Article Text

Abstract
Objectives Gut microbiota is a key component in obesity and type 2 diabetes, yet mechanisms and metabolites central to this interaction remain unclear. We examined the human gut microbiome’s functional composition in healthy metabolic state and the most severe states of obesity and type 2 diabetes within the MetaCardis cohort. We focused on the role of B vitamins and B7/B8 biotin for regulation of host metabolic state, as these vitamins influence both microbial function and host metabolism and inflammation.
Design We performed metagenomic analyses in 1545 subjects from the MetaCardis cohorts and different murine experiments, including germ-free and antibiotic treated animals, faecal microbiota transfer, bariatric surgery and supplementation with biotin and prebiotics in mice.
Results Severe obesity is associated with an absolute deficiency in bacterial biotin producers and transporters, whose abundances correlate with host metabolic and inflammatory phenotypes. We found suboptimal circulating biotin levels in severe obesity and altered expression of biotin-associated genes in human adipose tissue. In mice, the absence or depletion of gut microbiota by antibiotics confirmed the microbial contribution to host biotin levels. Bariatric surgery, which improves metabolism and inflammation, associates with increased bacterial biotin producers and improved host systemic biotin in humans and mice. Finally, supplementing high-fat diet-fed mice with fructo-oligosaccharides and biotin improves not only the microbiome diversity, but also the potential of bacterial production of biotin and B vitamins, while limiting weight gain and glycaemic deterioration.
Conclusion Strategies combining biotin and prebiotic supplementation could help prevent the deterioration of metabolic states in severe obesity.
Trial registration number NCT02059538.
- obesity
- micronutrients
- nutrition
- intestinal bacteria
- diabetes mellitus
Data availability statement
Data are available in a public, open access repository. Data are available on reasonable request. metacardis https://www.ebi.ac.uk/ena/browser/view/PRJEB41311, https://www.ebi.ac.uk/ena/browser/view/PRJEB38742, https://www.ebi.ac.uk/ena/browser/view/PRJEB37249, mouse experiments; 16S data, https://www.ebi.ac.uk/ena/browser/view/PRJEB42967, mouse experiments; nanopore data, https://www.ebi.ac.uk/ena/browser/view/PRJEB42966, Code paper analyses, https://git.ummisco.fr/ebelda/metatransitionbiotingut.git.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Disclaimer: this video summarises a scientific article published by BMJ Publishing Group Limited (BMJ). The content of this video has not been peer-reviewed and does not constitute medical advice. Any opinions expressed are solely those of the contributors. Viewers should be aware that professionals in the field may have different opinions. BMJ does not endorse any opinions expressed or recommendations discussed. Viewers should not use the content of the video as the basis for any medical treatment. BMJ disclaims all liability and responsibility arising from any reliance placed on the content.
What is already known on this subject?
Gut microbiota influences metabolic health by producing many metabolites including bacteria-derived B vitamins. Impaired bacterial production of B vitamins can affect both microbial community functions and host metabolism and inflammation. However, these aspects have not yet been explored in the context of severe obesity and dysbiosis.
Rodent and clinical studies have shown altered serum and tissue biotin status in obesity and metabolic diseases.
The obesogenic diet is known to induce severe dysbiosis, but it is not known whether it also leads to concomitant alteration of bacterial metabolism and reduced biotin availability to the host.
What are the new findings?
Metagenomic analysis in a large European population, MetaCardis, with severe obesity and metabolic alteration shows that the loss of microbial cell loads observed in these conditions is linked to altered bacterial potential of biosynthesis and transport of biotin in humans.
The altered bacterial biotin status is also associated with metabolic and inflammation phenotypes in severe obesity.
High fat diet-induced obesity leads to altered microbial and host biotin status in mice.
Germ free, antibiotic-treated SPF mice and human to mice gut microbiota transfer experiments in mice demonstrate the contribution of gut microbiota in host serum biotin status.
Bariatric surgery-induced weight loss improves microbial biotin metabolism and host biotin status.
Oral biotin and prebiotic supplementation in mice improve body weight, glucose metabolism and biotin microbial status.
How might it impact on clinical practice in the foreseeable future?
Increase awareness about considering both gut microbiota and host biotin status in clinical management of metabolic diseases.
A concomitant management of gut dysbiosis via gut-focused therapies (eg, prebiotics), and B vitamin availability, including biotin, appears interesting to prevent obesity from transitioning to a more severe metabolic state.
Introduction
Severe obesity, defined by a body mass index (BMI) ≥35 kg/m2, has increased 11-fold for men (2.3%) and 3-fold for women (5.0%) worldwide from 1975 to 2014,1 and is responsible for heavy burden due to its associated cardiometabolic complications.2 Tissue alterations (eg, altered metabolism and increased inflammation) and systemic low-grade inflammation are well-known components linking severe obesity to type 2 diabetes (T2D). Increasing evidence has revealed the gut microbiota as a factor contributing to these phenomena. From overweight to severe obesity, the prevalence of reduced gut microbial gene richness3 increases along with the severity of metabolic complications.4 We recently showed that the enrichment of Bacteroides 2 (bact 2) enterotype in severe obesity is associated with inflammatory markers in a subset of the European MetaCardis cohort.5 Thus, obesity and glucose metabolism deterioration are characterised by metagenomic species signatures,4 6 whose functional potential still needs to be better deciphered.
The gut microbiota can directly or indirectly influence metabolic health by producing many metabolites,7 including bacteria-derived B vitamins. It was suggested that impaired bacterial production of B vitamins can affect both microbial community functions8 and host metabolism and inflammation9 10 while vitamin B7/biotin has been scarcely explored in severe obesity.11
In the microbiota, the homoeostasis of biotin metabolism and recycling is key for proper bacterial growth and function. Indeed, gut bacteria form complex ecosystems consisting of trophic webs with complementary metabolic features including vitamin cross-feeding; essential for the growth of bacterial species not able to synthesise a vitamin; for example, auxotrophic biotin transporters.12 Using vitamins from external sources is energetically more advantageous than synthesising them. Actually, producing one biotin molecule (eg, prototrophic eucaryotes) requires more than six enzymes and seven ATP equivalents.13 Interestingly, biotin biosynthesis has been described as a signature of Bacteroides-enriched microbiome composition.14 This is in agreement with an enrichment of Bacteroides lineages in prokaryotes prototrophic for biotin biosynthesis described in different genomic surveys of B-vitamin metabolism.8 15 Moreover, increase in the abundance of biotin biosynthesis potential of the microbiome has been described in altered health conditions including obesity and T2D.16 However, these analyses were solely based on relative abundance metagenomic profiles. Yet, as observed in severe obesity, Bacteroides-enriched microbiome, which is generally defined from relative abundance profiles, is strongly linked to low microbial cell density,5 17 which has a strong and significant impact on the quantification of metagenomic features and the subsequent association with clinical phenotypes.17 We can hypothesise that the abundance of metagenomic features like biotin biosynthesis potential, strongly enriched in Bacteroides lineages, could be severely impacted by the cell density of the ecosystem. Thus, evaluating the absolute quantification of biotin production and transport potential of the microbiome by considering microbial cell density could shed light about microbial biotin status in severe obesity.
It is unknown whether biotin bacterial metabolism is altered in severe obesity and whether a dysbiotic gut microbiota profile can affect the biotin status of the host. While neglected, biotin is key for host physiological functions including carbohydrate and lipid metabolism.18 Biotin is a cofactor of carboxylases which play key roles in fatty acid synthesis and mitochondrial oxidation, including in the human adipose tissue.19 In humans with moderate obesity and T2D, serum biotin can be found decreased compared with controls,20 independently of the host genetic background.20 Whether the obesogenic diet, known to induce severe dysbiosis also leads to concomitant altered bacterial metabolism and reduced biotin availability to the host is unknown.
Biotin supplementation was suggested to improve glucose metabolism in T2D subjects and in rodents,18 21–23 and to impact adipose tissue metabolism. Murine models of oral biotin supplementation showed metabolic amelioration and improved tissue expression of some biotin-dependent carboxylases.24 25 However, it has yet to be deeply explored in the context of westernised diet-induced obesity or in populations with severe obesity.
Taking into account dysbiosis and loss of microbial cells, we thus aim to characterise bacterial biotin metabolism potential in severe obesity and investigate its importance for host metabolic state, by cross-sectionally analysing the faecal microbiota of 1545 subjects with obesity at different stages of metabolic deterioration.5 We further gained deeper insights into bacterial biotin and host cross-talks by performing a series of murine experiments, including the effect of correcting biotin status while acting on gut microbiota dysbiosis.
Results
Quantitative metagenomic association revealed altered biotin synthesis and transport potential in severe obesity
We compared patients with severe obesity (SOB, BMI ≥35 kg/m²; n=608) to those with overweight or grade I obesity (MOB, BMI 25–34.9 kg/m²; n=299) and to lean non-obese individuals (NOB, BMI <25 kg/m²; n=638). Within these BMI groups, we also further investigated the impact of the increase in severity of glucose metabolism alterations (metabolically healthy group (MH; n=458), metabolically unhealthy group (MUH; n=430) and T2D group (T2D; n=657)) (online supplemental discussion for cohort description, see online supplemental tables S1-S4).
Supplemental material
Supplemental material
Supplemental material
This stratification by disease severity (BMI and glucose metabolism deterioration) was the variable explaining the highest non-redundant fraction of microbiome composition (n=1545, stepwise distance-based redundancy analyses (dbRDA) with genus-level relative abundance profiles of mOTU abundance data, R2=3.17, false discovery rate (FDR)=1.0×10−4), followed by BMI (dbRDA, R2=0.53, FDR=1.0×10−4), metformin intake (dbRDA, R2=0.52, FDR=1.0×10−4), percentage of fat mass (dbRDA, R2=0.48, FDR=1.0×10−4), serum triglycerides (dbRDA, R2=0.36, FDR=7.0×10−4) and plasma glucose (dbRDA, R2=0.16, FDR=1.82×10−2) (figure 1A). Statins were also included among the variables with non-redundant effect on microbiome variation (dbRDA, R2=0.16, FDR=2.1×10−2, figure 1A).5

Functional features associated with the severity of obesity in metabolic health groups: effect of bacterial cell load. (A) Major variables explaining the microbiome compositional variation in the MetaCardis cohort subset (distance-based redundancy analyses, dbRDA; genus-level Bray-Curtis dissimilarity), either independently (univariate effect sizes in black) or in a multivariate model (cumulative effect sizes in grey). The cut-off for significant non redundant contribution to the multivariate model is represented by the red line. (B) Gene richness distribution across obesity groups stratified by metabolic health status. (**P value<0.05 in Kruskal- Wallis test controlled for country of recruitment and age, FDR<0.05 pairwise Wilcoxon rank-sum tests controlled for country of recruitment and age). The dash line represents the threshold that stratifies individuals as High vs. Low gene count (HGC/LGC) based on the median of gene richness in healthy German population (n=91) which exhibit gene richness bimodality. (C) Microbial cell counts distribution across obesity groups stratified by metabolic health status. (**P value<0.05 in Kruskal-Wallis test controlled for country of recruitment, FDR<0.05 pairwise Wilcoxon rank-sum tests controlled for country of recruitment). (D) Estimated marginal means and confidence intervals of log-transformed absolute abundances of microbiome biotin biosynthesis and consumption potential across obesity groups adjusted by statin intake and stratified by the metabolic health status. (E) Estimated marginal means and confidence intervals of log-transformed absolute abundances of biotin producers (eg, prokaryotic organisms harboring all biotin biosynthesis genes from pimelate precursor and no biotin biosynthesis transport genes), biotin transporters (prokaryotic organisms with no biotin biosynthesis genes) and biotin producers and transporters (prokaryotic organisms with all biotin biosynthesis genes from pimelate and biotin transport genes) across obesity groups adjusted by statin intake and stratified by the metabolic health status (*FDR<0.05 on linear regression models of feature abundance by obesity status adjusted by statin intake, P-adj<0.05 on pairwise Tukey tests between obesity states). BMI, body mass index; HbA1c, haemoglobin A1c; HDL, high-density lipoprotein; MH, metabolically healthy; MUH, metabolically unhealthy; T2D, type 2 diabetes.
