeting peptides identified by phage display may also target CAFs in other types of tumours characterised by particularly enhanced desmoplasia and could be used in cancer diagnosis.
Given currently limited therapeutic options for patients with PDAC, this study provides new perspectives on PDAC therapy using a stroma-targeted delivery system composed of NO donors and therapeutic proapoptotic proteins with the potential to be translated into a safe and promising PDAC treatment.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers, with a 5-year survival rate of less than 5%.1 PDAC frequently exhibits an abundant desmoplastic stroma, which substantially blocks the penetration of therapeutics, including small molecule chemotherapy and macromolecular therapeutic agents (ie, tumour necrosis factor-related apoptosis-inducing ligand, TRAIL), into the tumour.2 Thus, there is an urgent need to identify new therapeutic strategies for PDAC.
The use of strategies to eliminate the extracellular matrix (ECM) barrier in combination with standard chemotherapy has shown beneficial outcomes in patients with PDAC in preclinical and clinical trials.3–6 For example, enzymatic ECM-degradation strategies involving the administration of collagenase or hyaluronidase were used to deplete the tumour stroma and increase drug delivery.7 However, unwanted toxicity or mortality was observed after the systemic administration of ECM-degrading enzymes.7 In addition, enzymatic depletion of the ECM may endow PDAC tumours with enhanced aggressiveness, suggesting that the development of a desmoplastic stroma-modulating agent without systemic toxicity and protumour effects could be a potential therapeutic strategy for the treatment of PDAC.8 The recent study of carcinoma-associated fibroblast (CAF) diversity—most notably inflammatory CAFs expressing proinflammatory cytokines and myofibroblastic CAFs mainly producing ECM components—has yielded new insights for the development of novel approaches to ‘normalise’ CAFs toward a less active state to sensitise PDAC to chemotherapy and immunotherapy.9 10 Given that activated pancreatic stellate cells (PSCs) are major sources of CAFs in PDAC, it may be possible to modulate the desmoplastic and inflammatory tumour microenvironment (TME) by reprogramming activated PSCs, thus potentially providing a more effective therapeutic approach than enzymatic ECM depletion.
Nitric oxide (NO) plays a role in downregulating fibroblast activation and preventing tissue desmoplasia to inhibit the progression of various fibrotic diseases.11–13 In addition, NO was reported to activate the apoptosis pathway by regulating antiapoptotic BCL-2 family members and the tumour suppressor p53.14 15 Based on these previous findings, we hypothesised that the reprogramming of activated PSCs by inhibiting the expression of fibrogenic genes and the suppression of the antiapoptotic pathway with NO would deplete the desmoplastic tumour stroma, facilitate the tumour penetration of anticancer agents and restore apoptosis sensitivity in PDAC.
As it preferentially triggers apoptosis in cancer cells, TRAIL-based therapy is a potential anticancer therapeutic.16 17 However, clinical trials for recombinant TRAIL or TRAIL receptor agonists have shown only a moderate therapeutic benefit for patients with advanced PDAC.16 18 19 The efficacy of TRAIL therapy is limited due to the short half-life of recombinant TRAIL and its low bioavailability in tumour tissues due to the stromal barrier.20 Second, PDAC cells constitutively overexpress antiapoptotic proteins such as Bcl-2 and Bcl-xL, leading to resistance to TRAIL-induced apoptosis.21 This study aimed to develop a TRAIL-based nanotherapy that not only eliminated the ECM barrier to increase TRAIL delivery into tumours but also blocked antiapoptotic mechanisms to overcome TRAIL resistance in PDAC. To achieve this aim, we developed nanoparticle (NPs), which we termed TRAIL-NO@Nanogel, consisting of a silk fibroin (SF) hydrogel core loaded with TRAIL and a shell composed of lipids and poly(lactic-coglycolic) acid (PLGA) loaded with a synthetic NO donor dinitrosyl iron complex (dinitrosyl iron complexes (DNIC); Fe(μ-SEt)2(NO)4).22–24 Due to its excellent versatility, biocompatibility, biodegradability and low immunogenicity, SF was selected here to carry TRAIL in the nanoscale hydrogel core.25 26 To achieve targeted delivery to the desmoplastic stroma in PDAC, we adopted an in vitro–in vivo combinatorial phage display technology to identify novel peptide ligands that target tumour-associated PSCs in an orthotopic PDAC model. Phage display-identified tumour stroma-targeting peptides were conjugated to TRAIL-NO@Nanogel to enhance the delivery of TRAIL and NO to PDAC. As illustrated in figure 1, tumour stroma-targeted TRAIL-NO@Nanogel decreased desmoplasia and improved TRAIL delivery in orthotopic PDAC models. Additionally, we demonstrated the feasibility of reprogramming the desmoplastic stroma and downregulating the expression of the antiapoptotic proteins Bcl-2 and Bcl-xL via tumour stroma-targeted NO delivery, which thereby improved the efficacy of TRAIL therapy.

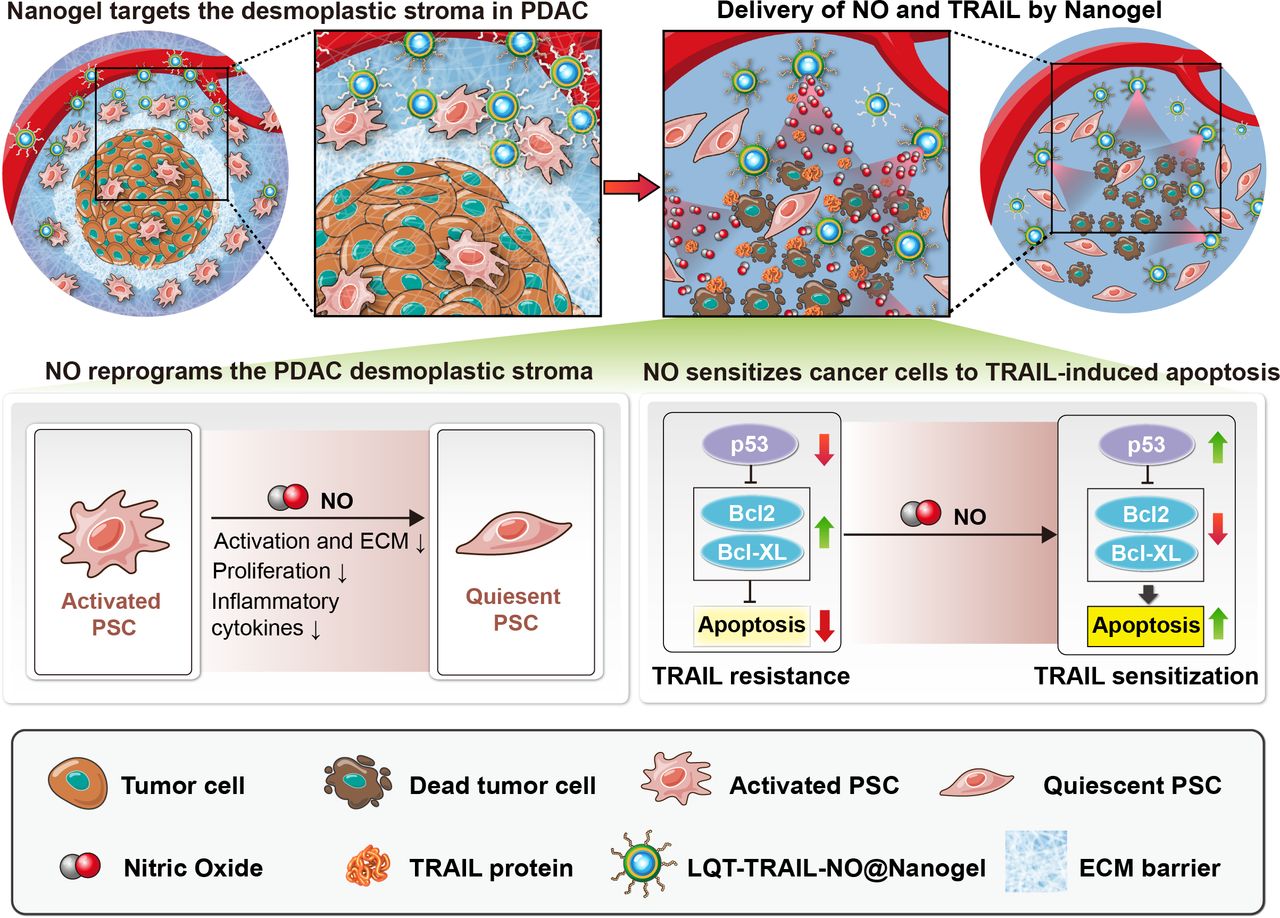
