Article Text

Abstract
Objective The human gut fungal community, known as the mycobiome, plays a fundamental role in the gut ecosystem and health. Here we aimed to investigate the determinants and long-term stability of gut mycobiome among middle-aged and elderly adults. We further explored the interplay between gut fungi and bacteria on metabolic health.
Design The present study included 1244 participants from the Guangzhou Nutrition and Health Study. We characterised the long-term stability and determinants of the human gut mycobiome, especially long-term habitual dietary consumption. The comprehensive multiomics analyses were performed to investigate the ecological links between gut bacteria, fungi and faecal metabolome. Finally, we examined whether the interaction between gut bacteria and fungi could modulate the metabolic risk.
Results The gut fungal composition was temporally stable and mainly determined by age, long-term habitual diet and host physiological states. Specifically, compared with middle-aged individuals, Blastobotrys and Agaricomycetes spp were depleted, while Malassezia was enriched in the elderly. Dairy consumption was positively associated with Saccharomyces but inversely associated with Candida. Notably, Saccharomycetales spp interacted with gut bacterial diversity to influence insulin resistance. Bidirectional mediation analyses indicated that bacterial function or faecal histidine might causally mediate an impact of Pichia on blood cholesterol.
Conclusion We depict the sociodemographic and dietary determinants of human gut mycobiome in middle-aged and elderly individuals, and further reveal that the gut mycobiome may be closely associated with the host metabolic health through regulating gut bacterial functions and metabolites.
- intestinal microbiology
- molecular epidemiology
Data availability statement
Data are available in a public, open access repository. The raw data of the ITS2 sequencing, 16S rRNA sequencing and metagenomic sequencing are available in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb (accession number CNP0002114, CNP0000829 and CNP0001510, respectively). Other data sets generated during and/or analysed during the current study are available from the corresponding author upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The human gut fungal community, known as the mycobiome, is integral to the gut ecosystem and of putative health consequence.
The composition and long-term stability of the gut mycobiome in middle age and later life are not known.
What are the new findings?
Considerable gut fungal composition is temporally stable while modulated by age, long-term habitual diet and host physiological states.
The gut mycobiome is interdependent on bacterial taxonomy and function, where Saccharomycetales spp may interact with gut bacterial diversity to influence insulin resistance.
Bidirectional mediation analyses indicate that bacterial function or faecal histidine may causally mediate an impact of gut fungi on blood cholesterol.
How might it impact on clinical practice in the foreseeable future?
Gut mycobiome features may be new interventions or therapeutic targets for human metabolic diseases.
Introduction
Fungi, as eukaryotes, are ancestrally and ecologically intrinsic to terrestrial life with multiple roles, extending to the regulobiotic.1–3 As we appreciate more and more how socioecological we are, the place of fungi in our biology and health is increasingly evident.3 4 The human gut fungal community,5 6 referred to as the gut mycobiome,7 comprises less than 0.1% of the microbial cells in the gut ecosystem,8 but changes in its composition are linked to a number of diseases including IBD, IBS and colorectal cancer.9 Geography, urbanisation, ethnicity and diet play a role in shaping the gut mycobiome composition.6 10 11 While the gut mycobiome may be individual-specific, it may undergo age-associated and personal behaviourally related changes.12 13 Presently, little is known about the profiles and long-term stability of the gut mycobiome among adults as they age.
Gut bacteria are associated with human metabolic diseases like type 2 diabetes,14 hypertension15 and obesity.16 The gut fungal and bacterial communities are unavoidably connected and likely to influence each other in various ways which interdigitate with host metabolic pathways. The modes of cross-kingdom interaction include mutualism, commensalism, amensalism, parasitism and competition.7 These may be affected by physical association, molecular dialogue and an environmental milieu constituted by gut ecology.17 This ecosystem subserves the functionality of the colorectum in transiting food residue, fermentation and nourishing the gut itself. While some associations between the gut mycobiome and metabolic disease have been reported in animal models18 19 and in limited human studies,20 21 the potential for gut fungal and bacterial cohabitation to modulate metabolic health in humans remains unclear.
In this study, we have profiled gut fungal and bacterial community structure and the faecal metabolome in a population-based Guangzhou Nutrition and Health Study (GNHS) cohort in China (n=1244). We then characterised the long-term stability and determinants of the fungal community among these middle-aged and elderly adults. To gain biological insight, we examined the ecological links between gut fungi, bacteria and the faecal metabolome. Finally, we hypothesised that gut fungi could interact with bacteria to modulate metabolic health.