Severe obesity was characterised by a significantly lowered microbial gene richness in metabolic groups except for T2D individuals not treated by metformin (figure 1B; FDR<0.05 NOB vs SOB groups). Individuals from this group were significantly under-medicated for T2D in comparison with other groups (Χ2 P=7.57×10−3; online supplemental figure S1). Severe obesity was characterised by significantly lowered microbial cell density in MH and T2D individuals treated by metformin (figure 1C; FDR<0.05 NOB vs SOB groups).
Supplemental material
Supplemental material
We estimated the absolute quantification of microbiome biotin biosynthesis and consumption potential using quantitative microbial profiles (QMP). QMP evaluation was derived from gene abundances of the Integrated Gene Catalogue (IGC) of the human gut microbiome26 and from the microbial cell density of this population (see methods). QMP of biotin transport potential significantly decreased with severe obesity in metabolic groups, except T2D individuals not treated by metformin (figure 1D; FDR<0.05 NOB vs SOB groups). Similarly, QMP of biotin production potential tends to decrease with increasing obesity severity in the MH group, this decrease being statistically significant in T2D individuals treated by metformin (figure 1D; FDR<0.05 NOB vs SOB groups).
We confirmed this observation when the microbiome production and consumption potential was estimated using annotations of B-vitamin biosynthesis and transport phenotypes in prokaryotic organisms.15 The total absolute abundance of biotin auxotrophs (eg, bacteria with no biotin biosynthesis genes) and strict biotin prototrophs (eg, bacteria with biotin biosynthesis genes from pimelate and no biotin transport genes) decreased with increasing obesity severity within MH individuals and T2D individuals treated by metformin groups (figure 1E, FDR<0.05 NOB vs SOB groups), with similar association observed in bacterial groups able to produce and consume biotin.
We compared the absolute quantification of biotin production and consumption potential based on gut microbiome genes with the absolute abundances of different bacterial subgroups based on biotin biosynthesis and transport gene content.15 The absolute biotin biosynthesis potential strongly correlates with the abundance of bacterial species with complete biotin biosynthesis pathway and no gene involved in biotin transport (online supplemental figure S2A; P1 group, r=0.909, p<0.05). This group is dominated by Proteobacteria and Bacteroidetes lineages (online supplemental figure S2B: 49.32% and 43.58% of the 296 organisms in these groups, respectively). In contrast, the absolute biotin consumption potential strongly correlates with the abundance of bacterial species with no biotin biosynthesis genes and harbouring the biotin transport gene BioY from the energy coupling factor transport system27 (online supplemental figure S2A); r=0.90, p<0.05). This group was dominated by Firmicutes (online supplemental figure S2A: 77.93% of the 618 organisms in this group). Finally, these abundance profiles of biotin auxotrophs and prototrophs across obesity groups could be extended to other B vitamins (online supplemental figure S3).
Supplemental material
Supplemental material
This analysis suggests that the loss of microbial cell loads across aggravated metabolic disease states is associated with altered synthesis and transport potential of B vitamins, including biotin.
Microbial biotin functional modules associate with altered metabolism and inflammation in severe obesity
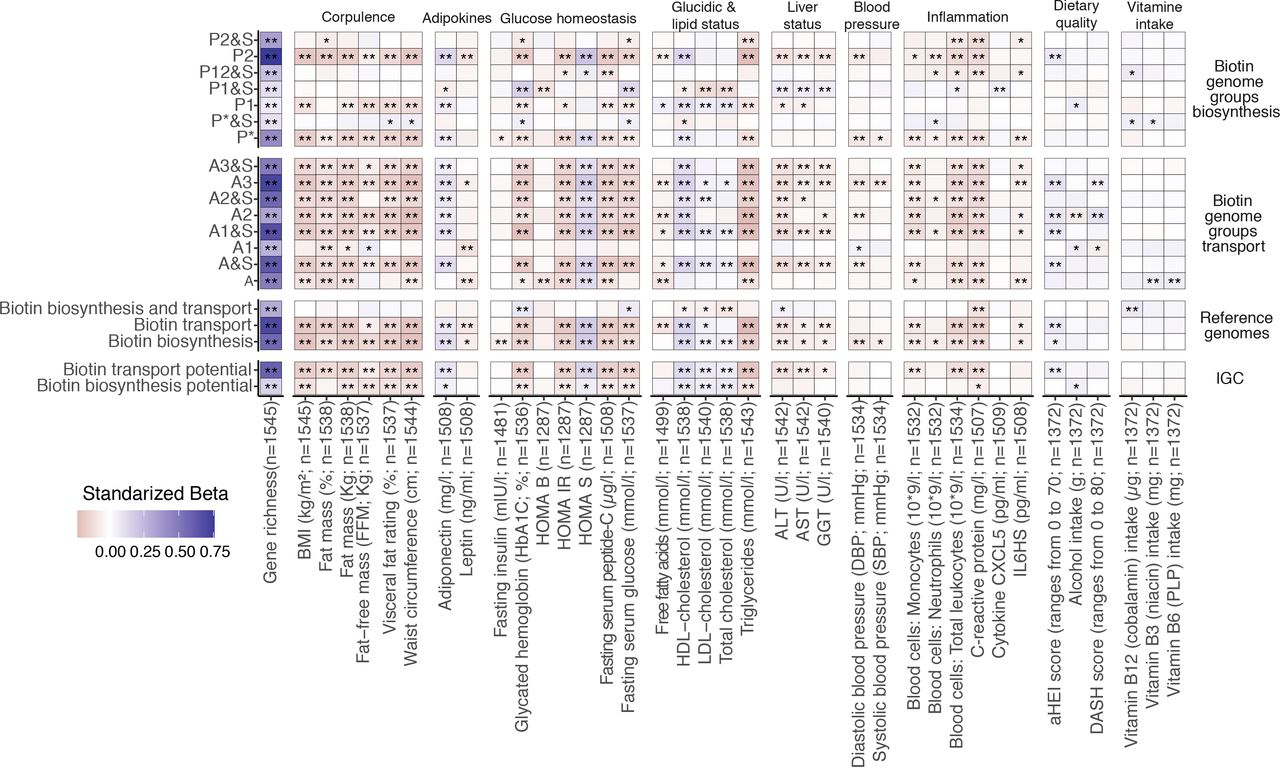
We then hypothesised that the potential functional alterations in bacterial biotin metabolism might be related to host’s metabolic and inflammation phenotypes. We performed linear regression analyses of log10-transformed QMP abundances of metagenomic signatures related to biotin transport and biosynthesis potential (eg, absolute IGC gene abundances, groups of biotin producers and transporters based on reference genomes), against 53 clinical and nutritional variables. The absolute abundances of groups of biotin auxotrophs (A groups) and strict de novo biotin producers from fatty acid intermediates (P1 group) and from pimelate precursor (P*, P2 groups) were negatively associated with corpulence variables (BMI, fat mass (%), waist circumference, visceral fat rating), glucose tolerance and metabolic-related variables (fasting plasma glucose, glycated haemoglobin (HbA1c), C peptide, triglycerides and insulin resistance and sensitivity surrogates), as well as the inflammatory status (C reactive protein (CRP), interleukin 6 (IL-6), total leucocytes, neutrophils and monocytes counts) (figure 2, standardised beta coefficients <0, FDR <0.05). Absolute abundances of biotin transport and production showed a positive association with microbial gene richness, insulin sensitivity surrogates (HOMA-S), and cholesterol-related variables including high-density lipoprotein (HDL)-cholesterol (figure 2, standardised beta coefficients>0, FDR<0.05). These associations were reproduced with overall estimates of microbial biotin production and transport from IGC gene abundances and reference genomes (figure 2). This association analysis supports an interplay between host metabolism, inflammatory state and the disturbed homoeostasis of bacterial biotin metabolism in severe obesity.

Association between microbiome biotin status and host metabolic and inflammation markers in the MetaCardis subcohort. Heatmap indicating adjusted associations between log-10 transformed QMP abundance profiles of metagenomic signatures regarding biotin production and transport with clinical and lifestyle factors. The y-axis represents independent variables and the variables in the x-axis are the dependent variable (n=1545 individuals). These models were adjusted for the country of recruitment and age. (*P value<0.05; **FDR<0.05. Clinical and lifestyle variables for which no association with FDR<0.05 was found are not included in the heatmap). The color tones correspond to effect sizes represented by standardized beta coefficients from the adjusted linear regression models. Biosynthesis and transport genome groups were defined according to the nomenclature defined in Rodionov et al.15 Briefly, these included 3 groups of strict biotin producers (P1, P2, P* groups) harboring all 4 genes common to the different pathway variants of biotin biosynthesis from pimelate (P2) or pimeloyl-ACP (P1, P*). This also included 8 groups of strict biotin auxotrophs A&S/A groups; microorganisms not capable of biotin production 45 and with (A&S groups) or without (A groups) genes involved in biotin transport) with different levels of incompletion in the 4 core biotin biosynthesis genes (harboring from 1 to 3 biosynthetic genes at most), and 4 groups of biotin producers that also harbors genes coding for biotin transport (P&S groups). ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; CXCL5, C-X-C motif chemokine ligand 5; GGT, γ-glutamyltransferase; HDL, high-density lipoprotein. LDL, low-density lipoprotein.
Altered host biotin in severe obesity
We examined whether the potential perturbation of bacterial biotin metabolism in severe obesity also translated in host systemic biotin levels. We quantified serum biotin in 212 lean and SOB subjects with low or high gene richness and in 17 subjects with more severe obesity from the independent Microbaria bariatric study4 cohort (mean BMI ±SD=24.7±3.1 in MetaCardis lean group, 41.2±4.9 in MetaCardis SOB group, 44.5±5.1 in Microbaria SOB group). We found a moderate but significant decrease in serum biotin in the SOB group (figure 3A. FDR=3.0×10−2) with a higher reduction in Microbaria group (figure 3A, FDR=1.12×10−3). We also assessed biotin deficiency using recommended and published thresholds.28 We found that 78.09% of the MetaCardis SOB group (88.24% for the Microbaria group) showed suboptimal (serum biotin: 200–400 ng/L) or deficiency levels (serum biotin: <200 ng/L) whereas only 19.65% of lean subjects were below this level (figure 3B). We observed an increase in the urinary excretion of 3-hydroxyisovaleric acid, an early marker29 of biotin deficiency; in agreement with host biotin deficiency. This marker was positively associated with BMI (n=1545 individuals) with (R2=5.6×10−2, p=7.9×10−14) or without (R2=3.0×10−3, p=3.6×10−2) adjustments for diabetes status, metformin, statin and biotin intake (linear regression; data not shown). The disease severity groups explained the highest fraction of variance in the urinary concentration of 3-hydroxyvaleric acid in a multivariate model with 28 other clinical and nutritional variables (figure 3C; eta squared=1.5×10−2; p =1.2×10−2). Yet, the systemic biotin concentration differences found across the BMI spectrum are unlikely explained by differences in biotin intake. Indeed, biotin intake derived from ubiquitous food sources (eg, predominantly meat, fish, poultry, egg, dairy and vegetables30 was similar in lean and SOB, and even slightly higher during obesity and T2D) (figure 3D).