Schematic showing the mechanism by which tumour stroma-targeted TRAIL-NO@Nanogel suppresses PDAC progression in mice. NO released from tumour stroma-targeted TRAIL-NO@Nanogel remodels the fibrotic tumour microenvironment of desmoplastic PDAC. (1) NO released from NPs modified with tumour stroma-targeting peptides identified by phage display suppresses PSC activation, reduces ECM production and increases tumour perfusion in PDAC. (2) NO reprogrammes the desmoplastic stroma and overcomes TRAIL resistance, sensitising PDAC tumours to TRAIL therapy. (3) Co-delivery of TRAIL and NO by tumour stroma-targeted TRAIL-NO@Nanogel efficiently suppresses tumour growth. ECM, extracellular matrix; NO, nitric oxide; NPs, nanoparticle; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cells; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand.
Results
Reprogramming of activated PSCs and downregulation of antiapoptotic proteins in PDAC cells by NO
Activated PSCs are key mediators of the production of ECM components in the stromal compartment in PDAC.27 To explore the mechanism by which NO regulates PSC activation, PSCs activated with TGFβ were treated with 0.5 µM DNIC for 24 hours, and the messenger RNA (mRNA) expression of a panel of 84 key genes involved in fibrosis was examined with an RT2 Profiler PCR array (figure 2A, see online supplemental table S1). Most fibrosis-related genes, including genes encoding ECM-remodelling enzymes, TGFβ signalling factors and inflammatory cytokines, were downregulated after DNIC treatment, indicating that NO inhibited PSC activation. We next performed qRT-PCR to validate the mRNA expression levels of markers for myofibroblast activation (ACTA2, COL1A1 and TGFB1). Similar to the results of the PCR array analysis, the qRT-PCR results indicated that NO decreased the mRNA expression of ACTA2, COL1A1 and TGFB1 in activated PSCs (figure 2B). Furthermore, we observed significant dose-dependent decreases in α-smooth muscle actin (SMA) and collagen I protein expression and in the expression of downstream TGFβ signalling molecules (NF-κB and IκBα) and profibrotic Akt activation after treatment with DNIC (figure 2C, see online supplemental figure S1).
Supplemental material

Reprogramming of activated PSCs and downregulation of antiapoptotic proteins in PDAC cells by NO. (A) The mRNA expression levels of a panel of 84 key fibrosis-associated genes in primary, culture-activated human PSCs after 24 hours of treatment with DNIC (0.5 µM) were measured with an RT2 Profiler PCR array. The results are expressed as the fold change relative to the corresponding level in the untreated control group (n=2). (B) Expression levels of myofibroblast activation markers in primary, culture-activated human PSCs after 24 hours of treatment with DNIC (0.5 µM) were measured by RT-qPCR. The results are expressed as the fold change relative to the corresponding level in the untreated control group (n=5). (C) Western blotting was used to analyse α-SMA and collagen I protein expression as well as downstream TGFβ signalling activation (phospho-AKT, phospho-NF-κB and phospho-IκBα levels) in primary, culture-activated human PSCs treated with or without increasing concentrations of DNIC. The experiments were repeated two times independently. (D) Expression levels of proinflammatory cytokines in primary, culture-activated human PSCs in a coculture system with PDAC cells (2×105 AsPC-1 cells) after 24 hours of treatment with DNIC (2 µM) were measured by RT-qPCR. The results are expressed as the fold change relative to the corresponding level in the untreated control group (n=3). (E) Western blotting was used to analyse p53, Bcl-xL and Bcl-2 expression in AK4.4 cells. The experiments were repeated two times independently. (F) Dox (2 or 4 µM) in combination with DNIC (2 µM) significantly enhanced the induction of apoptosis in murine AK4.4 and human AsPC-1 PDAC cells, as detected using annexin V staining (n=3). (G) Recombinant TRAIL (2000 ng/mL) in combination with DNIC (2 µM) significantly enhanced the induction of apoptosis in murine AK4.4 and human AsPC-1 PDAC cells, as detected using annexin V staining (n=4). DNIC, dinitrosyl iron complexes; ECM, extracellular matrix; IL, interleukin; NO, nitric oxide; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cells; SMA, smooth muscle actin; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand.
In addition to the profibrotic phenotype, paracrine factors secreted from cancer cells activate PSCs toward a proinflammatory phenotype.9 To investigate the interactions between PDAC cells and PSCs, we cultured PSCs in the presence of PDAC cells in Transwell inserts. We found that several proinflammatory cytokines, interleukin (IL)1A, IL1B, IL6, IL11, CXCL1, CXCL2 and CSF3, were upregulated in PSCs in a coculture system with PDAC cells, as determined by qRT-PCR (figure 2D). Exposure to the NO donor DNIC significantly decreased the mRNA expression of proinflammatory cytokines in PSCs in a coculture system with PDAC cells (figure 2D), indicating that NO also normalised the inflammatory phenotype of PSCs.
Finally, we observed reductions in the expression of the antiapoptotic proteins Bcl-2 and Bcl-xL and increased expression of the tumour suppressor p53 in PDAC cells after DNIC treatment, indicating the ability of NO to activate the apoptosis pathway (figure 2E, see online supplemental figure S2). To assess the impact of NO on chemotherapy-induced or TRAIL-induced anticancer effects, we detected the apoptosis of murine AK4.4 cells and human AsPC-1 PDAC cells treated with DNIC or chemotherapeutic agents (Dox or Gem)/or TRAIL for 16 hours using an annexin V-binding assay and found that the combination of DNIC and chemotherapeutic agents/or TRAIL induced a significant increase in apoptosis compared with single treatment (figure 2F,G, see online supplemental figure S3). Taken together, the results indicate that NO acts as an antifibrotic and anti-inflammatory effector and sensitises cancer cells to anticancer therapeutics.