Results
Composition and long-term stability of the human gut mycobiome
We studied 1244 participants with a mean age of 64.9 years as a subset of the GNHS cohort (online supplemental figures S1 and S2A and online supplemental table S1). Based on the internal transcribed spacer 2 (ITS2) sequences, a taxonomic profile of the gut mycobiome was determined (online supplemental figure S1). There was a total of 204 gut fungal genera (online supplemental tables S2 and S3) where Ascomycota, Basidiomycota and unclassified Fungi sp were the most dominant phyla with a mean relative abundance of 77.0%, 5.7% and 17.0%, respectively (figure 1A–C). After 5% prevalence filtering, 26 gut fungal genera were identified as a group of core taxa, including Saccharomyces, Candida, Aspergillus, Malassezia, etc (figure 1A and online supplemental table S3). Through literature review, we showed that these gut fungal genera might have potential environmental determinants or sources (online supplemental table S4). We found similar abundance ratios of Ascomycota to Basidiomycota in the discovery GNHS cohort as in the Human Microbiome Project cohort12 and the Danish cohort22 (online supplemental figure S2B). Some genera were consistently present, with 40 genera detected in all three cohorts (online supplemental figure S2C and online supplemental table S5).
Supplemental material
Supplemental material

Composition and variation of human gut mycobiome. (A) Taxonomic tree based on ITS2 sequencing data (n=1244, baseline) showing the main fungal representatives. The colour of each node is consistent with the colour of the corresponding fungal phylum node located in the lower left corner of the graph. The nodes from the inner to the outer circles represent the fungal taxa from the phylum to the species level. The triangles on the outermost ring represent the core genera of the study. (B) The relative abundance of fungal phyla among individuals. The X axis represents the individual ID number.’Fungi sp.’ here represents unidentified fungal phylum. (C) PCoA plots of Bray-Curtis distance of gut mycobiome composition from ITS2 sequences. Each dot represents an individual. The heatmap colour indicates the relative abundance of each phylum, where red indicates high abundance and blue indicates low abundance. (D) Long-term alterations of the gut mycobiome in the population from baseline to follow-up (n=184). Sankey plots displayed the proportions of gut fungal genera with significant (q<0.05) increased or decreased abundance or no significant alteration (stable). The median follow-up time was 3.2 years. The q values (false discovery rate adjusted p value) were calculated using the Benjamini-Hochberg method. PCoA, principal coordinate analysis.
Long-term stability of the gut mycobiome was determined in follow-up after ~3.2 years in 184 participants (online supplemental table S6) with ITS2 fungal data that showed non-significant alterations over time in 11 genera of the Ascomycota (eg, Pichia, Alternaria and Wickerhamiella) (figure 1D and online supplemental table S7), which indicates that the relative abundances of these genera were temporally stable. Saccharomyces, Candida and Aspergillus were abundant at baseline and remained the most prevalent taxa at the follow-up visit (online supplemental figure S3A,B). There were seven core fungal genera repeatedly detected in the same individual at both time points in over 40% of the participants (online supplemental figure S3C). For the dominant fungal phyla, this probability was over 85%. However, eight genera had a significant increased change in relative abundance, Penicillium and Candida being examples, while 5 out of 26 genera showed a decreased abundance (paired Wilcoxon test, q<0.05) (online supplemental table S7). These changes may be partly attributable or contribute to increasing age, as in a metagenomic study with age-associated alterations in the gut mycobiome, particularly Aspergillus and Penicillium.13
Compared with gut bacteria, the gut mycobiome has sparser characteristics. Gut bacterial composition was profiled using the 16S rRNA sequencing for 1244 participants. The gut bacteria had a larger α-diversity than gut fungi (online supplemental figure S4A), which was similar to other microbiomic reports in healthy individuals.12 23 However, gut mycobiome composition varied greatly between individuals (online supplemental figure S4B), suggesting that gut fungi may be more personalised.