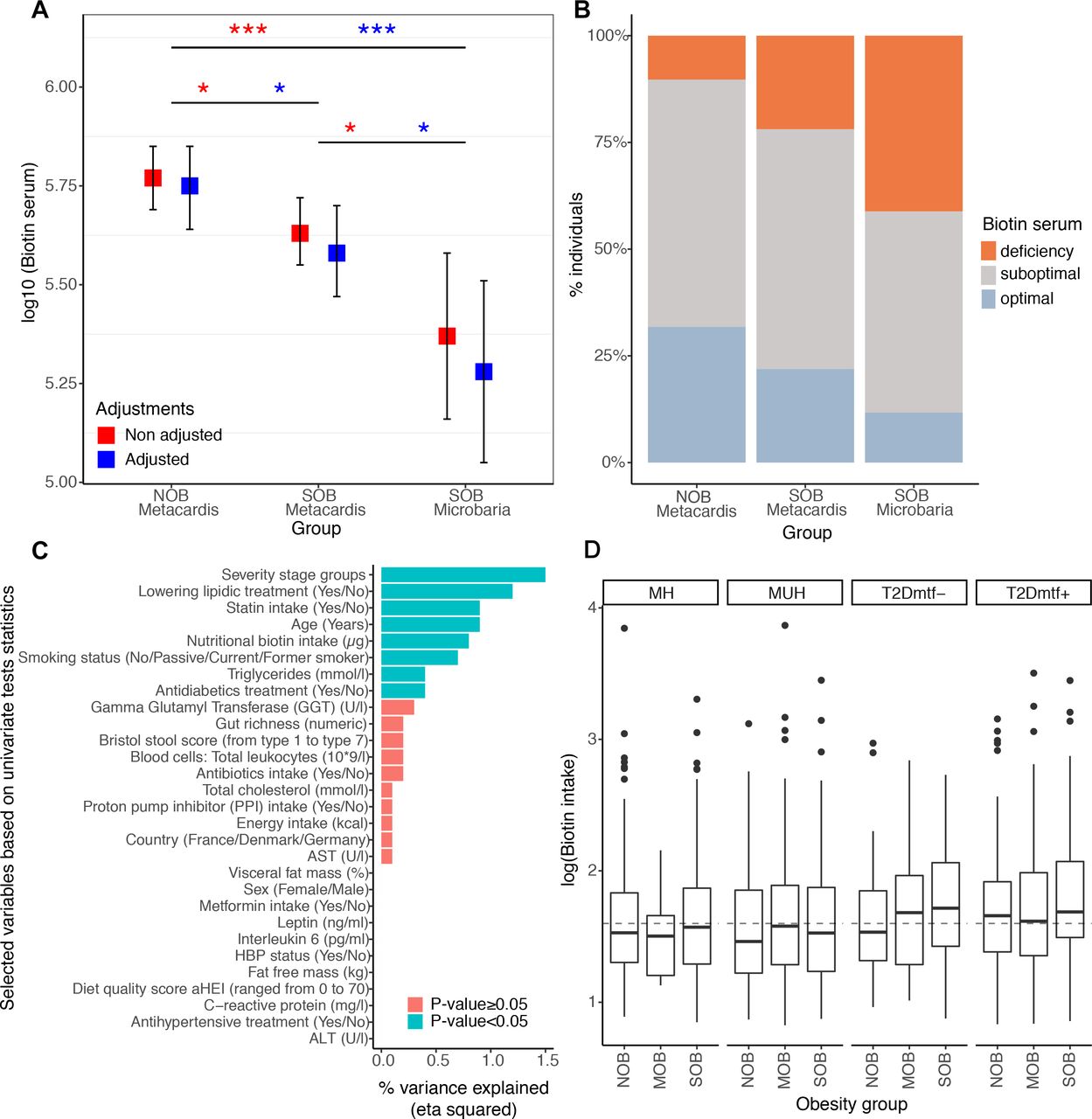
Systemic and nutritional biotin profiles across obesity groups in MetaCardis subcohort. (A) Differences of biotin serum levels between obesity groups in 212 individuals from the MetaCardis subcohort (n=107 (NOB), n=105 (SOB)) and 17 more severely obese individuals of the Microbaria study (*P value<0.05; ***P value<0.001). Significant differences were observed with non-adjusted and adjusted (for diabetes status, metformin, statin and biotin intakes) Generalized Linear Models and lsmeans function, with P-value adjustment for multiple comparisons with Benjamini-Hochberg method. Biotin serum was log10 transformed to enable a normal distribution of the biotin variable (NOB vs. SOB (MetaCardis and Microbaria) Cohen’s D effect size=0.91. NOB vs. SOB MetaCardis Cohen’s effect size D =0.18). (B) Distribution of biotin deficiency status between obesity groups according to the following thresholds:28 deficiency (<200 ng/l), suboptimal levels (200-400 ng/l), optimal levels (>400 ng/l). Significant differences were observed with Chi-2 tests (P-value=1.0x10-2). (C) Association between clinical covariates and biotin status defined by the urinary metabolite 3-hydroxyisovaleric acid. Horizontal bars correspond to the variance in 3-hydroxyisovaleric acid explained by each clinical covariate (measured by the eta squared statistic derived from a multivariate ANCOVA model, n=1545). Statistical significance is indicated for a global model containing all the variables. ALT: Alanine Aminotransferase, AST: Aspartate Aminotransferase, GGT: Gamma-Glutyl Transferase, HBP: high-blood pressure. (D) Differences in log10 transformed nutritional biotin intake (μg/day) across obesity groups stratified by metabolic health status (n=284 (NOB-MH), n=130 (NOB MUH), n=51 (NOB-T2Dmtf-), n=173 (NOB-T2Dmtf+), n=13 (MOB-MH), n=81 (MOB-MUH), n=41 (MOB-T2Dmtf-), n=164 (MOB-T2Dmtf+), n=161 (SOB-MH), n=219 (SOB-MUH), n=85 (SOB T2Dmtf-), n=143 (SOB-T2Dmtf+)). No significant differences in biotin intake were observed across study groups (FDR>0.05; non-parametric pairwise univariate tests controlled by country or statin intake). Dashed line represents the recommended daily biotin intake according to the European Food Safety Authority (40μg/day).50 ; MH, metabolically healthy; MUH, metabolically unhealthy; T2D, type 2 diabetes.
We next hypothesised that biotin metabolism may be altered in biotin-dependent host tissues, such as the adipose tissue. Biotin is covalently linked to many carboxylases and this binding is catalysed by holocarboxylase synthetase (HLCS) in the mitochondrion and cytosol, which is important for tissue metabolism.19 Using a previously published data set of individuals with severe obesity,31 we observed negative associations between BMI and subcutaneous adipose tissue gene expression of biotin-dependent carboxylases like acetyl-CoA carboxylase (ACACA), propionyl-carboxylases (PCCA, PCCB), 3-methylcrotonyl-CoA carboxylase (MCC2), pyruvate carboxylase (PC) or HLCS20 (qPCR- data, Spearman correlations between −0.42 and −0.70, all p<0.05; n=24; online supplemental figure 4). The expression of these genes was negatively correlated with the expression of inflammatory genes, such as the chemokine CC-Chemokin ligand 2 (CCL2), IL-18 binding protein, metallopeptidase 9, secreted phosphoprotein 1 and the tumour necrosis factor (TNF) receptor superfamily member 1A (online supplemental figure 4). Altogether, a relative deficit in systemic and adipose tissue biotin status was observed in severe obesity.
Supplemental material
Obesogenic diet alters microbiota and host biotin status
We next evaluated whether an obesogenic condition brought by a high-fat diet (HFD) induces altered microbial biotin metabolism and host biotin availability in mice. HFD (60% kcal provided by lipids)-induced obesity was associated with significantly lowered plasma biotin levels, despite the two-fold higher intake of biotin in HFD compared with chow-fed animals (0.65 vs 0.26 µg of biotin/day for these diets respectively). After 4 weeks of HFD-feeding, mice displayed a decrease in plasma biotin compared with chow-fed animals (figure 4A, left, Chow vs HFD: p=7.32×10−2), that reached significance after thirteen weeks of HFD-feeding (figure 4A, right, Chow vs HFD: p=3.72×10−3). As seen in humans, we found a significant reduction in the expression of biotin carboxylases in epididymal adipose tissue from those mice (online supplemental figure 5A). At the microbiome level, following 4 weeks of HFD, there was a significant drop in the abundance of biotin producers (with biotin biosynthesis potential from pimelate and no biotin transport gene) and biotin producers and transporters (with biotin biosynthesis and transport genes) in parallel with an increase in the abundance of biotin transporters (bacteria without biotin biosynthesis genes) (figure 4B; FDR=4.66×10−4, 9.32×10–3, 4.66×10–4, respectively; Wilcoxon rank-sum (WRS) tests). Similar results were found after thirteen weeks of HFD feeding (figure 4B; FDR<0.05 in all groups). Importantly, taking into account total bacterial abundance (measured by qPCR), as a proxy of bacterial load, confirmed the decrease in biotin producers potential after thirteen weeks of HFD feeding (online supplemental figure 5E, FDR<0.05).The increase in the abundance of biotin transporters was explained by the large increase of Lactococcus lactis after HFD feeding (online supplemental figure 5B), which has been described as a food contaminant in different studies.32 Yet, similar results were observed when L. lactis was excluded from the microbiome profiles (online supplemental figure 6).
Supplemental material
Supplemental material

HFD-induced obesity in mice leads to depletion of biotin serum levels together with depletion of bacterial biotin production lineages. (A) Plasma biotin concentration of age-matched Chow-fed and HFD-fed C57BL6/J mice after 4 (left panel) and 13 weeks (right panel) (**P value<0.01; Chow n=7 for day 35 and day 90, HFD n=5 for d35 and n=8 for d90, Wilcoxon rank-sum test) (B) Relative abundance profiles of biotin producers (bacteria with all biotin biosynthesis genes from pimelate and no biotin transport gene), biotin transporters (bacteria with no gene involved in biotin biosynthesis) and biotin producers+transporters (bacteria harboring biotin biosynthesis and transport genes) in these same mice at baseline (day 1), day 35 and day 90 (*P value and FDR<0.05 pairwise Wilcoxon rank-sum test). (C) Serum biotin concentration of germ-free (GF) and conventionally raised (CONV-R) C57BL6/J mice (*P value<0.05, C57BL6/J GF n=7 and CONV-R n=5; Wilcoxon rank-sum tests). (D) Plasma biotin concentration and (E) total bacterial 16S rRNA gene load measured by qPCR in chow-fed mice with (n=7) and without (n=8) large spectrum antibiotics (100mg/kg of vancomycin and 200 mg/kg of ampicillin, neomycin and metronidazole) diluted in water for 14 days (*P value<0.05; Wilcoxon rank-sum test). (F) Beta-coefficients obtained with multivariate linear regression models between diet, phenotype and the abundances of biotin production and transport inferred from 16S data and serum biotin in a same global model with all covariates (*P value<0.05) from fecal transfer experiments in mice from panels g and h. (G) Serum biotin levels of Swiss Webster mice colonized with faecal slurries of 4 subjects from the MetaCardis subcohort (2 NOB; 2 SOB). Mice were colonized for 28 days and were fed either chow (NOB, n=16; SOB, n=12) or western diet (NOB, n=17; SOB, n=17) (*P value and FDR<0.05; ***P value<0.001 and FDR<0.05; Wilcoxon rank-sum test). (h) Abundance of biotin production module inferred from PICRUSt functional profiles of 16S rRNA gene amplicon data of mice from panel F (**P value<0.05; Wilcoxon rank-sum test). CONV-R, conventionally raised; GF, germ-free; HFD, high-fat diet; ns, not significant; WD, Western diet.