In vitro/in vivo combinatorial biopanning of the Ph.D.−12 phage display peptide library in a murine orthotopic PDAC model in vivo and in human PSCs in vitro. (A) Schematic diagram of the method used for in vitro–in vivo combinatorial phage display biopanning. A phage display random peptide library was intravenously injected into orthotopic murine PDAC models generated using AK4.4.28 One hour after injection, bound phages were isolated from PDAC tumours and amplified for subsequent rounds of biopanning. After the first round of in vivo selection, we performed three cycles of in vitro biopanning using human PSCs. Enriched phages from the third in vitro biopanning round were randomly selected for amplification, and the affinity of each selected phage for PSCs was evaluated by ELISA and compared with that of a negative control helper phage. The gene encoding the PDAC tumour stroma-specific oligopeptide displayed on the selected phage was amplified by PCR, cloned and sequenced. (B) Identification of phages capable of binding primary, culture-activated human PSCs. PDAC stroma-bound phage clones were selected by ELISA. A control phage without an insert was used as the negative control (control phage). The red line indicates a threshold binding affinity >2.5-fold greater than that of the negative control helper phage threshold. (C) primary culture-activated human PSCs were incubated with representative positively selected phage clones expressing different sequences. The binding affinities were measured using phage ELISA (n=3). The red line indicates a threshold binding affinity >2.5-fold greater than that of the negative control helper phage threshold. (D) Verification of the tumour-homing abilities of phage clones in the orthotopic PDAC model. PDAC (AK4.4) orthotopic tumour-bearing mice were intravenously injected with phage clones. One hour later, the organs/tumours were harvested, and the phages were recovered and titrated using the plaque assay (n=3–6). Two-tailed Mann-Whitney U test, *p<0.05 compared with the negative control helper phage. (E) Representative images of immunofluorescence) staining to detect phage clones in PDAC. Red, phage (anti-M13 antibody); green, α-SMA; blue, nuclei (DAPI). Scale bars, 20 µm. All data are shown as the mean±SE of the mean (SEM). PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cells; SMA, smooth muscle actin.
Identification of tumour stroma-targeting peptides by in vivo–in vitro combinatorial phage display
Targeted delivery of NO to tumour-associated PSCs in PDAC may serve as a way to modulate the desmoplastic tumour stroma and reduce the ECM while avoiding off-target side effects due to non-specific targeting. Therefore, we aimed to develop a tumour stroma-targeted carrier by modifying tumour-associated PSC-targeted ligands. We exploited an in vivo–in vitro combinatorial phage display strategy to identify stroma-targeting peptides that selectively recognise tumour-associated PSCs (figure 3A). To isolate PDAC stroma-targeted phages, we intravenously injected a phage display random peptide library into orthotopic murine PDAC models generated using AK4.4.28 One hour after injection, bound phages were isolated from PDAC tumours and amplified for subsequent rounds of biopanning. After the first round of in vivo selection, we performed three cycles of in vitro biopanning using human PSCs (figure 3A). We observed that phage titers increased with each cycle of biopanning (see online supplemental figure S4). Enriched phages from the third in vitro biopanning round were randomly selected for amplification, and the affinity of each selected phage for PSCs was evaluated by ELISA and compared with that of a negative control helper phage (figure 3B,C). Using a threshold binding affinity >2.5-fold greater than that of the negative control helper phage threshold, among 150 phage clones, 55 clones efficiently bound PSCs (figure 3B,C, see online supplemental figure S5). Sequencing of the 55 clones that bound PSCs demonstrated that they displayed the same consensus peptide motifs (table 1). We also observed that the enriched phages not only recognised activated PSCs but also moderately bound to malignant PDAC cells (figure 3B,C, see online supplemental figure S5). In contrast, the PDAC stroma-targeted phages did not exhibit substantial affinity toward normal mouse hepatocytes (FL83B) (see online supplemental figure S6). Finally, from these 55 phage clones, we selected 7 for further assessment based on their sequence conservation and ELISA results (table 1).