Determinants of variation in gut mycobiome structure
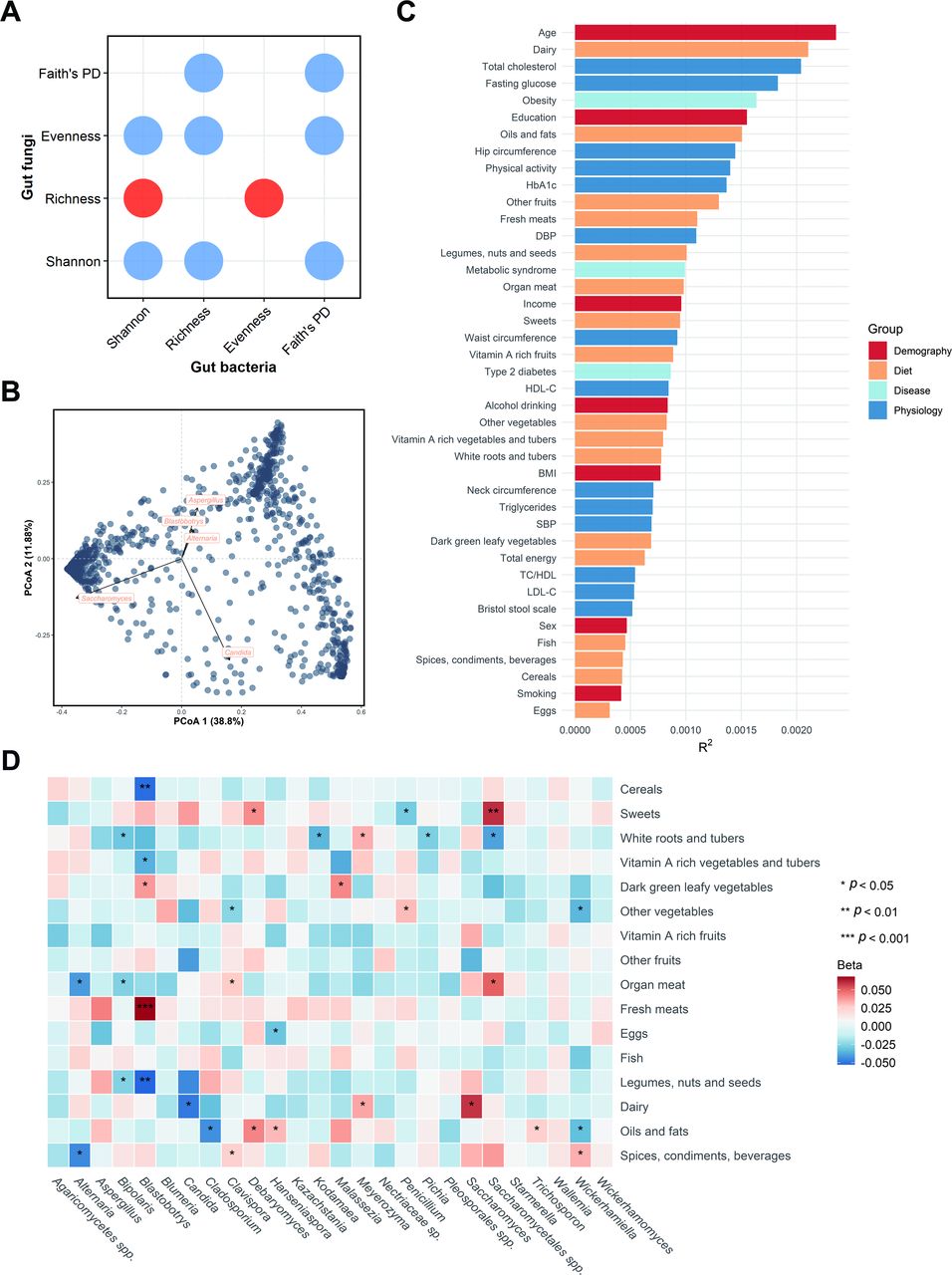
Procrustes analysis revealed a strong correlation between the data matrix of the gut fungi and bacteria (p=0.009) (online supplemental figure S5). There were negative associations for most of the α-diversity indices between the two kingdoms (figure 2A), suggesting the ecobalance between gut fungal and bacterial community. At the genus level, five were identified as the top contributors to the gut fungal community variation, including Saccharomyces, Candida and Aspergillus, followed by Alternaria and Blastobotrys (figure 2B).

Determinants of gut fungal structural variation. (A) Dot plot shows the associations between gut fungal and bacterial α-diversity indices. The red indicates a positive association, while blue indicates a negative association. (B) Top five contributors to fungal community variation was determined by envfit analysis (p<0.001), plotted on the two first PCoA dimensions. (C) The effect sizes of host factors on human gut mycobiome were evaluated by permutational multivariate analysis of variance (Adonis, permutations=999, n=1188, baseline). The bars were coloured according to metadata categories. (D) Associations between long-term dietary habits and gut mycobiome (n=1239). Beta and p values were calculated in a linear regression model adjusted for age, gender, BMI, total energy, smoking status, drinking status, income level, education, physical activity and Bristol stool scale. The quartiles of dietary variables were z-score transformed. ‘sp‘ or ‘spp‘ here represents unidentified fungal genus. BMI, body mass index; DBP, diastolic blood pressure; HbA1c, glycated haemoglobin; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; PCoA, principal coordinate analysis; PD, phylogenetic diversity; SBP, systolic blood pressure; TC:HDL, total cholesterol:high-density lipoprotein cholesterol ratio.
We correlated 7 demographic factors, 14 physiological traits, 16 dietary intakes and 3 diseases with the overall variation in the gut mycobiome. Age, dairy intake and total cholesterol were significantly associated with the gut fungal composition at a nominal p value of <0.05 (Bray-Curtis distance, PERMANOVA) (figure 2C and online supplemental table S8). Age had the largest explanatory power for the interindividual variation in gut mycobiome, which further suggests that some fungal taxa alter with time. For instance, compared with middle-aged participants, Blastobotrys and Agaricomycetes spp were depleted, while Malassezia was enriched in the elderly (p<0.05, online supplemental table S9). Spearman correlation analysis was used to examine the association between fungal α-diversity and host factors. None of the signals surpassed the conservative threshold of q<0.05. Notwithstanding, gut fungal richness was positively associated with the total energy intake, various fruits consumption and Bristol stool scale, and was negatively associated with smoking status at a nominal p value of <0.05 (online supplemental figure S6). The patterns of significance for different factors varied among middle-aged and elderly participants with total energy intake positively associated with fungal α-diversity in middle-aged individuals but not in the elderly (online supplemental figure S6).