Gut microbiota contributes to host serum biotin
We evaluated whether the gut microbiota contributes to biotin circulating levels. First, serum biotin in chow diet-fed germ-free (GF) mice was significantly lower than in conventionally raised (CONV-R) C57BL/6 J mice (figure 4C; C57BL/6J p=1.01×10−2). Second, we administered a combination of large spectrum antibiotics33 during 14 days to C57BL/6 J mice fed a standard chow diet, which led to a major decrease in plasma biotin (figure 4D; p=3.11×10−4). We confirmed by qPCR that the faecal bacterial load was decreased by the antibiotic treatment (figure 4E; p=2.0×10−4). Third, to determine if different configurations of the human gut microbiota differentially impact biotin levels, we colonised Swiss Webster GF mice using stools from two subjects with severe obesity (SOB) and two lean subjects (NOB) who harboured increased amounts of bacterial biotin producers in comparison to the SOB subjects. To consider the effect of the diet, we fed the mice either a chow diet or a western diet (WD 40% kcal from fat). In linear regression analyses with diet, donor phenotype and the abundance of biotin biosynthesis and transport modules (from 16S rRNA gene sequencing data), we found that the diet had the strongest impact on biotin plasma levels (figure 4F; p=3.2×10−4). However, within the two chow-fed groups, we observed a significant decrease in biotin plasma levels in mice receiving the gut microbiota from SOB patients (figure 4G; p=1.0×10−2). We reproduced the decrease in serum biotin levels in mice under obesogenic diet (WD), this decrease being significant in the groups receiving the gut microbiota of NOB patients (figure 4G; p=4.1×10−4). For the same biotin dietary intake, the composition of the gut microbiota influences biotin circulating levels. 16S sequencing of faecal samples from the colonised mice confirmed that the difference in biotin production potential observed in the donors’ faecal microbiota was transferred to the chow-fed mice (figure 4H; p value=4.39×10−3), but not to the WD-fed mice. These results pointed to a significant effect of this diet on the transfer of the microbiome functional potential from donors to mice, however we cannot exclude the contribution of obesity per se.
Gut microbiota and host biotin status improve after bariatric surgery
We explored gut microbiota biotin metabolism in the context of severe obesity management. We focused on bariatric surgery (BS), known to induce gut microbiota changes in addition to improved metabolism and inflammation.4 34 We performed an enterogastro anastomosis (EGA) in C57BL/6 J mice receiving either a chow diet or HFD.35 We observed that plasma biotin significantly increased 3 months’ (in chow-fed animals) and 1 month’s (in HFD-fed animals) post-BS in comparison to sham-operated animals (figure 5A, left: chow, p=1.33×10−3; right: HFD, p=7.34×10−2). The increase in plasma biotin post EGA was higher in the chow than in the HFD-fed animals, further suggesting the important contribution of the diet in modulating biotin levels. We sequenced the gut microbiota of the HFD-fed operated animals and observed a significant increase in the relative abundance of biotin producers harbouring, or not, biotin transport genes (figure 5B, FDR=5.83×10−4 in both groups). Similar observations were made taking into account total bacteria abundance (data not shown). Importantly, these observations were replicated in humans, where the number of patients with optimal biotin status 1 year after Roux-en-Y gastric bypass (RYGB) (from the Microbaria study) improved in comparison with baseline levels (figure 5C, p=2.42×10−2). We found a significant increase in biotin-producing bacteria after BS that paralleled the improvement in host biotin status (figure 5D; p=4.94×10−2 T0-M3, p=3.55×10−2 T0-M12). We replicated this increase in serum biotin in an independent group of patients 1 year after BS (figure 5E, p value=2.0×10−2). Unlike the animal models, patients after BS are supplemented by a combination of vitamins, including biotin (150 µg/day). We can speculate this oral supplementation contributes to improving the potential of bacterial biotin production and ameliorates the host biotin status at least during the first year post-BS; even though BS is associated with vitamin malabsorption.36
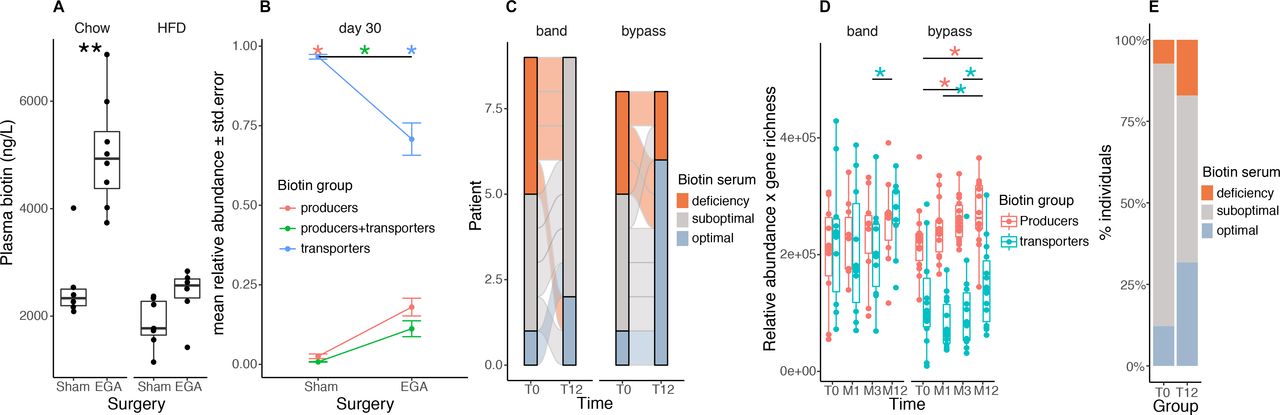
Biotin metabolism after bariatric surgery in mouse and human experiments. (A) Plasma biotin concentration of chow- or high-fat diet (HFD)-fed C57BL/6J mice with sham intervention (Sham) or bariatric surgery (Entero-gastro anastomosis, EGA).35 Blood was collected 1 month after surgery for the HFD group and 3 months after surgery for the Chow group (**P value<0.01 Wilcoxon rank-sum test; Chow-Sham n=6, Chow-EGA n=8, HFD-Sham n=7, HFD-EGA n=6). (B) Mean abundances of biotin producers (bacteria with all biotin biosynthesis genes from pimelate and no biotin transport gene), biotin transporters (bacteria with no gene involved in biotin biosynthesis) and biotin producers+transporters (bacteria harbouring biotin biosynthesis and transport genes) in sham and EGA mice of the HFD group 30 days after surgery (*FDR<0.05 pairwise Wilcoxon rank-sum test). (C) Distribution of biotin deficiency groups between baseline and month 12 in 17 individuals of the Microbaria study stratified by surgery group (n=9, gastric banding; n=8, Roux-en-Y gastric bypass) according to the following thresholds:28 deficiency (<200 ng/l), suboptimal levels (200-400 ng/l), optimal levels (>400 ng/l). P value=2.4x10-2 (bypass), P-value=1.1x10-1 (band); Fisher’s test. (D) Change of biotin producer and biotin transporter abundances (relative abundances multiplied by gene richness as a surrogate of microbial cell count to simulate QMP data) in 24 individuals of the Microbaria study stratified by surgery type (adjustable gastric banding, n=10; Roux-en-Y gastric, n=14) with metagenomics data at baseline, 1, 3, and 12 months after bariatric surgery (*P value<0.05; Wilcoxon signed-rank test). (E) Distribution of biotin deficiency groups at baseline (T0) and 12 months (T12) after bypass surgery in the BARICAN cohort (n=41; P value=2.0x10-2, Chi2 test). EGA, entero-gastro anastomosis; HFD, high-fat diet.
Combined prebiotics and biotin improve microbiota biotin and metabolic states
These results prompted us to examine a model of severe obesity through HFD-feeding in mice and to investigate the effects of biotin supplementation together with a gut microbiota modulation via a prebiotic (Fructo-oligosaccharides, FOS). Indeed, FOS is known to improve the metabolic status and at least partially correct the gut microbiota richness and composition induced by an obesogenic diet.37–39 In another experiment using FOS alone in parallel to HFD-feeding, we found increased plasma biotin levels and increased abundance of biotin producers considering total bacterial abundance or not, in comparison with HFD alone (online supplemental figure 5C-E). Despite the effect on circulating biotin levels, the sole administration of FOS was not able to restore the expression of biotin carboxylases in the epididymal adipose tissue (online supplemental figure 5A, discussion).