Sequences of phage clones isolated by in vitro–in vivo combinatorial biopanning of the Ph.D.−12 phage display peptide library in an orthotopic murine pancreatic ductal adenocarcinoma model in vivo and human PSCs in vitro

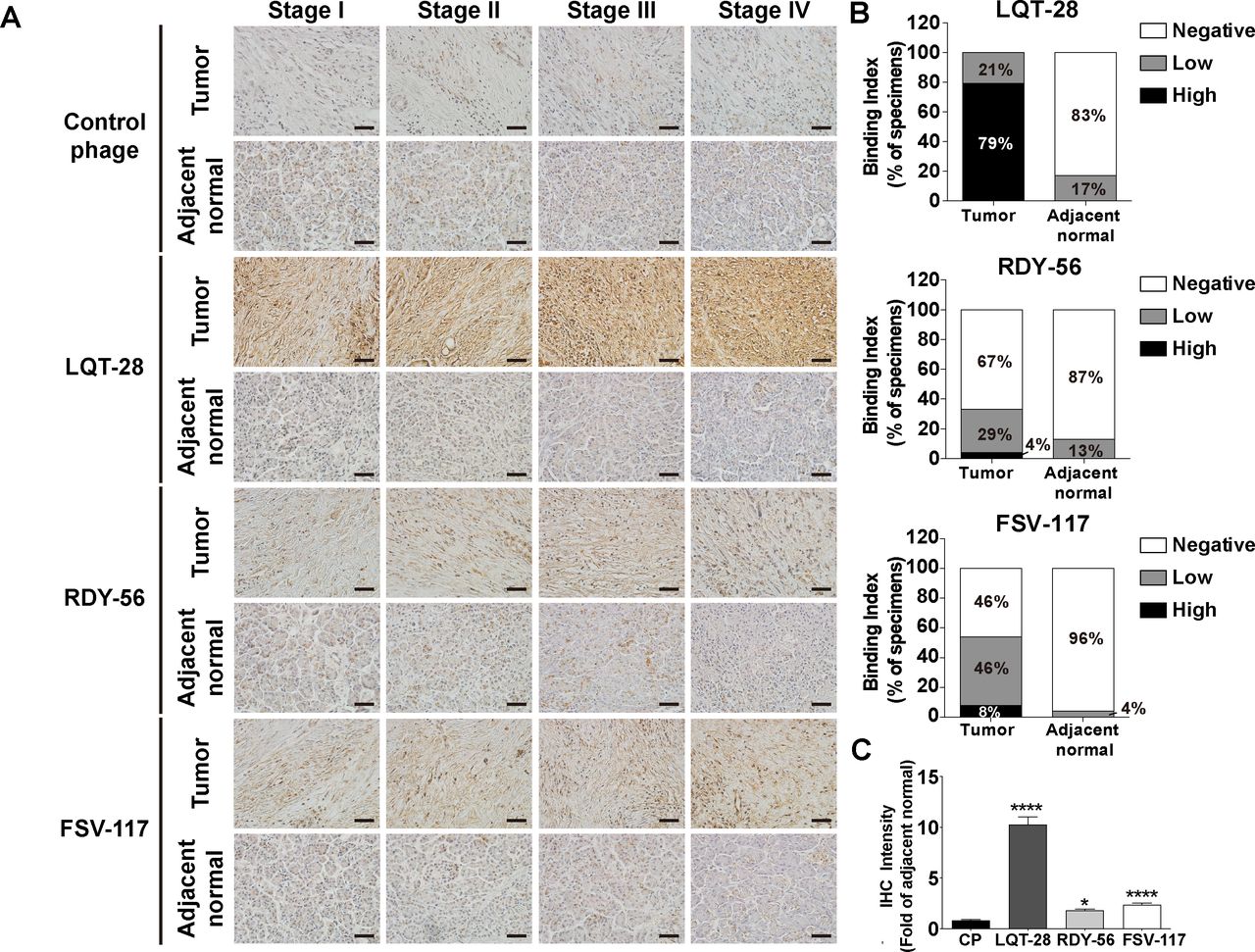
Binding of specific phage to biopsy specimens from patients with pancreatic cancer. (A) Biopsy specimens from patients with pancreatic cancer were incubated with LQT28, RDY56 or FSV117 peptide-displaying M13 phage or control helper phage and detected by anti-M13 phage antibody. Scale bar, 40 µm. (B) Extent of binding of LQT28, RDY56 or FSV117 peptide-displaying phage to tumour and adjacent normal tissues from 24 patients with pancreatic cancer (6 patients from each stage of patients with pancreatic cancer). (C) Quantification of fold change (tumour vs normal) in the IHC intensity of LQT28, RDY56 or FSV117 peptide-displaying M13 phage in biopsy specimens from patients with pancreatic cancer using Fiji. (n=120 section images of tumour and adjacent normal tissues from 24 patients with pancreatic cancer) all data are shown as the mean±SEM. *P<0.05, ****p<0.0001 compared with control helper phage. IHC, immunohistochemistry.
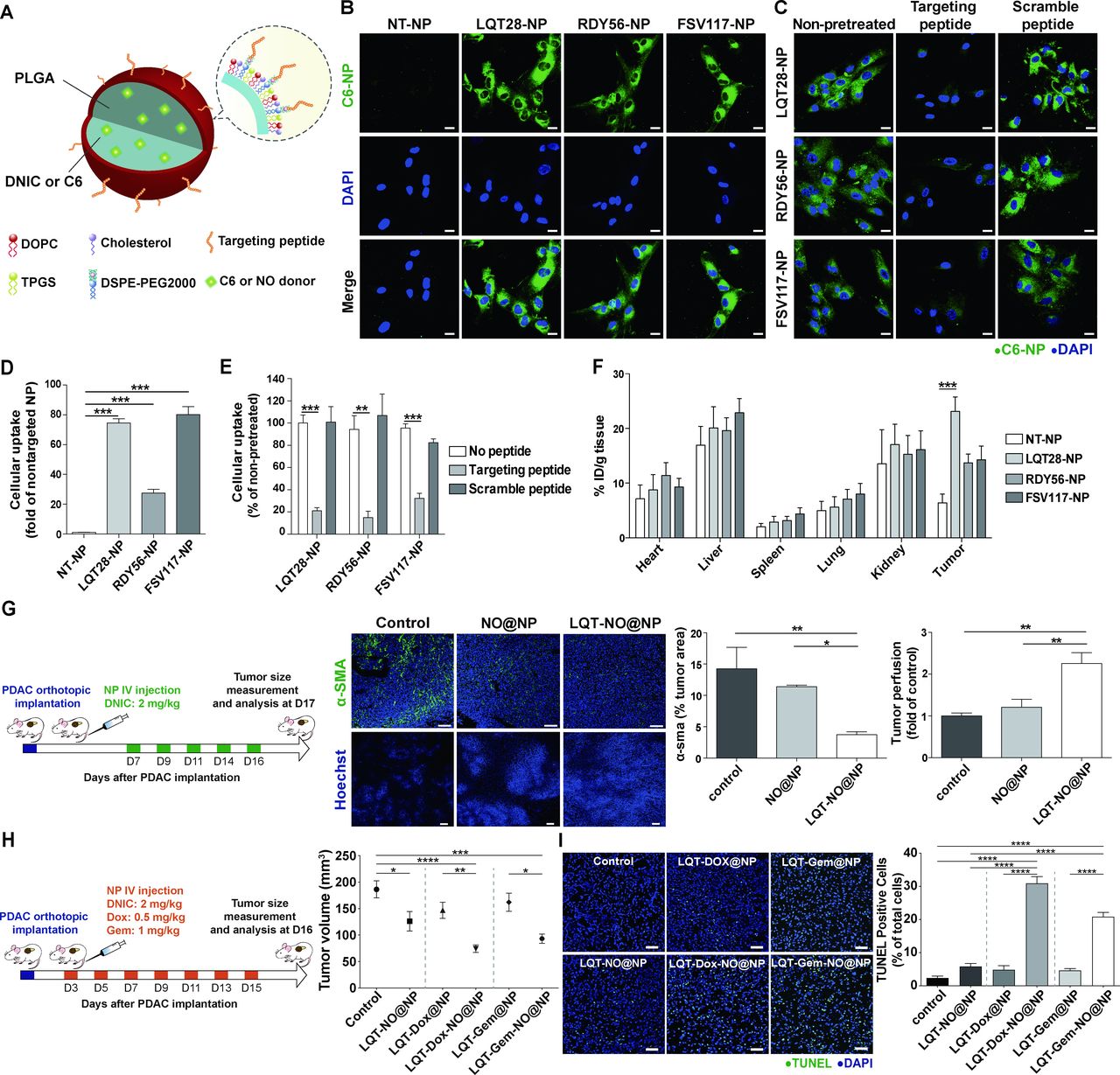
Tumour stroma-targeted lipid-PLGA NPs exhibited enhanced PDAC tumour uptake and NO delivery capability to reprogramme the desmoplastic tumour stroma in PDAC. (A) Structural schematic of NPs modified with tumour stroma-targeted peptides. (B) Primary, culture-activated human PSCs were treated with coumarin 6 (C6)-loaded NPs (0.175 µg/mL) modified with the tumour stroma-targeted peptide LQT28, RDY56 or FSV117 for 1 hour. Scale bars, 20 µm. Green, coumarin 6-loaded NPs; blue, nuclei (DAPI). (C) The uptake of NPs modified with the tumour stroma-targeted peptide LQT28, RDY56 or FSV117 into human PSCs was competitively inhibited by the addition of free corresponding stroma-targeted peptide. Cells were treated with free peptides for 30 mins prior to treatment with NPs and analysed for fluorescence signals by confocal microscopy. Scale bars, 20 µm. Green, coumarin 6-loaded NPs; blue, nuclei (DAPI). (D) The cellular uptake of NPs was imaged and quantified using a Zeiss LSM 780 confocal microscope (n=4). (E) The cellular uptake of NPs with or without pretreatment with free peptides was imaged and quantified using a Zeiss LSM 780 confocal microscope (n=4). (F) The tissue distributions of C6 in different formulations (n=7–15). NPs modified with LQT28 peptides showed enhanced tissue uptake in tumours 4- hour after intravenous administration in the orthotopic AK4.4 PDAC model. (G) Seven days after the implantation of AK4.4 PDAC cells and mice were treated with LQT-NO@NPs or non-targeted NO@NPs (DNIC: 2 mg/kg) on days 7, 9, 11, 14 and 16; tumours were then analysed on day 17 by immunostaining. Representative immunofluorescence images and quantification of α-SMA-positive cells and Hoechst 33 342-positive cells in PDAC tumours after treatment with LQT-NO@NPs or non-targeted NO@NPs in a murine orthotopic (AK4.4) PDAC model. Green, α-SMA-positive myofibroblasts; blue, DAPI (top panel) or Hoechst 33 342 (bottom panel) (n=5 section images from three mice). Scale bars, 100 µm. Mice were injected intravenously with 500 µg of Hoechst 33342 on day 17, after which the tumours were harvested. (H) Three days after the implantation of AK4.4 PDAC cells and mice were treated with DNIC (2 mg/kg) and/or Dox (0.5 mg/kg) or Gem (1 mg/kg) loaded in