We then investigated the links between long-term habitual dietary intakes and the gut mycobiome. The multivariable linear regression models indicated that habitual diet may be an important determinant of the mycobiome with most foods associated positively or negatively with specific fungi. The only foods not significantly associated with fungi were fruits and fish. For instance, dairy intake was positively associated with Saccharomyces and Meyerozyme but inversely associated with Candida (p<0.05) (figure 2D).
Ecological connectedness of gut fungi and bacteria
To explore the ecological links between gut fungi and bacteria, the Sparse Correlations for Compositional data (SparCC) algorithm was applied to construct the interkingdom and intrakingdom association networks. A total of 6346 significant associations were identified and 3538 were validated by the Spearman correlation analysis (q<0.05, online supplemental figure S7A), suggesting extensive taxonomic associations between gut fungi and bacteria. However, only 134 associations were cross-kingdom associations (online supplemental figure S7B), which indicates potentially greater intrakingdom communication than cross-kingdom. However, given the evolutionary resilience and ecological dominance of fungi, their regulobiotic position may be disproportionately consequential.
The association between fungi and bacteria was highly variable in taxonomy (figure 3A). The positive correlation of some bacteria (Bifidobacterium and Veillonella) with fungi (Blumeria and Saccharomyces) suggests that there may be a codependence for both microbes. The inverse correlation of other bacteria (Butyricimonas and Alistipes) with fungi (Hanseniaspora and Wickerhamiella) suggests that there may be competitive or inhibitory actions (q<0.05; figure 3A and online supplemental table S9). When α-diversity of the gut bacteria was considered, significant correlations were found with specific fungal taxa (online supplemental figure S8). Overall, there were more negative associations for richness, evenness and phylogenetic diversity with seven fungi (Wickerhamomyces, Starmerella, Meyerozyma, inter alia) and only two fungi (Saccharomyces and Agaricomycetes spp) with positive associations. Thus, gut fungi may have the potential to manipulate neighbouring bacterial communities or vice versa.

Ecological connections between gut fungi and bacteria. (A) Significant taxonomic associations between gut fungi and bacteria (|SparCC coefficient| >0.2, q<0.05; n=1244, baseline). The colour indicates the correlation coefficients estimated by the SparCC algorithm. The white spaces represent the non-significant associations (q>0.05). (B) Summary of the gut mycobiome-related bacterial KOs (n=1009, baseline). The top three bar plots show the number of gut mycobiome-related bacterial KOs enriched in the KEGG pathway, module (reaction) and brite (functional hierarchies), respectively. The bottom bar plot shows the number of bacterial KOs related to the specific fungal genus. Detailed statistics about the associations between specific gut fungi and bacterial KOs are listed in online supplemental table S11. The q values were calculated using the Benjamini-Hochberg method. ABC transporter, ATP-binding cassette transporter; KEGG, Kyoto Encyclopaedia of Genes and Genomes; KO, KEGG orthologue; TCA cycle, tricarboxylic acid cycle.
We hypothesised that gut fungi may be more related to bacterial gene function rather than taxonomy. Based on the metagenomics sequencing, the bacterial Kyoto Encyclopaedia of Genes and Genomes orthologues (KOs) were profiled using HUMAnN2 to assess the associations between core fungal taxa and the gene functions of gut bacteria. The KOs associated with the mycobiome were most enriched in metabolic pathways, followed by the biosynthesis of secondary metabolites and microbial metabolism in diverse environments, which indicates the potential ways of cross-kingdom communication between gut fungi and bacteria (q<0.05, figure 3B). When the KOs were mapped into the Brite hierarchical topologies, most of the significant KOs represented the bacterial enzymes, transporters, secretion systems and transcription factors. Interestingly, Saccharomycetales spp (unclassified genus) had the highest number (170) of associations with bacterial functions, but this taxon only had three significant associations with bacterial taxa (figure 3B and online supplemental tables S10 and S11).