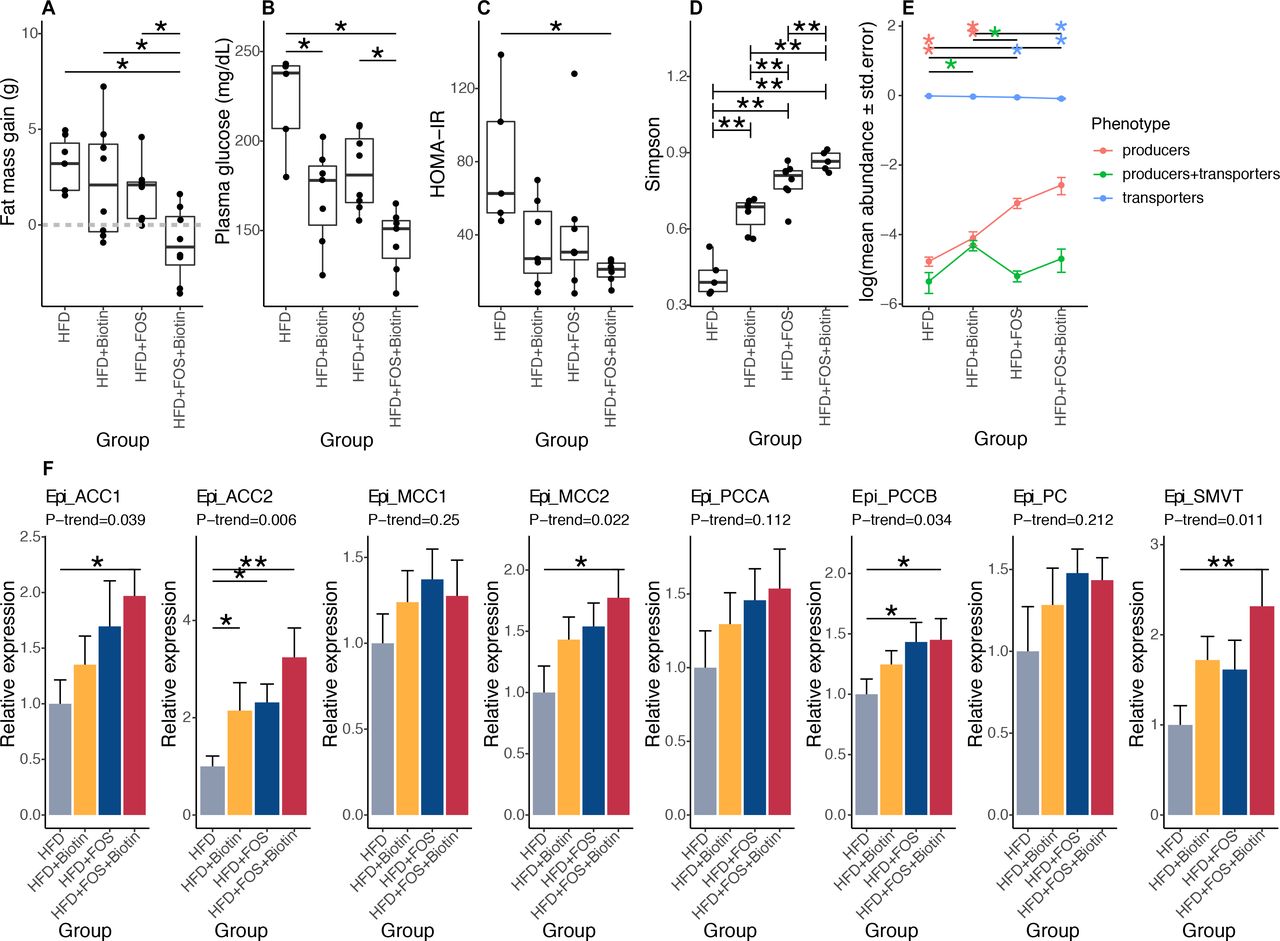
After 12 weeks of HFD leading to obesity (mean body mass was 37.9±5.1 g), C57BL/6J adult male mice were maintained under HFD and supplemented with FOS in the drinking water and/or biotin in the diet for eight additional weeks. In biotin supplemented groups, the circulating level of biotin statistically increased 136.6-fold compared with non-supplemented groups (data not shown). In animals with FOS and biotin, we observed a significant limitation of body weight gain, a reduction of fat mass accumulation (figure 6A), a decrease in fasting glucose, fasting insulin and an improved HOMA-IR (figure 6B,C, online supplemental figure S7A). Analyses of the gut microbiota showed a significant increase of bacterial diversity following the supplementation of FOS and biotin separately (FDR=5.05×10−3 in pairwise comparison of each one vs HFD group), further augmented by FOS and biotin combination (figure 6D; FDR=9.52×10−3). We highlighted a significant increase in the relative abundance of biotin producers and a significant decrease of biotin transporters mediated by the prebiotic supplementation (figure 6E; FDR=1.84×10−3 and 3.14×10–3, respectively, vs HFD group). Biotin supplementation alone led to the specific increase of the group of bacteria harbouring biotin biosynthesis and transport genes (figure 6E; FDR=2.82×10−2). Again, similar results were obtained taking into account total bacteria abundance (data not shown). Biotin supplementation per se may promote the increase of specific biotin producer lineages together with microbiome diversity. Moreover, both the producers and transporters of other B vitamins were similarly affected by the supplementation of both biotin and FOS considering total bacteria abundance (data not shown) or not (online supplemental figure S7B).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of biotin and FOS supplementation on host metabolism, biotin status and microbiome composition in established obesity in mouse experiments. (A) Fat mass gain of mice with established obesity, between day 82 (after twelve weeks of HFD and before treatments) and day 135 (after eight weeks of treatment by FOS and/or biotin) (A: HFD+FOS (n=10) vs. HFD (n=5); B: HFD+FOS vs. HFD+Biotin (n=9); C: HFD+Biotin vs. HFD; D: HFD+FOS+Biotin (n=5) vs. HFD; *P value<0.05, Kruskal-Wallis rank test with Dunn’s multiple comparison test) (B) Fasting glycaemia of these same animals measured after 6 weeks of treatment by FOS and/or biotin (*P value<0.05, Kruskal Wallis rank test with Dunn’s multiple comparison test). (C) HOMA-IR index calculated after 6 weeks of treatment by FOS and/or biotin (*P value<0.05, Kruskal Wallis rank test with Dunn’s multiple comparison test). (D) Simpson diversity distribution in different groups of mice with long-term established obesity (**P value<0.01 and FDR<0.05; pairwise Wilcoxon rank-sum test). (E) Mean abundances of biotin producers (bacteria with all biotin biosynthesis genes from pimelate and no biotin transport gene), biotin transporters (bacteria with no gene involved in biotin biosynthesis) and biotin producers+transporters (bacteria harbouring biotin biosynthesis and transport genes) in different groups of mice with long-term established obesity (*P value and FDR<0.05 pairwise Wilcoxon rank-sum test). (F) mRNA expression of biotin carboxylases (ACCA, ACCB, MCC1, MCC2, PCCA, PCCB, PC) and biotin transporter SMVT in epididymal adipose tissue of mice with long term established obesity supplemented with FOS and/or biotin after 20 weeks of total follow-up (Kruskal-Wallis rank test, with Dunn’s multiple comparison; *P value and FDR<0.05, **P value and FDR<0.01, pairwise comparisons and P-trend were calculated using linear contrast tests). HFD, high-fat diet; SMVT, sodium multi vitamin transporter.
The use of FOS and biotin also induced a significant increase of the expression of biotin carboxylases ACC1 (figure 6F, p-trend=1.50×10−2), ACC2 (p-trend=2.39×10−3), MCC2 (p-trend=2.13×10−2), PCCB (p-trend=2.87×10−2) and the biotin transporter sodium multi vitamin transporter (SMVT) (p-trend=6.38×10−3 ×10−2), and a trend towards an increase for PCCA (p-trend=8.16×10−2) in the epididymal adipose tissue. Finally, similar results were observed for body composition and glucose homoeostasis when biotin was administered alone in parallel to the development of obesity, independently of the administration mode of biotin either via food or via subcutaneous osmotic minipumps (online supplemental figure S7C,D).
Discussion
In the context of marked dysbiosis with altered bacterial cell abundance, low-grade inflammation and altered metabolism seen in severe obesity, we demonstrate the alteration of microbial biotin metabolism evaluated by the metagenomic quantification of bacterial producers and auxotrophs. Severe obesity is associated with decreased availability of microbial biotin which extended to other B vitamins. Moreover, low biotin availability in the host was evidenced by blood measurements showing suboptimal levels of systemic biotin. Another marker of relatively low biotin in the host was the decreased expression of biotin-dependent carboxylases evaluated by gene expression analyses in adipose tissue.
Since in our population, biotin intake through dietary analysis did not reveal major intake differences, we suggest that the particular decrease in transporters might be due to insufficient release by the producers which may also use biotin themselves or within the local bacterial ecosystem.
Human hosts lack the ability to produce biotin, which is, then, mainly absorbed from the diet in the jejunum, and to a lesser extent, in the distal intestine.40 Our results on axenic and antibiotics-treated mice suggest that the absence of gut microbiota negatively impacts circulating biotin levels.
Moreover, convergent experimental results in mice fed a HFD, emphasised the importance of diet quality in explaining altered biotin metabolism. Obesogenic diets led to a decrease in systemic biotin in mice, together with altered microbial biotin metabolism potential. In human populations, the degree to which biotin of bacterial origin contributes to circulating biotin levels remains unmeasured.
The origin of low biotin availability will also require further explorations. Here, correlation studies revealed an interplay between the altered microbial biotin metabolism and obesity related metabolic and systemic inflammation. We cannot exclude that metabolic and inflammatory perturbations in severe obesity contribute to altered biotin bacterial metabolism. The transport of biotin across the intestine epithelium might also be impaired. This transport requires the host transporter (SMVT (SLC5A6 gene).40 In published rodent experiments, it was shown that intestinal inflammation is negatively associated with biotin transport expression.41 TNF-alpha and interferon-gamma were able to inhibit SMVT expression in cultured intestinal epithelial cells.41 Moreover, colonic epithelial cells treated with LPS display decreased membrane expression of the SMVT transporter and lower colonic biotin uptake.42 While we could not explore this aspect in humans, we previously showed in patients with severe obesity that immune cells accumulate in the jejunum,43 a major absorption site of biotin. In unpublished observations, we found in patients with severe obesity that jejunal SMVT expression showed a trend of negative correlation with the expression of TNF-alpha by CD4 +lymphocytes and MAIT cells, a T cell subset known to be activated by gut microbial activity.41 Thus, the combination of increased intestinal inflammatory tone, together with the lowered number of biotin-producing bacteria, might be detrimental for the host biotin availability. Furthermore, it cannot be excluded that biotin requirements increase during the major expansion of fat mass in severe obesity, considering its importance for adipose tissue homoeostasis. The suboptimal availability of biotin to the host could be further amplified by decreased intestinal biotin absorption due to intestinal inflammation. In favour of this hypothesis, improved metabolism and inflammation concomitantly induced by BS did not only lead to improvement of microbial biotin metabolism but also to increased systemic biotin levels. However, better correction of biotin status might be considered in BS and notably gastric bypass since vitamin supplementation can be insufficient due to frequent malabsorption. Finally, in severe obesity, a vicious cycle for both the host and gut microbiota might occur, resulting in abnormal metabolic interactions, where molecular signals from the dysbiotic microbiota could contribute to the aggravation of host inflammation and tissue biotin deficiency.
As a consequence, we emphasise on the clinical importance of evaluating biotin deficiency in populations with severe obesity.
We are aware of some study limitations, which depends on metagenomic quantification of biotin production and consumption potential of the microbiome. Metagenomics data alone do not reflect the real biological activity of biotin biosynthesis and transport genes but provides a signature of the potential of the microbiome to achieve these biological functions based on the abundance of reference genes and bacterial species. Meta-transcriptomics and possibly meta-proteomics experiments would help further quantify the functional contribution of the microbiome to the host biotin status and determine which bacteria are important for circulating levels of biotin or other vitamins.
Finally, based on our results combining mouse and human investigations, we propose a concomitant management of gut dysbiosis via gut-focused therapies (eg, prebiotics) and improvement of vitamin availability which appears interesting to prevent obesity from transitioning to a more severe metabolic state. As such, biotin administration together with a prebiotic in mice with installed obesity limit weight gain and glucose metabolism deterioration while improving microbial metabolism. Interestingly, the supplementation improved bacterial metabolism of biotin together with other B vitamins. This pleads for the optimisation of vitamin B status in obesity and paves the way for future clinical investigations of biotin and prebiotic administration in humans with severe obesity.
Methods
Cross-sectional cohort description (MetaCardis)
The cross-sectional European MetaCardis study cohort consisted of 2214 participants that were recruited between 2013 and 2015 in the clinical departments of Pitié-Salpêtrière Hospital in Paris, France; in the Integrated Research and Treatment Center (IFB) Adiposity Diseases in Leipzig, Germany and in the Novo Nordisk Foundation Center for Basic Metabolic Research in Copenhagen, Denmark. Exclusion criteria included age above 75, history of abdominal cancer/radiation therapy on the abdomen, history of intestinal resection (except for appendectomy), acute or chronic inflammatory or infectious diseases (including hepatitis C virus VHC, hepatitis V virus VHB, and HIV), history of organ transplantation or receiving immunosuppressive therapy, severe kidney failure (modification of diet in renal disease (MDRD) glomerular filtration rate <50 mL (min 1.73 m²)−1), or drug or alcohol addiction. Study participants had to be free of any antibiotic use in the 3 months prior to inclusion. Subjects provided written informed consent and the study was conducted in accordance with the Declaration of Helsinki and is registered in clinical trial https://clinicaltrialsgov/show/NCT02059538. Detailed bioclinical information as well as gut microbial metagenomics sequences from faecal samples were obtained. Herein, we focused on a subset of 1545 participants based on BMI and glucose metabolism alterations.