lipid-PLGA NPs modified with LQT28 on days 3, 5, 7, 9, 11, 13 and 15; tumours were then analysed on day 16. Volumes of orthotopic PDAC tumours 16 days post implantation in treated and untreated (control) mice (n=7–8), DNIC: 2 mg/kg; Dox: 0.5 mg/kg; Gem: 1 mg/kg. (I) Representative immunofluorescence images and quantification of TUNEL staining after treatment with different formulations in a murine orthotopic (AK4.4) PDAC model, as described in (h) (n=8 section images from four mice). Scale bar, 50 µm. All data are shown as the mean±SEM. *P<0.05, **p<0.01, ***p<0.001. DNIC, dinitrosyl iron complexes; IV, intravenously; NO, nitric oxide; NP, nanoparticle; PDAC, pancreatic ductal adenocarcinoma; PLGA, poly(lactic-coglycolic) acid; PSC, pancreatic stellate cells; SMA, smooth muscle actin.

Delivery of TRAIL and NO by tumour stroma-targeted nanogels reduced collagen I production and enhanced apoptosis induction in PDAC/PSC 3D culture models. (A) Schematic illustration of the preparation of LQT-TRAIL-NO@Nanogel. The TRAIL-loaded SF hydrogel core was first synthesised by the solvent-mediated transformation of SF hydrogel. Using microemulsion technology, the SF hydrogel core was then coated with phospholipid DOPA to form a well-dispersed suspension in organic solvent. Next, the hydrophobic lipid-coated SF hydrogel core was formulated into lipid-PLGA NPs to form the TRAIL@Nanogel. To co-deliver NO and TRAIL into PDAC, DNIC was loaded into the TRAIL@Nanogel to form the TRAIL-NO@Nanogel. Finally, to achieve tumour stroma-targeted delivery of the TRAIL-NO@Nanogel into PDAC, the LQT28 peptide was conjugated on the surface of NPs to form the LQT-TRAIL-NO@Nanogel. (B) A representative scanning electron micrograph of the LQT-TRAIL-NO@Nanogel. Scale bar, 100 nm. (C) Kinetics of DNIC degradation and NO release from the NO@Nanogel under physiological conditions (pH 7.4). The NO@Nanogel was incubated in PBS, and DNIC degradation was measured at 360 nm using a UV spectrophotometer (n=9). The results are expressed as the percentage of the initial DNIC content. The release of NO was measured as the fluorescence intensity (excitation at 566 nm and emission at 596 nm) of the NO-specific probe DAR-1. The results are expressed as fold changes compared with time 0 (n=3). (D) Kinetics of cargo protein (FITC-BSA) release from the nanogel or NO@Nanogel under different pH conditions. Nanogel or NO@Nanogel loaded with FITC-BSA was incubated in PBS (pH 7.4) or acetic acid buffer (pH 5.5), and FITC-BSA release was measured using a plate reader at an excitation wavelength of 494 nm and an emission wavelength of 520 nm (n=3–6). *P<0.05 and ***p<0.001 compared with the nanogel at pH 7.4. (E) Serum concentration profiles of free-form FITC-labelled TRAIL in different formulations (n=3). (F) Representative immunofluorescence images of AsPC-1 PDAC cell/PSC 3D spheroid cultures after treatment with different formulations. Green, collagen I or TUNEL staining; blue, nuclei (DAPI). Scale bars, 50 µm. (G) Expression levels of collagen I in PSC 3D spheroid cultures consisting of PDAC tumour cells (murine AK4.4 or human AsPC-1 PDAC cells) and PSCs after 48 hours of treatment with TRAIL and/or DNIC loaded in different formulations were analysed and quantified by immunostaining (n=5–15). (H) Apoptosis induction in PSC 3D spheroid cultures consisting of PDAC tumour cells (murine AK4.4 or human AsPC-1 PDAC cells) and PSCs after 48 hours of treatment with TRAIL and/or DNIC loaded in different formulations was analysed and quantified by immunostaining TUNEL staining (n=5–17). All data are shown as the mean±SEM. *P<0.05, **p<0.01, ***p<0.001. 3D, three-dimensional; DNIC, dinitrosyl iron complexesn; FITC-BSA, fluorescein isothiocyanate-labelled bovine serum albumin; NO, nitric oxide; NPs, nanoparticles; PBS, phosphate-buffered saline; PDAC, pancreatic ductal adenocarcinoma; PLGA, poly(lactic-coglycolic) acid; PSC, pancreatic stellate cells; SF, silk fibroin; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand.