Gut mycobiome and the faecal metabolome
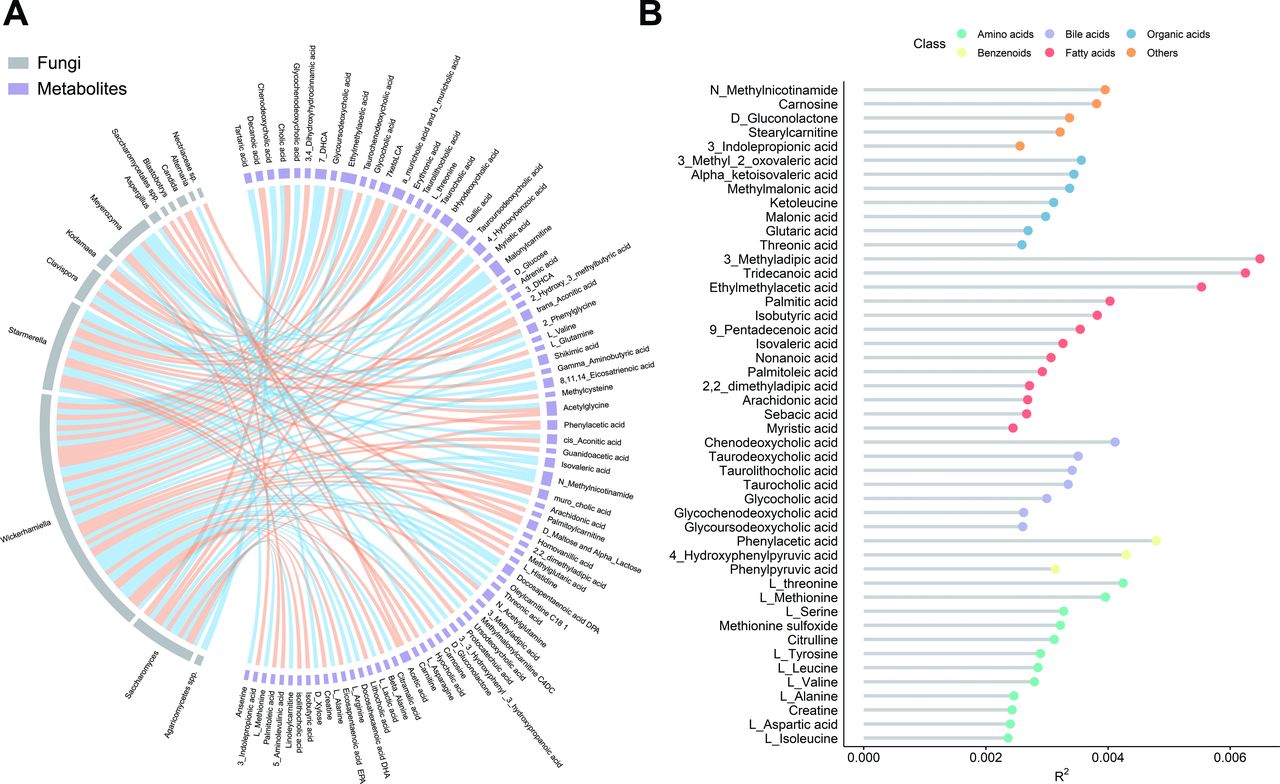
As with cross-kingdom associations, examining the faecal metabolites that were associated with gut mycobiome could facilitate mechanistic insight into the gut ecosystem. The absolute concentrations of 198 metabolites were targeted measured using the UPLC-MS/MS system in 913 participants. The Procrustes analysis demonstrated a strong cooperativity of the gut mycobiome and faecal metabolome (online supplemental figure S9A). There were 110 associations identified involving 13 gut fungi and 82 faecal metabolites that were significant at a 5% FDR (figure 4A and online supplemental table S12). Notably, about 60% bile acids (15 out of 25) were significantly associated with Wickerhamiella (figure 4B). For instance, Wickerhamiella was positively associated with chenodeoxycholic acid, cholic acid and glycochenodexycholic acid (q<0.01). Saccharomyces was positively associated with decanoic acid which is a medium-chain saturated fatty acid with antibacterial and an anti-inflammatory role (Spearman’s rho=0.18, q<0.001). Intriguingly, all these metabolites were associated with various fungi-related KOs (online supplemental table S13), which might support an interpretation of gut mycobiome and bacterial codependence or functional synergism.

Gut mycobiomic associations with faecal metabolome. (A) the significant (q<0.05) associations between gut fungi and faecal metabolites (n=913, baseline; orange, positive; blue, negative). (B) Proportion of variation in gut mycobiome explained by the faecal metabolites (PERMANOVA, q<0.2). The colours of dots indicate the different classes of the metabolites. The q values were calculated using the Benjamini-Hochberg method.
To further use faecal metabolite information in the understanding of gut mycobiomic composition and variation, correlation analyses between the abundances of faecal metabolites and gut fungal community characteristics were conducted. Overall, 47 faecal metabolites were associated with the fungal structure at p<0.05 (PERMANOVA, q<0.2; figure 4B). Among these metabolites, more than a quarter (13 out of 47) were fatty acids. The major contributors to the variance of the whole gut mycobiome were 3-methyladipic acid, tridecanoic acid and ethylmethylacetic acid. In addition, more than half (25 out of 47) of the above faecal metabolites were also associated with specific fungal taxa, such as carnosine, D-gluconolactone and L-threonine (online supplemental figure S9B). This permits a speculation that the faecal metabolites may play an elaborate role in the cross-talk between gut fungi, bacteria and general host biology with health implications.