We separated patients according to their BMI status (non-obese normal BMI (NOB BMI<25 kg/m²), overweight/moderately obese ((MOB), BMI 25–34.9 kg/m²)), severely obese, (severe obesity, SOB ≥35 kg/m2). Furthermore, since BMI presents some limits in particular in addressing obesity-specific comorbidities, we further defined three different groups based on the degree of metabolic disease severity, named ‘MH’ (MH state, defined by the absence of the metabolic syndrome (MS) and/or T2D), ‘MUH’ (MUH state, defined by the presence of the MS using the International Diabetes Federation (IDF) definition but the absence of T2D), and finally ‘T2D’ defined with a fasting glycaemia >6.9 mmol/L, and/or HbA1c ≥6.5% and/or the presence of anti-diabetic drugs. In the end, nine final study groups were obtained.
The MS was defined based on the IDF 2005 Consensus Worldwide Definition of the Metabolic Syndrome (http://www.idf.org/metabolic-syndrome), for example, waist circumference >94 cm in men and >80 cm in women, and any two of the following four factors: (1) triglycerides levels≥1.7 mmol/L or treatment for lipid abnormality (statin and/or fibrate or ezetimibe); (2) HDL cholesterol <1.03 mmol/L for European men and 1.29 mmol/L in European women and/or treatment for lipid abnormality; (3) abnormal blood pressure (BP): systolic BP ≥130 mm Hg and/or diastolic BP ≥85 mm Hg or patients taking antihypertensive treatments and (4) abnormal fasting plasma glucose ≥5.6 mmol/L or prevalent T2D. T2D status was defined using the American Diabetes Association definition (https://care.diabetesjournals.org/content/41/Supplement_1/S13): fasting glycaemia >6.9 mmol/L and/or 2 hours values in the oral glucose tolerance test >11 mmol/L and/or HbA1c ≥6.5% and/or use of any antidiabetic treatment.
Biochemical analyses
Biochemical analyses were conducted on blood samples collected after an overnight fast. Fasting glucose, total cholesterol, HDL cholesterol, triglycerides and HbA1c were measured using enzymatic methods, and kinetic assays based on coupled enzyme systems were used to measure alanine aminotransferase, aspartate aminotransferase and γ-glutamyltransferase. LDL cholesterol was calculated based on the Friedwald equation for French and Danish participants, and measured for German participants. Free fatty acids were measured via a photometric method (Diasys Diagnostic systems). A chemiluminescence assay (Insulin Architect, Abbott) permitted to measure serum insulin and C-peptide at a fasting state and at 30 and 120 min during an oral glucose tolerance test, and serum leptin was measured using the Human Leptin Quantikine ELISA Kit (R&D Systems). Adiponectin was measured using an ELISA sandwich assay (HMW & Total Adiponectin ELISA kit by the American Laboratory Products Company (ALPCO)).
Levels of high-sensitivity CRP (hs-CRP) were measured using an IMMAGE automatic immunoassay system (Beckman-Coulter). Blood concentrations of hs-IL-6 were measured using the Human IL-6 Quantikine HS ELISA Kit (R&D Systems), and concentrations of sCD14 were measured using the Human CD14 Quantikine ELISA Kit (R&D Systems). A Luminex assay (ProcartaPlex Mix&Match Human 13-plex, eBioscience) was used to measure the following cytokines: Interferon gamma-induced protein 10, C-X-C motif chemokine ligand 5 (CXCL5), CCL2, Eotaxine, IL-7, Macrophage migration inhibitory factor, Macrophage inflammatory protein 1β, stromal cell-derived factor 1, and Vascular endothelial growth factor A. Values <the 1st standard were replaced by 1/2 lower limit of quantification (LLoQ), and values below the limit permitting extrapolation were replaced with LLoQ/4. Blood cell counts (leucocytes, monocytes, neutrophils and immune cells) were measured using flow cytometry.
Biotin serum levels were quantified in serum samples from 212 individuals of this MetaCardis subset. Samples were selected among French individuals specifically chosen from the extremes of health status (NOB; n=107) and obesity state (SOB; n=105) excluding also individuals with coronary artery disease. Among the 381 samples retained from eight study groups (NOB-MH, n=87, NOB-T2D, n=107, SOB-MH, n=86, SOB-T2D, n=101), 27 samples in the extreme of gene richness distribution for each group were selected for biotin serum quantification. Biotin was measured from serum samples with competitive ELISA by MDbioscience (http://www.mdbiosciences.com/). The urinary excretion of 3-hydroxyisovaleric acid was quantified by ultra-performance liquid chromatography coupled to tandem mass spectrometry.
Collection of anthropometric and clinical data
Weight and height were assessed during the clinical inclusion visit according to standardised procedures using the same scales and units. Body fat mass and fat-free mass were measured via bioelectrical impedance analysis. Systolic and diastolic BP were measured using a mercury sphygmomanometer (measures were taken three times on each arm, and the mean of the last two measurements in the right arm was used for analyses). Moreover, a detailed list of prescribed medications as well as the patient’s medical history were gathered.
Collection of lifestyle and dietary data
Data on physical activity were collected using the Recent Physical Activity Questionnaire. This questionnaire, which assesses physical activity practices over the last 4 weeks, has been validated against doubly labelled water.44 Dietary data were collected via a Food-Frequency Questionnaire (FFQ) that was adapted to the cultural habits of each of the countries of recruitment. The MetaCardis FFQ was based on the validated European Prospective Investigation of Cancer-Norfolk FFQ and the content was based on several relevant European FFQs. Portion size and nutrient composition were derived from national food consumption surveys and food composition databases. A validation study against repeated 24-hour dietary records among 324 French MetaCardis participants has indicated an acceptable relative validity level, with a mean correlation coefficient of 0.503 for micronutrients.45
Biotin intake
Although there is no extensive validated food composition database for biotin, we have estimated biotin intake based on values concerning the biotin contents by two independent sources. The first concerned selected foods published by Staggs et al.30 The second concerned estimated biotin intake with a recent comprehensive nutrient database FooDB (https://foodb.ca/) added to the Danish national food database (DTU food database: https://frida.fooddata.dk/) which contains more complete biotin contents for 1029 foods. Importantly, we found that the FFQ based estimation of biotin intake based on the derivation of foods contents using these two methods (published values30 vs online data FooDB or DTU food database) were well correlated (with a mean correlation coefficient of 0.75). Consequently, for each individual, we decided to average the dietary intake of biotin estimated by the two methods to adjust our analyses. This allowed us to rank individuals based on dietary biotin intake and to adjust our analyses on nutritional intakes.
Metagenomic sequencing
Total faecal DNA was extracted following the International Human Microbiome Standards guidelines (SOP 07 V2 H) and sequenced using ion-proton technology (ThermoFisher Scientific) resulting in 23.3±4.0 million (mean±SD) 150 bp single-end reads per sample on average. Reads were cleaned using Alien Trimmer (V.0.2.4) in order to remove resilient sequencing adapters and to trim low quality nucleotides at the 3’ side (quality and length cut-off of 20 and 45 bp, respectively). Cleaned reads were subsequently filtered from human and potential food contaminant DNA (using human genome RCh37-p10, Bos taurus and Arabidopsis thaliana with an identity score threshold of 97%).
Faecal microbial load
Microbial loads of faecal samples were determined as described previously.17 Briefly, 0.2 g frozen (−80°C) aliquots were dissolved in physiological solution to a total volume of 100 mL (8.5 g/L NaCl; VWR International, Germany). Subsequently, the slurry was diluted 1000 times. Samples were filtered using a sterile syringe filter (pore size of 5 µm; Sartorius Stedim Biotech, Germany). Next, 1 mL of the microbial cell suspension obtained was stained with 1 µL SYBR Green I (1:100 dilution in DMSO; shaded 15 min incubation at 37°C; 10 000 concentrate, Thermo Fisher Scientific, Massachusetts, USA). The flow cytometry analysis was performed using a C6 Accuri flow cytometer (BD Biosciences, New Jersey, USA).46 Fluorescence events were monitored using the FL1 533/30 nm and FL3 >670 nm optical detectors. In addition, also forward and sideward-scattered light was collected. The BD Accuri CFlow (V.1.0.264.21) software was used to gate and separate the microbial fluorescence events on the FL1/FL3 density plot from the faecal sample background. A threshold value of 2000 was applied on the FL1 channel. The gated fluorescence events were evaluated on the forward/sideward density plot, as to exclude remaining background events. Instrument and gating settings were kept identical for all samples (fixed staining/gating strategy).46 Based on the exact weight of the aliquots analysed, cell counts were converted to microbial loads per gram of faecal material.
Metagenomics data processing
Gene abundance profiling was performed using the 9.9 million gene integrated reference catalogue of the human microbiome.26 Filtered high-quality reads were mapped with an identity threshold of 95% to the 9.9 million-gene catalogue using Bowtie (v2.2.6) included in the METEOR software. A gene abundance table was generated by means of a two-step procedure using METEOR. First, the uniquely mapping reads (reads mapping to a single gene in the catalogue) were attributed to their corresponding genes. Second, shared reads (reads that mapped with the same alignment score to multiple genes) were attributed according to the ratio of their unique mapping counts. The gene abundance table was processed for rarefaction and normalisation and further analysis using the MetaOMineR (V.1.2) R package. To decrease technical bias due to different sequencing depth and avoid any artefacts of sample size on low abundance genes, read counts were rarefied. The gene abundance table was rarefied to 10 million reads per sample by random sampling of 10 million mapped reads without replacement. The resulting rarefied gene abundance table was normalised according to the FPKM strategy (normalisation by the gene size and the number of total mapped reads reported in frequency) to give the gene abundance profile table.
Microbial gene richness (gene count) was calculated by counting the number of genes that were detected at least once in a given sample, using the average number of genes counted in 10 independent rarefaction experiments.
For quantification of functional modules metagenome reads mapped to the IGC gene catalogue after rarefaction to 10M reads per sample, were binned by functional category as per the annotations of the IGC previously carried out within the MOCAT2 framework (https://mocat.embl.de/). Functional potentials at each class of annotations (eg, Kyoto Encyclopedia of Genes and Genomes KEGG KOs, modules) were summed within each annotation term. Biotin production potential of the microbiome was computed as the mean abundances of biotin biosynthesis modules from KEGG database (M00123, M00577, M00573). Biotin consumption potential of the microbiome was derived from the abundance of KEGG module M00581.
Quantitative abundance profiling
Relative metagenomic profiles (RMP) of taxonomic and functional metagenomic features were transformed to QMP by multiplication of relative proportions to an indexing factor proportional to the microbial cell densities of the samples (load), defined as the sample load divided by the median load over the entire MetaCardis cohort.
Enterotyping
Enterotyping (or community typing) based on the Dirichlet multinomial mixtures approach was performed in R, with package Dirichlet Multinomial, as described by Holmes et al on a subsample (n=2022) of the n=2214 MetaCardis cohort (clinical and metagenomics data available).