The PDAC stroma-homing potential of the selected phages was further characterised using an in vivo homing assay in which the phages were intravenously injected into orthotopic murine PDAC (AK4.4) tumour-bearing mice. Ultimately, we identified three phage clones (LQT28, RDY56 and FSV117) that showed greater homing ability to the tumours than the negative control helper phage (figure 3D). Additionally, these three phage clones showed a significantly increased ability to home to tumours compared with normal organs, including the brain, heart and lungs (figure 3D). All three selected phage clones (LQT28, RDY56 and FSV117) were highly enriched in the tumour tissues compared with the control phage clone and colocalised with activated fibroblasts (α-SMA positive) in PDAC tumours (figure 3E, see online supplemental figure S7). The data confirmed the specificity of these phages for PDAC tumour stroma.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Delivery of TRAIL and NO by tumour stroma-targeted nanogels reduced collagen I production, increased apoptosis induction and suppressed tumour growth in both murine and human orthotopic PDAC models. (A) Schematic illustration of the LQT-TRAIL-NO@Nanogel treatment protocol. After the implantation of PDAC cells, mice were treated intravenously with various NP formulations encapsulating the NO donor DNIC and/or TRAIL on days 3, 5, 7, 9, 11, 13 and 15; tumour volume was measured on day 16. (B) Representative H&E, Masson’s trichrome staining and immunofluorescence images showing the results of collagen I, α-SMA and TUNEL staining in orthotopic murine PDAC (AK4.4) tumours after treatment with various formulations. Blue, nuclei (DAPI). Scale bars, 50 µm. (C–D) LQT-TRAIL-NO@Nanogel significantly reduced collagen I production (C) and α-SMA expression (D) in orthotopic PDAC tumours, as indicated by immunofluorescence staining for collagen I and α-SMA (n=7 section images from four mice). (E) Volumes of orthotopic PDAC tumours 16 days after implantation in treated and untreated (control) mice (AK4.4 PDAC model, n=10; AsPC-1 PDAC model, n=5). (F) LQT-TRAIL-NO@Nanogel significantly enhanced the induction of apoptosis in orthotopic PDAC tumours, as indicated by TUNEL staining (AK4.4 PDAC model, n=7 section images from four mice; AsPC-1 PDAC model, n=6 section images from three mice). (G) LQT-TRAIL-NO@Nanogel significantly prolonged the overall survival in orthotopic AK4.4 PDAC tumour model (n=8). ****P<0.0001 compared with untreated (control) mice, *p=0.0118 compared with LQT-NO@Nanogel, *p=0.0161 compared with LQT-TRAIL@Nanogel and *p=0.0340 compared with TRAIL-NO@Nanogel. Comparison of survival curves was performed using a log-rank Mantel-Cox test (two-sided). All data are shown as the mean±SEM. *P<0.05, **p<0.01, ***p<0.001. DNIC, dinitrosyl iron complexes; NO, nitric oxide; NP, nanoparticle; PDAC, pancreatic ductal adenocarcinoma; SMA, smooth muscle actin; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand.
The binding specificity of the LQT28, RDY56 and FSV117 peptide-displaying M13 phage to pancreatic cancer specimens from patients with pancreatic cancer was further investigated for potential clinical applications. The three selected phages were able to recognise tumour tissues in the pancreatic cancer specimens more efficiently than their normal counterparts (figure 4A–C). Among the three phages, a high positive rate of LQT28 phage binding to tumour tissues derived from patients with pancreatic cancer was observed (79% of specimens with high interaction) (figure 4A,B). Immunoreactivity against the control helper phage was not observed in tumour tissues (figure 4A,C).
Enhanced PDAC uptake of NPs modified with tumour stroma-targeting peptides
We next examined whether nanocarrier modification with phage display-identified PDAC stroma-targeting peptides would achieve selective delivery into the PDAC stroma (figure 5A). In this study, we used lipid-PLGA NPs to encapsulate therapeutic agents (NO donor DNIC, chemotherapeutics and TRAIL-loaded hydrogel). By using coumarin 6 loaded within the lipid-PLGA NPs as a tracer molecule (figure 5A), we found that the uptake of NPs modified with the LQT28, RDY56 or FSV117 peptide into activated PSCs was significantly enhanced compared with that of non-targeted NPs (PEG-NPs) (figure 5B,D). The uptake of NPs modified with the LQT28, RDY56 or FSV117 peptide was significantly inhibited by the addition of free corresponding stroma-targeting peptides (figure 5C,E), indicating that the cellular uptake was ligand dependent. We further evaluated whether modification of NPs with PDAC stroma-targeting peptides would enhance NP uptake into orthotopic PDAC tumours in vivo (figure 5F). Four hours after intravenous injection of NPs modified with the LQT28, RDY56 or FSV117 peptide into orthotopic murine PDAC (AK4.4) tumour-bearing mice, NP uptake into the PDAC tumours was greater than that of non-targeted control NPs (figure 5F). Among the three peptides, LQT28 achieved the highest accumulation in tumours.
Thus, LQT28 peptide was selected as a targeting ligand for PDAC stroma. A tumour stroma-targeted nanoscale NO carrier (LQT-NO@NP) was assembled via encapsulation of NO donor DNIC into lipid-PLGA NPs modified with the LQT28 peptide (figure 5A). The size of the LQT-NO@NPs was 143 nm, and the zeta potential was −23.7 mV. The encapsulation efficiency of DNIC was approximately 75%.
Reduction in tumour desmoplasia by the tumour stroma-targeted NO carrier
We next examined whether targeted delivery of NO into the PDAC stroma by LQT-NO@NPs would suppress tumour desmoplasia and increase tumour perfusion in a murine (AK4.4 cells) orthotopic PDAC model (figure 5G). Compared with non-targeted NO@NP treatment and the untreated control, intravenously injected LQT-NO@NPs suppressed PDAC-associated fibrosis, as indicated by a reduction in the α-SMA+ area (figure 5G), and thus increased tumour perfusion, as determined by measuring the Hoechst 33342+ area (figure 5G), in PDAC tumours. Given the potential of LQT-NO@NPs to improve tumour perfusion, we next examined the ability of LQT-NO@NPs to enhance the therapeutic efficacy of the small-molecule anticancer drug (Dox or Gem) in PDAC. Chemotherapeutic agents (Dox or Gem) were loaded into LQT-NO@NPs (LQT-Dox-NO@NPs or LQT-Gem-NO@NPs), which were used to treat orthotopic PDAC tumour-bearing mice as described above, and LQT-Dox-NO@NP or LQT-Gem-NO@NP treatment resulted in a significant reduction in tumour volume (figure 5H) and an increase in the number of apoptotic cells (figure 5I) in tumours compared with control and tumour stroma-targeted NPs loaded with only chemotherapeutic agents (LQT-Dox@NPs or LQT-Gem@NPs). Thus, the enhanced induction of apoptosis by LQT-Dox-NO@NPs or LQT-Gem-NO@NPs might be mediated by the ability of NO to enhance the tumour perfusion of the therapeutic agent and directly affect the expression of apoptosis-related molecules.