Pathways from gut mycobiome to metabolic health
We sought to explore the potential relevance of the human gut mycobiome to metabolic health by performing two linear regression models (see online supplemental methods). These models were performed between 26 core fungi and 11 metabolic risk factors adjusted for age and gender. Accordingly, 12 nominally significant associations from the abundance-based models and 26 from the presence-based models were identified (p<0.05, figure 5A and online supplemental table S14). For example, we observed that the relative abundance of Penicillium was positively associated with fasting glucose, and the presence of Malassezia and Kazachstania was positively associated with fasting glucose and waist circumference, respectively. These findings were replicated in the independent replication cohort (figure 5A, online supplemental figure S10 and online supplemental table S15). Notably, the relative abundance of Saccharomyces was inversely associated with fasting glucose but positively associated with high-density lipoprotein cholesterol (figure 5A), suggesting that Saccharomyces might be a beneficial fungal taxon for human metabolic health. In line with this, an animal study showed that Saccharomyces boulardii could reduce blood glucose and triglycerides in mice with diabetes.24 Even so, none of the direct associations surpassed the conservative threshold of q<0.05.

Associations between gut mycobiome and metabolic traits. (A) Heatmap of cross-sectional associations between gut fungi abundance and presence (left: continuous variable, right: binary variable) and metabolic traits. Linear regression models were adjusted by age and gender. The total number of participants in each analysis was 1211 for HDL cholesterol, LDL cholesterol, triglycerides, TC and TC:HDL, 1244 for BMI, 1210 for fasting glucose, 1212 for HbA1c and HOMA-IR, 1016 for insulin and 1235 for waist circumference. Triglycerides, fasting glucose, TC:HDL and insulin were log-transformed. The bold square represents the significant associations replicated. (B) Forest plot shows the interaction of gut fungi with bacterial Shannon index on HOMA-IR. Associations were expressed as the difference in metabolic traits (in SD unit) per 1 SD of bacterial richness. Linear regression models were adjusted for age and gender. Insulin was log-transformed. Associations were stratified by the presence of gut fungi. The error bars represent CIs. (C) Hypothesised potential mechanisms linking the gut fungi, bacteria and insulin sensitivity. Based on the various associations in the present study, we showed that the presence of Saccharomycetales spp may increase the abundance of SCFA-producing bacteria, sush as Clostridium sensu stricto 1, Faecalitalea and Megamonas, which leads to the production of more SCFAs (acetic acid and butyric acid) in human gut. These changes may lead to the improvement of insulin sensitivity. This figure was created with BioRender.com. HbA1c, glycated haemoglobin; HDL, high-density lipoprotein; HOMA-IR, homeostatic model assessment of insulin resistance; LDL, low-density lipoprotein; SCFA, short-chain fatty acid; TC:HDL, total cholesterol:high-density lipoprotein cholesterol ratio.
We hypothesised that the gut mycobiome could modify the associations between bacterial α-diversity and metabolic health. With a linear regression model, we explored the interaction of core fungal taxa with bacterial α-diversity indices on the metabolic traits. Three significant interactions were observed repeatedly in the replication cohort (figure 5B and online supplemental figure S10). A noteworthy example was the significant interaction of Saccharomycetales spp with bacterial α-diversity on homeostatic model assessment of insulin resistance (HOMA-IR) (p value for interaction <0.05, figure 5B), as well as insulin (online supplemental figure S11A). Per 1 SD difference in the Shannon index was associated with a lower HOMA-IR only among individuals with Saccharomycetales spp but not those without this fungus. Intriguingly, changes in the relative abundance of this fungus between baseline and follow-up were also inversely associated with parallel changes in total and low-density lipoprotein (LDL) cholesterol (p<0.05, online supplemental table S16).
To further understand the potential mechanisms of the aforementioned interactions, we looked for associations between Saccharomycetales spp, bacterial taxa and short-chain fatty acids (SCFAs). SCFAs are gut bacteria-derived metabolites, produced in the host as energy substrates for the colonic epithelium and peripheral tissues, that can be a pathway to metabolic disorders.25 We observed that Saccharomycetales spp had a positive association with Clostridium sensu stricto 1, Faecalitalea and Megamonas (p<0.05, figure 5C). All these bacterial taxa are SCFA producers.26–28 Consistent with this, Saccharomycetales spp was positively associated with acetic acid and butyric acid (p<0.008, online supplemental table S12). Nevertheless, more mechanistic work is needed to understand the relationship among gut Saccharomycetales spp, bacteria, SCFAs and glucose homeostasis.