Quantification of B vitamin biosynthesis and transport bacterial groups
To carry out an in-depth profiling of the bacterial metabolism of biotin and other B vitamins, we quantified the abundance of 15 different groups of bacteria with different phenotypes in terms of biotin biosynthesis and transport capabilities based on their gene content defined in the study of Rodionov et al.15 Briefly, 2126 reference genomes were retrieved from PATRIC database using PATRIC command line API (https://www.patricbrc.org/). Human-filtered metagenomic reads from 1545 samples were mapped against these 2126 reference genomes using NGLESS. Reads with >95% identity and >45% alignment length were retained and reference genome abundances were computed with the dist1 option of NGLESS, where first the genome abundances are computed from unique mapped reads and then are corrected by the multiple mapped reads weighted by the coverage of unique mapped reads. The genome abundance table was processed for rarefaction and RPKM normalisation with MetaOMineR (V.1.2) R package. The raw genome abundance table was rarefied to 2 million reads per sample and the rarefied genome abundance table was normalised according to the FPKM strategy (normalisation by genome size and the genome abundances reported in frequency). The abundance of the 15 biotin species groups was computed as the sum of the abundances of individual bacterial genomes. Similar approach was followed to compute the total abundances of biotin producers (bacteria with all biotin biosynthesis genes from Pimelate and no biotin transport gene), biotin transporters (bacteria with no gene involved in biotin biosynthesis) and biotin producers+transporters (bacteria harbouring biotin biosynthesis and transport genes). From this abundance table (RMP), absolute abundances (QMP) were computed by multiplying relative abundances by the ratio between the sample microbial load and the median value of microbial load throughout the MetaCardis cohort.
Analyses of microbial diversity and community structure
Microbial gene richness (the number of genes from the IGC gene catalogue with non-zero abundance) was calculated after rarefying the data to 10 million reads per sample. Differences in microbial gene richness across disease groups (metabolic health groups within different obesity states with T2D individuals stratified by metformin treatment; obesity groups within metabolic health status) were evaluated with Kruskal-Wallis tests controlled for potential confounder of country of study with independence_test function of the COIN R package (options ytrafo=rank, teststat = ‘quad’). Pairwise differences between all study groups within each comparison were evaluated with WRS test adjusted for the same potential cofounders (independence_test; options ytrafo=rank, teststat=’scalar’) and corrected for multiple testing using the Benjamini-Hochberg FDR. To identify features with non-redundant explanatory power on microbiota variation, first bdRDA was carried out on genus-level community ordination (PCoA based on Bray-Curtis beta-diversity matrix) with 26 patient variables including disease severity group, obesity parameters (BMI, %fat mass, waist to hip ratio), biological markers of metabolic impairment (plasma glucose, HDL-cholesterol, triglycerides, HbA1c, uric acid), clinical markers of metabolic impairment (systolic and diastolic BP, hypertension status), diabetes status and medications consumed by at least 10% of the individuals with capscale function of the vegan R package. 18 covariates were identified (capscale p<0.05), which were subsequently filtered to identify the ones with non-redundant explanatory power with the env2fit function of the vegan R package.
Animal experiments
Experiments with GF mice and faecal microbiota transfer
The 12–15 weeks female CONV-R and GF Swiss Webster and C57BL/6 J mice were fed an autoclaved chow diet (5021, LabDiet) and water ad libitum. CONV-R mice were housed in individually ventilated cages (Green line Sealsafe plus, Tecniplast, Buguggiate, Italy) and GF mice were housed in flexible film isolators; all mice were housed in groups of maximum five mice per cage under a strict 12 hours light cycle. GF status was tested by culturing faecal bacteria anaerobically and aerobically, and by PCR of the bacterial 16S rRNA gene with universal primers 27F (5’-GTTTGATCCTGGCTCAG-3’) and 1492R (5’-CGGCTACCTTGTTACGAC-3’). PCR reactions were performed with a 5 min preincubation at 95°C followed by 30 cycles of 30 s at 94°C, 45 s at 52°C, and 90 s at 72°C and then kept at 72°C for 7 min.
For faecal microbiota transfer, female Swiss Webster GF mice were colonised at the age of 10–13 weeks for 4 weeks with faeces from two subjects with SOB and two subjects NOB who had higher amounts of microbiota biotin producers in comparison to SOB subjects. Colonised mice were kept in individually ventilated cages (ISOcage N System, Tecniplast, Buguggiate, Italy) with a maximum of 5 mice per cage. 500 mg of frozen stools were suspended in 5 mL of LYHBHI medium (Brain-heart infusion medium supplemented with 0.5% yeast extract and 0.1% cellobiose, 0.1% maltose, 0.05% cysteine and 0.25% hemin47 containing 20% glycerol). Faecal slurries were prepared in a Coy chamber, aliquoted in 1 mL volumes, and stored at −80°C until use. The mice were randomised into four groups and colonised twice by oral gavage with 200 µl slurry after a 4-hour fasting; the second gavage was administered 2 days after the first dose. Colonised mice from each donor were fed ad libitum either an autoclaved chow diet (5021, LabDiet) or an irradiated WD (40% kcal fat, TD.09683, Harlan Teklad).
Serum samples were collected from the cava vein after a 5-hour fasting.
Analyses of Illumina 16S V4 amplicons from faecal transfer experiments were carried out with QIIME2 (https://qiime2.org/). Quality filtering of raw reads, amplicon reconstructions, chimaera removal and reconstruction of Amplicon Sequence Variants were carried out with the DADA2 plugin of QIIME2. Taxonomic classification of ASV was carried out with the Naïve Bayes Classifier of QIIME2 trained against GreenGenes 13_8 database. Raw ASV tables were rarified to 30 000 counts per sample to avoid biases associated with differences in sequencing depth between samples after inspection of rarefaction curves. Functional profiling from rarefied ASV tables were carried out with PICRUSt2.48
Animals
Male C57BL/6 J mice were obtained from Charles River Laboratories (France) and Janvier Labs (France). Adult male C57BL/6NTac were obtained from Taconic Biosciences (Denmark). Mice were housed in groups of 5 and maintained on a 12-hour light-dark cycle with ad-libitum access to water. They had access either to a standard chow diet (A04-10, Safe Diets, Germany) or a HFD, D12492, 60% kcal fat, Research Diets, USA. The experiments were carried out in accordance with the recommendations of Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. The experiments were approved by the local committee and received permission from the French ‘Ministère de la Recherche’.
Body weight and composition
Body weight and food intake were monitored once a week in all animal models. Whole body composition was assessed weekly by Nuclear Magnetic Resonance (Minispec LF50, Bruker, USA), which quantified body fat, lean tissue and free body fluid. Mice were weighed before being inserted into the instrument for analysis (2 min/mouse).
Antibiotic administration
Adult male C57BL/6NTac control mice (n=16) were gavaged daily gavage either with water (n=8) or with a solution of broad-spectrum antibiotics (100 mg/kg of vancomycin and 200 mg/kg of ampicillin, neomycin and metronidazole)33 diluted in sterile water (n=8) for 14 days.
EGA surgery
Fourteen C57BL/6J animals underwent an EGA. As a control group, thirteen animals underwent a laparotomy (Sham operated animals). The EGA procedure was performed as previously described.35 In brief, C57Bl6 mice on HFD or a standard Chow diet (A04-10, Safe Diets, Germany) undergoing surgery were fasted for 6 hours and anaesthetised with 2% isoflurane (Abbott, Rungis, France) and air/oxygen. Analgesia was delivered intraperitoneally 30 min before surgery (Buprenorphine, 0.03 mg/kg, Axience SAS, France) and at the end of the procedure (Ketoprofen, Merial, 1%, diluted 1/100, 150 µL per mouse). The procedure consisted in a pyloric sphincter’s ligature, followed by an entero-gastric anastomosis allowing the exclusion of the duodenum and the proximal jejunum of the alimentary tract. Sham-operated mice (simple laparotomy) underwent the same duration of anaesthesia as EGA mice. In both animal groups, the laparotomy was repaired in two layers. All mice were maintained on a standardised post-operative protocol to monitor pain, body weight and hydration, subcutaneous injection of saline serum and additional analgesia was given as necessary. The mice had access to water and diet right after the surgery.
Biotin supplementation
Two groups of animals (male C57BL/6J, 8 weeks old) were implanted with osmotic pumps (#2006, 200 µL, 0.15 µL/hour, Alzet, USA) filled with either biotin (Sigma Aldrich, USA) diluted at a concentration of 75 mg/mL in vehicle solution (water +5% DMSO; pH adjusted to 7.5) or the vehicle alone. Pumps were implanted subcutaneously in mice anaesthetised by isoflurane.
Other groups of animals were fed a biotin-supplemented HFD custom diet containing 97.7 mg of biotin/kg of diet (Research Diets, USA).
FOS treatment
The administration of a prebiotic, (FOS, Orafti P95, Germany) was done in the drinking water of the animals. This compound was obtained from Bénéo and incorporated into drinking water at a dose of 10% w/w. The drinking solution was filtered on 0.22 µm and changed every 2 days to avoid spoilage.
Fasting glucose and insulin measurements
Animals were fasted for 6 hours then weighed and glycaemia was measured from tail blood.
Plasma insulin concentration was measured by a commercially available ELISA kit (Mouse Ultrasensitive Insulin ELISA, ALPCO, USA) in blood samples collected after 6 hours fasting, at the same time fasting glycaemia was measured. qPCR on tissues
Total RNA was extracted from tissues using RNeasy Mini Kit (QIAGEN, Germany). Then, the RNA was reverse-transcribed using random primers and M-MLV (Promega, USA). We performed qPCR with StepOne System (Applied BiosystemsTM, USA) using Fast SYBR Master Mix (Life Technologies, USA). Each sample was normalised to housekeeping genes, 18S and rplp0. Relative fold changes in gene expression were calculated by the ∆∆CT method using the equation 2(-∆∆CT). Sequences used are summarised in the table 1 below.
Primer sequence used for adipose tissue gene expression
Plasma/serum biotin quantification in mice
Depending on the experimental design, blood was obtained either from retro-orbital bleeding under general anaesthesia (isofluorane 3%) or from the tail vein after 6-hour fasting in EDTA-coated tubes. The blood was then centrifuged at 13 000 g during 3 min to collect plasma. For the experiments using serum, blood was collected under anaesthesia (isofluorane) by cannulation of both the cava and porta veins after opening the peritoneal cavity. The blood was kept in a standing position for 30 min and then centrifuged at 1500 g for 10 min at 4°C to collect serum. Biotin level was estimated in the plasma/serum following the instructions of the IDK Biotin ELISA kit (K8141, Immundiagnostik). Plasma/serum was diluted to 1:10 and 1:20.
Faecal DNA extraction
Faeces from mice were collected weekly and immediately stored at −80°C on usage. Extraction was then performed using the PureLink Microbiome DNA Purification kit (Invitrogen) following the provider’s protocol. DNA was assessed both by Qubit 4 Fluorometer (Invitrogen) for the quantity and NanoDrop (Thermofisher) for the quantity and quality.
Faecal DNA sequencing (Nanopore)
Extracted faecal DNA was sequenced by the MinION from Oxford Nanopore Technologies (ONT). The DNA library was prepared with the Ligation Sequencing Kit (ref SQK-LSK 109, ONT), with multiplexing so that up to 12 samples were sequenced at the same time (ref EXP-NBD104, ONT). The ZymoBIOMICS Microbial Community DNA Standard (#D6306, Zymo Research) was used as an internal control for each sequencing run.
Total faecal bacterial abundance (qPCR)
Faecal DNA was diluted to 0.1 ng/µL and qPCR using primers targeting the 16S rRNA gene were performed with StepOne System (Applied BiosystemsTM, USA) using Fast SYBR Master Mix (Life Technologies, USA).