Characterisation of a nanogel for co-delivery of NO and TRAIL
Given the potential of NO to eliminate the ECM barrier and mediate the expression of apoptosis-related molecules, we next examined the ability of NO to enhance the therapeutic efficacy of TRAIL in PDAC. The preparation and proposed structure of a tumour-targeted nanogel coencapsulated with an NO donor and recombinant TRAIL are depicted in figure 6A. We first encapsulated recombinant TRAIL in a nanoscale lipid-coated SF hydrogel core in a water-in-oil microemulsion. The hydrophilic head of the phospholipid could interact with SF at the interface. The lipid chains were hydrophobic such that the lipid-coated SF hydrogel cores were soluble in a non-polar solvent such as chloroform. The solvent-mediated transformation of SF hydrogel was initiated at the interface of the microemulsion to encapsulate recombinant TRAIL. The hydrophobic TRAIL-loaded SF core and NO donor DNIC were formulated into lipid-PLGA NPs to generate the TRAIL-NO@Nanogel (figure 6A). To achieve tumour stroma-targeted delivery of TRAIL and NO, we further modified the surface of the NPs with the PDAC stroma-targeting LQT28 peptide to generate an LQT-TRAIL-NO@Nanogel (figure 6A). Scanning electron microscopy (figure 6B) and dynamic light scattering analyses showed that the LQT-TRAIL-NO@Nanogel formed well-dispersed spheres with an average diameter of 166.4±10.3 nm, a polydispersity index of 0.2–0.3 and a nearly neutral zeta potential (0.310±0.694). The TRAIL and DNIC encapsulation efficiencies of the LQT-TRAIL-NO@Nanogel were 97.3%±0.3% and 41.2%±6.9%, respectively. The cumulative release of NO over time was then assessed using the NO-specific fluorescent probe 4,5-diamino-N,N,N′,N′-tetraethylrhodamine (DAR-1). Continuous release of NO from the nanogel occurred over a period of 2–24 hours and was accompanied by the decomposition of DNIC (figure 6C), suggesting that the nanogel could support the continuous release of NO. Using fluorescein isothiocyanate-labelled bovine serum albumin as a tracer protein cargo, we found that the release of protein cargoes from the nanogel under physiological conditions (pH 7.4) was slower than that under acidic conditions (pH 5.5) (figure 6D), suggesting that the decomposition of PLGA and the SF cores in the NPs was pH dependent. The addition of the NO donor did not significantly affect the protein release profile (figure 6D). Thus, these data suggest that the nanogel offers controlled and sustained release of NO and protein cargoes (ie, TRAIL) from NPs. Next, we evaluated the pharmacokinetics of FITC-labelled TRAIL with or without loading in the nanogel in FVB/NJNarl mice (figure 6E). We found that LQT-TRAIL-NO@Nanogel significantly prolonged the circulation of TRAIL compared with free TRAIL after intravenous administration (figure 6E). The results suggest that the nanogel increases the stability of TRAIL and extends its half-life in the blood circulation.
Reduction in collagen I production and increase in apoptosis induction via LQT-TRAIL-NO@Nanogel
To examine the ability of LQT-TRAIL-NO@Nanogel to remodel the fibrotic TME of desmoplastic PDAC, we evaluated changes in collagen deposition in three-dimensional (3D) spheroid cultures consisting of PDAC tumour cells (murine AK4.4 cells or human AsPC-1 PDAC cells) and PSCs after treatment with different formulations for 48 hours (figure 6F,G, see online supplemental figure S8). NO delivered by tumour stroma-targeted NPs (LQT-NO@Nanogel or LQT-TRAIL-NO@Nanogel) significantly suppressed collagen I expression compared with NO delivered by non-targeted TRAIL-NO@Nanogel or NPs loaded with TRAIL alone (LQT-TRAIL@Nanogel) (figure 6F,G, see online supplemental figure S8).
We next evaluated whether NO and TRAIL co-delivery by LQT-TRAIL-NO@Nanogel could enhance TRAIL-induced cell death (figure 6F,H, see online supplemental figure S8). The increase in the percentage of apoptotic cells in the 3D spheroid cultures following treatment with LQT-TRAIL-NO@Nanogel was substantially greater than that after treatment with NPs loaded with each agent alone or with the non-targeted TRAIL-NO@Nanogel (figure 6F,H, see online supplemental figure S8). Taken together, the results indicate that LQT-TRAIL-NO@Nanogel could efficiently induce cancer cell apoptosis in PDAC/PSC 3D culture models via NO-mediated ECM reduction and apoptosis sensitisation.
Remodelling of the fibrotic TME and inhibition of tumour growth by LQT-TRAIL-NO@Nanogel
Next, we examined whether the administration of LQT-TRAIL-NO@Nanogel would reduce the amount of desmoplastic tumour stroma in murine (AK4.4 cells and KPC001 cells) and human (AsPC-1 cells) orthotopic PDAC models.29 30 NPs loaded with recombinant TRAIL and/or DNIC (TRAIL: 4 mg/kg, DNIC: 2 mg/kg every other day) were intravenously administered to mice bearing orthotopic PDAC tumours starting on the third day after tumour implantation, and alterations in the desmoplastic TME were evaluated by Masson’s trichrome and immunofluorescence staining after 2 weeks of treatment (figure 7A). When delivered using tumour stroma-targeted Nanogel modified with LQT28 peptides, both the combination of TRAIL and NO (LQT-TRAIL-NO@Nanogel) and NO alone (LQT-NO@Nanogel) significantly suppressed tumour desmoplasia, as indicated by reduced collagen I expression and reduced infiltration of α-SMA-positive myofibroblasts in PDAC tumours, compared with that observed after treatment with non-targeted TRAIL-NO@Nanogel and the untreated control (figure 7B–D, see online supplemental figures S9–S11). As TRAIL receptor 2 (DR5) expression was found to be upregulated in activated stellate cells, TRAIL may trigger activated PSC apoptosis and thus reduce the ECM.31 Thus, we observed that treatment with TRAIL alone (LQT-TRAIL@Nanogel) moderately alleviated tumour desmoplasia (figure 7B–D, see online supplemental figures S9–S11).
We evaluated whether co-delivery of NO led to increased anticancer activity of TRAIL in orthotopic PDAC models. Compared with treatment with tumour stroma-targeted NPs loaded with either TRAIL or NO alone (LQT-TRAIL@Nanogel or LQT-NO@Nanogel) or with non-targeted TRAIL-NO@Nanogel, treatment with both TRAIL and NO (LQT-TRAIL-NO@Nanogel) resulted in a significant reduction in tumour volume and an increase the number of apoptotic cells in tumours in all AK4.4, KPC001 (murine) and AsPC-1 (human) cell PDAC models (figure 7B, E and F, see online supplemental figures S11 and S12). Furthermore, the combination of TRAIL and NO treatment (LQT-TRAIL-NO@Nanogel) also significantly increased overall survival in the orthotopic murine PDAC(AK4.4) model compared with either TRAIL or NO alone (LQT-TRAIL@Nanogel or LQT-NO@Nanogel) or with non-targeted TRAIL-NO@Nanogel (figure 7G).
To assess treatment safety, we evaluated safety parameters in FVB/NJNarl mice. Tumour stroma-targeted LQT-TRAIL-NO@Nanogel was well tolerated in animal safety experiments, with no changes in hepatic enzyme levels (aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase and γ-glutamyltransferase), renal function indexes (blood urea nitrogen and creatinine) or serum methemoglobin levels noted, as these values were comparable to those of untreated mice (table 2).
Assessment of LQT-TRAIL-NO@Nanogel toxicity
In this study, we showed that encapsulation of the NO donor DNIC into a PEGylated PLGA-coated SF nanogel and modification of the nanogel with tumour stroma-targeting peptides enhanced the stability of the NO donor, enabled the steady release of NO and facilitated targeted NO delivery into the desmoplastic stroma of PDAC tumours. This NO-producing stroma-targeted nanogel suppressed PSC activation, reduced ECM production, increased tumour perfusion and sensitised PDAC to TRAIL. By taking advantage of the therapeutic effects of NO, which can reprogramme the desmoplastic stroma and achieve apoptosis sensitisation in cancer cells, this combination therapy—co-delivery of TRAIL and NO by the stroma-targeted nanogel—efficiently suppressed tumour growth in both murine and human PDAC models. Thus, this stroma-targeted NO and TRAIL co-delivery system has great potential to improve the efficacy of TRAIL therapy and possibly that of other chemotherapies or targeted therapies for future clinical application in PDAC.