To evaluate whether gut bacterial function can mediate the fungal impact on host metabolic health, bidirectional mediation analysis was conducted using baseline gut mycobiome and fungi-related bacterial KOs/faecal metabolites, together with any longitudinal change of host phenotypes between baseline and follow-up visit. This showed that the presence of Pichia probably contributed to a decrease in LDL cholesterol by increasing the bacterial functional levels, such as exoribonuclease II (K01147), deoxyribodipyrimidine photolyase-related protein (K06876) and putative hemin transport protein (hmuS, K07225) (all pmediation <0.007, figure 6). Specifically, hmuS protein levels explained 26% of the total effect of Pichia on LDL cholesterol, while the inverse mediation analyses revealed that hmuS could not influence the LDL cholesterol levels through Pichia. We also found that Pichia may decrease the total cholesterol levels through faecal histidine (pmediation <0.05, online supplemental figure S12). Of interest in ageing biology, the positive association between the Saccharomycetales spp and Pichia decreased from middle age to later life (online supplemental figure S13A). Support for the biological meaningfulness of this finding comes from the longitudinal data where this concurrence weakened from baseline to the follow-up visit (online supplemental figure S13B). Taken together, these findings represent gut mycobiome interactions by gut bacterial taxonomy and function relevant to metabolic health.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mediation linkages among the gut mycobiome, KEGG orthology and metabolic traits. pIDE and pinv-IDE were estimated by the bidirectional mediation analysis. The metabolic traits were the longitudinal change between the baseline and follow-up visits, while the gut mycobiome and bacterial gene were from the baseline (n=284). The red arrowed lines indicate the gut fungal effects on the changes of phenotype mediated by bacterial gene function with corresponding mediation pIDE. Inverse mediation was performed to check whether gut bacterial gene function can influence the human phenotype through gut mycobiome. The percentage number in the centre of each figure are effect ratios reflecting the proportion of the total effect of the independent variable on the dependent variable that is explained by the mediator, that is, 28%. The figure was created with BioRender.com. IDE, indirect effect; inv-IDE, inverse indirect effect; LDL-C, low-density lipoprotein cholesterol.
Discussion
This work depicts the gut mycobiome in a broad microbiomic context, for an ethnogeographically defined cohort defined by age, long-term habitual diet and physiological status. Ascomycetes were the dominant group of gut fungi and temporally stable even after 3.2 years. Interactions were identified between the gut mycobiota and bacterial α-diversity in relation to metabolic health, which could be replicated in an independent cohort. Of indicative and metabolic interest, Pichia may influence LDL cholesterol through specific bacterial functions.
Previous research has shown that Ascomycota and Basidiomycota are two dominant phyla in the GI tract, which is consistent with our findings.7 The present study identified 26 core fungal taxa, 8 of which have been listed as the most commonly detected fungi in gut mycobiomic studies,29 including Candida, Saccharomyces, Penicillium, Aspergillus, Malassezia, Cladosporium, Debaryomyces and Trichosporon.30 Notably, we found through literature review that these gut fungal genera have various environmental counterparts which indicate the potential origins or sources of the fungi and reflect that human health may be inherently affected by the environments that we traverse. Many fungal taxa displayed long-term stability even after 3.2 years in 184 participants, indicative of a core mycobiome, within its total. A fundamental question is which fungi are truly colonising the human gut? In 2018, a study in 100 healthy individuals argued that fungi do not routinely colonised the GI tracts of healthy adults.31 However, core fungal taxa were identified in the present study and could be observed at both time points in the discovery and replication cohort over 3 years apart. We also found some fungi were temporally stable in the same individual. It seems that the gut mycobiome may play a role in the long-term stability of the gut ecosystem. To our knowledge, the present study is the first investigation into the difference in gut mycobiome between the elderly and younger individuals. We not only found that Blastobotrys was decreased from baseline to follow-up longitudinally, but also revealed that this genus had a depletion in the elderly compared with the middle-aged individuals cross-sectionally. Blastobotrys adeninivorans can assimilate a wide spectrum of carbon and nitrogen sources, which makes it a dominant yeast in the fermentation of Pu-erh tea.32 Further studies are needed for a more comprehensive profiling of mycobial alterations through the full human life cycle.
The influence of diet on human gut mycobiome composition, and in association with the bacterial microbiome, appears likely, extensive and complex, especially for specific food categories.11 33 Previous work has shown that long-term habitual dairy consumption is associated with a higher bacterial α-diversity.34 Interestingly, dairy consumption made the largest contribution to the variation of fungal community among the 16 food categories in our study. Moreover, dairy consumption was positively associated with Saccharomyces but inversely associated with Candida. Previous studies reveal anti-inflammatory effects of Saccharomyces35 36, which were negatively associated with the Framingham 10-year risk score.21 However, some species of Candida can infect humans, such as Candida albicans37 and C. parapsilosis.38 We found that bacterial α-diversity was positively associated with Saccharomyces and negatively associated with Candida. Whether coherent with this or not, gut bacterial α-diversity seems to be a good indicator of a ‘healthy gut’.39 Perhaps dairy consumption contributes to a healthier diet in terms of gut ecohealth by modulating gut microbial relationships.
We have comprehensively analysed the ecological connections between gut fungi and bacteria. There are broad associations between gut fungi and bacterial taxa, gene functions and functional products, that is, metabolites, to be found in our study. Specifically, the human gut mycobiome is closely related to various metabolic pathways and the biosynthesis of secondary metabolites by bacteria. Moreover, since fungal signalling may be via volatile organic compounds (VOCs), recognition and diagnosis of such phenomena may be more successful with exhaled breath than with faecal tests.40 Saccharomyces was positively associated with decanoic acid (capric acid). This metabolite is a VOC fatty acid which can be produced by S. boulardii.41 It exemplifies many ecological interactions involving fungi mediated by VOCs.42 Decanoic acid can, in turn, modulate fatty acid biosynthesis by bacteria.43 Given the likely underestimation of gut microbial VOC physiology, its pursuance should be worthwhile.
Microbiomic ecology is fundamental for the optimisation of human health, for which Saccharomycetales spp is proving an exemplar.44 There is a significant interaction with Saccharomycetales spp for bacterial α-diversity on HOMA-IR. The lower value of HOMA-IR indicated a higher degree of insulin sensitivity.45 In a population-based cohort study, bacterial α-diversity measures were lower among participants with higher measures of HOMA-IR in both the NFBC1966 cohort (n=506) and in the Twins UK cohort (n=1140).46 This is in line with the present study among the Saccharomycetales spp carriers. SCFAs are used as energy sources, contributing to the whole-body energy metabolism and glucose homeostasis.25 47 Because of the positive associations between the Saccharomycetales spp and SCFA producers, the bacterial community should have more potential to produce SCFAs and increase insulin sensitivity in Saccharomycetales spp carriers. The wide associations between gut mycobiota and bacterial biosynthesis of secondary metabolites support such a hypothesis. In addition, the mediation analyses indicate that gut bacterial function can mediate fungal effects on host metabolic health. The elevated LDL cholesterol is a major cause of cardiovascular diseases.48 49 Madeeha et al demonstrated that Pichia kudriavzevii had cholesterol assimilation capability ranging from 66.7% to 80.4% after 72 hours of incubation at 37°C.50 Here, Pichia may modulate cholesterol turnover through the interaction with bacterial function and faecal histidine. The present observational findings need the support of intervention studies for confirmation. In the meantime, this is an example of how integrative cross-kingdom microbiomics might open novel opportunities to address metabolic disease risk and management.
The present study has limitations. First, the findings may or may not be extrapolatable to other ethnicity groups. Second, the study is observational, and associations are not proof of causation. Nevertheless, the multiomics and longitudinal analyses (with causal mediation analyses) can infer potential causality and improve ecobiological understanding, amenable to experimental pursuit.
Conclusion
These findings show that the gut mycobiome is an essential component of the gut ecosystem, interactive with the bacterial community and contribute to the overall human metabolic health.
Data availability statement
Data are available in a public, open access repository. The raw data of the ITS2 sequencing, 16S rRNA sequencing and metagenomic sequencing are available in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb (accession number CNP0002114, CNP0000829 and CNP0001510, respectively). Other data sets generated during and/or analysed during the current study are available from the corresponding author upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the ethics committees of the School of Public Health at Sun Yat-sen University (2018048) and Westlake University (20190114ZJS0003). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank all study participants of the Guangzhou Nutrition and Health Study. We thank Westlake University Supercomputer Centre for assistance in data storage and computation. We acknowledge the support from Westlake Education Foundation and Westlake Intelligent Biomarker Discovery Lab at the Westlake Laboratory of Life Sciences and Biomedicine. We thank Guoqing Zhang for valuable discussion.
References
Footnotes
MS, YF and H-lZ are joint first authors.
Twitter @TienHuynhAU, @Ju-Sheng Zheng@zheng_jusheng
MS, YF and H-lZ contributed equally.
Contributors J-SZ, MLW and YC designed the study; H-LZ, J-JX, YL and ZM collected the data; MS and YF performed the data analysis; YF, ZJ, WG, TH and MLW contributed to the discussion; MS, YC and J-SZ drafted the manuscript; J-SZ, MLW and TH revised the manuscript; all authors read, revised and approved the final manuscript. J-SZ acts as guarantor for this study and publication.
Funding This work was supported by the National Natural Science Foundation of China (82073529, 81903316, 81773416 and 82103826); Zhejiang Ten-thousand Talents Program (2019R52039); Westlake Multidisciplinary Research Initiative Centre (MRIC20200301); the Zhejiang Provincial Natural Science Foundation of China (LQ21H260002); the China Postdoctoral Science Foundation (2020M681945); and the 5010 Programme for Clinical Research (2007032) of the Sun Yat-sen University (Guangzhou, China). The funders were not involved in the study design, implementation, analysis or interpretation of data.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.