Discussion
Tumour stroma-targeted delivery systems have been developed by modifying ligands that recognise receptors expressed on myofibroblasts.32–34 Miao et al used anisamide, a ligand of the sigma receptor overexpressed in both tumour cells and CAFs, conjugated to a PEGylated gene carrier to successfully deliver genetic cargoes into orthotopic PDAC tumours.35 Plasma membrane serine protease fibroblast activation protein (FAP) is another potential target in CAFs. Carriers modified with a single-chain variable fragment that recognises FAP were shown to efficiently deliver chemotherapeutics or immunotoxins into CAFs and deplete FAP-expressing stromal cells, thus altering the TME to achieve anticancer effects.36 37 Although the sigma receptor and FAP are highly expressed on cancer cells and fibroblasts, respectively, these receptors are also expressed by other normal cells, such as pericytes and epithelial cells. To overcome this issue of non-specific targeting, in this study, we identified novel tumour stroma-targeting peptides by in vitro–in vivo screening of phage libraries in which the first round of in vivo screening was conducted in mice bearing orthotopic PDAC tumours. After enrichment of the phages that homed to PDAC tumours, we performed three rounds of in vitro screening in human PSCs to capture peptides capable of targeting both PDAC tumours and human PSCs for future experimental and clinical applications. The tumour stroma-targeting peptides identified by phage display may also target CAFs in other types of tumours characterised by particularly enhanced desmoplasia.
The potential limitations of stromal depletion therapy are the possibility of increasing cancer aggressiveness and metastasis.8 The ECM component collagen I acts as a bimodal molecule that regulates cell division and cell growth.38 An overly abundant ECM can function as a barrier to suppress pancreatic cancer progression and metastasis.39 CAFs may also suppress tumour growth in pancreatic cancer by mediating anticancer immunity.40 Non-selective depletion of the ECM or elimination of CAFs in PDAC was found to increase PDAC aggressiveness and metastatic capability.8 In this study, we established tumour stroma-targeted NPs to deliver NO into the PDAC tumour stroma, resulting in the reprogramming of activated PSCs and the alleviation of tumour desmoplasia. Rather than promoting metastasis as reported for other stromal depletion agents, our previous studies showed that the delivery of NO could suppress metastasis by inhibiting the migration/invasion of cancer cells and epithelial–mesenchymal transition in cancer cells.41 More studies are needed to examine the metastasis inhibition potential of NO to serve as a promising adjuvant treatment for PDAC.
In summary, our newly developed nanoscale TRAIL therapy includes the following beneficial elements: (i) a tumour stroma-targeting peptide that enhances the uptake of NPs into the PDAC stroma; (ii) an SF hydrogel core to load protein cargoes such as TRAIL that also allows the pH-responsive release of protein cargoes; and (iii) DNIC—a synthetic NO donor loaded in a shell composed of lipids and PLGA to reprogramme the desmoplastic stroma and promote apoptosis induction by TRAIL in PDAC. This combination therapy—the co-delivery of TRAIL and NO by a stroma-targeted nanogel that remodels the fibrotic TME and suppresses tumour growth—has the potential to be translated into a safe and promising treatment for desmoplastic tumour types such as PDAC.
Method and materials
Additional materials and methods are included in online supplemental information (see online supplemental materials and methods).
Preparation of the LQT-TRAIL-NO@Nanogel
SF aqueous solutions were prepared as previously described.42 The TRAIL-DNIC@Nanogel was prepared through a water-in-oil single emulsion. SF solution (1 wt %) was first mixed with TRAIL protein. DOPA (74 µL, 35 mM) was added to the oil phase of cyclohexane and Igepal-520 (7:3, v/v). The hydrophilic phase was added dropwise to the oil phase. The emulsions were then mixed for 40 min to form the condensed cores of SF nanogel/TRAIL. Later, 3 mL of 100% Ethanol (EtOH) was added to disrupt the emulsion, and the mixture was centrifuged at 20133 g for 15 min. After removing the supernatant solution, the SF nanogel/TRAIL cores were collected and washed two times with 100% EtOH to remove residual organic solvents. The SF nanogel/TRAIL cores were suspended in chloroform, vortexed and sonicated. A mixture of free lipids and DNIC (DOPC:DOTAP:DSPE-PEG2000:DSPE-PEG2000-MAL:cholesterol:DNIC:PLGA=1:1:1:0.1:2:0.75:0.025 molar ratio) was added to the SF nanogel/TRAIL cores and then dried under N2. After evaporating the chloroform, 250 µL of water was added to form the TRAIL-DNIC@Nanogel. Peptide modification was achieved by conjugation of the thiol group on the peptides with maleimide on DSPE-PEG2000. The unconjugated maleimide groups were blocked by cysteine to avoid non-specific binding. The LQT-TRAIL-DNIC@Nanogel was centrifuged at 25 000 relative centrifugal force (rcf) for 20 min at 25°C to collect the NPs.
Statistics
Statistical analyses were performed using GraphPad Prism V.6. Student’s t-tests or Mann-Whitney U tests were used for comparisons between two groups according to the data distribution. One-way analysis of variance followed by Tukey’s post-hoc test was used for comparisons of three or more groups. Values were normally distributed, and the variance was similar between compared groups. P<0.05 was considered statistically significant.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All relevant data supporting the key findings of this study are available within the article and the supplementary files.
Ethics statements
Patient consent for publication
Ethics approval
We performed immunostaining for analysing binding affinity of phages to human pancreatic tumour samples, obtained under the reviewed and approved protocols of the Institutional Review Board of Taipei Veterans General Hospital (IRB2017-01-016C, 2021-07-041BC).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
H-CH, Y-CS and C-PL contributed equally.
Contributors H-CH and Y-CSu conceived and designed the experiments and analysed the data; C-PL, P-HC, Y-TT, B-WL, F-FH, Y-TC, Y-HL, HTH and Y-CSh performed the experiments; DW, H-TC, C-CH, I-JL and H-CW contributed materials and analysis tools; T-TL and JW supervised experiments; YC was the principal investigator of the study responsible for study concept and design. Approval of the final version of the manuscript: all authors.
Funding This study was supported by the Ministry of Science and Technology (108-2221-E-007-104-MY5, 110-2628-E-007-007), the National Tsing Hua University, Taiwan (Grant No. 110Q2709E1, 110Q2705E1, 110Q2711E1), the National Health Research Institutes (NHRI-EX110-11015BI) and the ‘Frontier Research Center on Fundamental and Applied Sciences of Matters’ from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE 110QR001I5) and the Ministry of Science and Technology (MOST 110-2634-F-007-023).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.