Bioinformatic data processing (Nanopore)
Nanopore fast5 reads (343 206 reads per sample on average) were basecalled and demultiplexed with Guppy (V.2.1.3). Quantification of biotin and B vitamin bacterial groups was carried out mapping Nanopore fastq files against the same set of 2126 reference genomes mentioned above coming from the study of Rodionov et al15 using minimap2 aligner with the map-ont option optimised for Nanopore sequencing data. SAM files product of minimap2 alignment were used as input for NGLESS to compute the abundances of reference genomes with the dist1 option as described. The genome abundance table was processed for rarefaction and RPKM normalisation with MetaOMineR (V.1.2) R package. The raw genome abundance tables were rarefied to 40 000 mapped reads per sample and the rarefied genome abundance table was normalised according to the FPKM strategy (normalisation by genome size and the genome abundances reported in frequency). The abundance of the 15 biotin species groups was computed as the sum of the abundances of individual bacterial genomes. Similar approach was followed to compute the total abundances of biotin producers (bacteria with all biotin biosynthesis genes from pimelate and no biotin transport gene), biotin transporters (bacteria with no gene involved in biotin biosynthesis) and biotin producers+transporters (bacteria harbouring biotin biosynthesis and transport genes). Absolute metagenomic abundances were obtained by multiplying the relative metagenomic abundances by the total bacterial abundance obtained by qPCR of the 16S rRNA gene.
Microbiome data and biotin measures from independent BS cohorts
We quantified serum biotin and performed functional metagenomics from faecal samples collected from patients with severe obesity involved in a BS programme (Microbaria) as reported elsewhere.4 Briefly, we included women with SOB at Pitié-Salpêtrière Hospital Nutrition department, Paris. Patients were assigned for adjustable gastric banding or RYGB following international BS guidelines. Clinical, anthropometric, and biological evaluations were obtained at baseline (T0) and during follow-up at 1 (T1), 3 (T3) and 12 months (T12) postsurgery. Faeces were collected at each visit. No patient had received antibiotic treatment for 3 months prior to BS, nor had any past history of acute or chronic gastrointestinal diseases and were weight stable before the intervention. All subjects signed the informed written consent and the protocol was registered at ClinicalTrial.gov (Microbaria Study, NCT01454232). Biotin serum levels were quantified in serum samples from 17 individuals from Microbaria cohort at baseline and 1 year after BS (34 samples overall). Biotin was measured with competitive ELISA by MDbioscience (http://www.mdbiosciences.com/). The biotin serum for the additional cohort of BS patients (BARICAN (CNIL No. 1222666; P050318 Les Comités de Protection des Personnes approval 24 November 2006, NCT01655017, NCT01454232)), (n=41 patients at each time T0: baseline, T12: 1 year after surgery) was measured using the IDK Biotin ELISA kit (K8141, Immundiagnostik). Measurements were performed on non-diluted and 1:2 diluted serum.
Quantification of biotin producers and transporters were carried out over 24 individuals from the Microbaria cohort with metagenomic data on baseline, 1, 3 and 12 months after BS. Human filtered SOLiD reads from these individuals were downloaded from the ENA repository and mapped over the set of 2126 reference genomes mentioned above coming from the study of Rodionov et al15 using bowtie allowing a maximum of 3 mismatches and keeping only the best alignments in each strata.49 SAM alignment was used as input for NGLESS to compute the abundances of reference genomes with the dist1 option as described above. The genome abundance table was processed for rarefaction and RPKM normalisation with MetaOMineR (V.1.2) R package. The raw genome abundance tables were rarefied to 1 million reads per sample and the rarefied genome abundance table was normalised according to the FPKM strategy (normalisation by genome size and the genome abundances reported in frequency). The abundance of biotin producers and transporters was computed as the sum of the abundances of individual bacterial genomes in each group and these abundances were multiplied by the gene richness in order to have an estimate of the absolute (QMP) metagenomic abundances, based on the positive correlation described between microbial diversity and microbial cell density17 and observed in the MetaCardis cohort (Spearman’s r=0.34, p=2.74×10−44, n=1545 individuals).
Adipose tissue gene expression and blood inflammation markers
For 43 obese patients who underwent BS, subcutaneous adipose tissue gene expression measures were available from microarray and/or qPCR-based methods (42 patients had microarray data, of which 24 patients also had available qPCR data; 25 patients had qPCR data, and 1 of these patients did not have available microarray data). For these gene expression measurements on subcutaneous adipose tissue samples, RNA was extracted from needle biopsies taken before surgery. Blood inflammation markers were measured using a multiplex immuno-assay (Myriad-Rules-Based Medicine (RBM)) among n=161 subjects with severe obesity, before BS. Missing values for blood inflammation markers (n=8 for CCL2, n=5 for IL-18, n=4 for CEACAM1, n=3 for CXCL9, n=1 for ANGPT1 and n=1 for SPP1) were imputed using the LLoQ and a beta law.
Microarray experiments
Total RNA was extracted from stored tissues by using the RNeasy total RNA Mini kit (QIAGEN) with one-column DNase digestion. RNA quality and concentration were assessed by using an Agilent 2100 Bioanalyzer (Agilent Technologies). An Illumina RNA amplification kit (Ambion) was used according to the manufacturer’s instructions to obtain biotin-labelled complementary RNA from 250 ng total RNA. Hybridisation processes were performed with Illumina Human HT-12 V.4.0 Expression BeadChips (Illumina). Hybridised probes were detected with cyanin-3-streptavidin (1 mg/mL; Amersham Biosciences, GE Healthcare) and scanned by using an Illumina BeadArray Reader. Raw data were extracted with GenomeStudio V.2011.1 software by using the default settings and without any additional normalisation. The target genes on which this study focused were selected as biotin-related genes (SMVT, HLCS, BTD, ACACA, PCCB, PC, ACACB, PCCA, MCCC2), as well as selected inflammation-related genes (A2M, ADIPOQ, ANGPT1, CAECAM1, CCL13, CCL19, CCL2, CCL23, CCL5, CCL8, CXCL10, CXCL12, CXCL9, DCN, FTH1, IGFBP3, IGFBP4, IGFBP5, IGFBP6, IL18, IL18BP, MMP2, MMP9, PECAM1, SERPINE1, SFTPD, SPP1, TIMP1, TNFRSF11B, TNFRSF1A, TNFRSF1B, TNFSF113B, VCAM1).
qPCR experiments
For the above-mentioned selected biotin-related genes, subcutaneous adipose tissue gene expression before and after BS was also measured by qPCR. For each target gene whose expression was measured by qPCR, measured CTs (eg, cycle threshold values) were transformed to ΔCTs by subtracting mean CT values observed at baseline (eg, before BS). CT values observed for the reference gene hypoxanthine phosphoribosyltransferase (HRPT1) were also transformed to ΔCTs in the same manner. Next, ΔCTs for HRPT1 were subtracted from ΔCTs for the target genes, and the resulting value was then squared. For a better visualisation in boxplot figures, these values were moreover transformed to Z-scores. 43,43
Statistical analysis
All statistical analyses were carried out using R statistical analysis software V.3.3.2. and statistical tests were two sided. All reported measures of correlation in the article are non-parametric Spearman correlations. R2 are expressed as %.
All p values were corrected for multiple testing when appropriate using the Benjamini-Hochberg method or Dunn’s method (for mice models). Only p value or FDR <0.05 was reported as significant. Regarding adjustments in the models, when appropriate, we have adjusted on country of recruitment and age because they are identified as confounding factors for gut richness. Sensitivity analyses were performed for all models regarding confounding covariates and did not change the results.
Association between log-transformed absolute abundances of microbiome biotin biosynthesis and production variables (from IGC gene abundances and from reference genomes) and obesity status were evaluated by linear regression models adjusted by statin intake to take into account the impact of this variable on microbiome composition.5 Pairwise comparison of estimated marginal means of the statin-adjusted linear regression between obesity groups was carried out with emmeans V.1.4.6 R package (Russell Lenth (2020). emmeans: Estimated Marginal Means, aka Least-Squares Means. R package V.1.4.6. https://CRAN.R-project.org/package=emmeans) with p value adjustment with Tukey method.
Data availability statement
Data are available in a public, open access repository. Data are available on reasonable request. metacardis https://www.ebi.ac.uk/ena/browser/view/PRJEB41311, https://www.ebi.ac.uk/ena/browser/view/PRJEB38742, https://www.ebi.ac.uk/ena/browser/view/PRJEB37249, mouse experiments; 16S data, https://www.ebi.ac.uk/ena/browser/view/PRJEB42967, mouse experiments; nanopore data, https://www.ebi.ac.uk/ena/browser/view/PRJEB42966, Code paper analyses, https://git.ummisco.fr/ebelda/metatransitionbiotingut.git.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by Ethics Committee CPP Ile-de France, Ethics Committee at the Medical Faculty at the University of Leipzig, and the Ethical Committees of the Capital Region of Denmark. The study design (observational cohort study) complied with all relevant ethical regulations, aligning with the Declaration of Helsinki and in accordance with European privacy legislation. The experiments were approved by the Ethics Committee on Animal Care and Use in Gothenburg, Sweden as well as in Sorbonne University, Paris, France. All animal experiments were conducted in accordance with the principles of the ‘Guide to the Care and Use of Experimental Animals’.
Acknowledgments
We thank the subjects for their participation in the MetaCardis study and particularly patient associations (Alliance du Coeur and CNAO) for their input and interface, as well as Dr Dominique Bonnefont-Rousselot (Department of Metabolic Biochemistry, Pitié-Salpêtrière hospital) for the analysis of plasma lipid profiles. We also thank Manuela Krämer, Robert Jakubowicz and Anna Hallén for work on the germ-free mice and sample preparation. We thank Amélie Lacombe and the PreclinICAN platform for help on mice experiments. We thank the nurses, technicians, clinical research assistants and data managers from the Clinical investigation platform at the Institute of Cardiometabolism and Nutrition for patient investigations, at the CRNH (Centre de recherche en Nutrition Humaine CRNH-Ile de France) and, the Clinical Investigation Center (CIC) from Pitié-Salpêtrière Hospital for investigation of healthy controls. Quanta Medical provided regulatory oversight of the clinical study and contributed to the processing and management of electronic data. We also thank supports from Assistance Publique-Hôpitaux de Paris (AP-HP), the promoter of the clinical investigation (MetaCardis and Microbaria studies), SFN (Société Française de Nutrition), Fondation Cœur et Artères, F-CRIN-FORCE network for support and INSERM via ITMO.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @jeandbdt, @A_Myridakis, @BackhedLab
EB, LV, VT, GF, SA and KEA contributed equally.
JR and KC contributed equally.
Correction notice This article has been corrected since it published Online First. The author's name, Edi Prifti, has been corrected.
Collaborators The MetaCardis consortium Collaborators: Rohia Alili, Renato Alves, Ehm Astrid Andersson Galijatovic, Olivier Barthelemy, Jean-Philippe Bastard, Jean-Paul Batisse, Magalie Berland, Randa Bittar, Hervé Blottière, Frederic Bosquet, Rachid Boubrit, Olivier Bourron, Mickael Camus, Dominique Cassuto, Cecile Ciangura, Jean-Philippe Collet, Arne Dietrich, Morad Djebbar, Angélique Doré, Line Engelbrechtsen, Leopold Fezeu, Sebastien Fromentin, Philippe Giral, Marianne Graine, Caroline Grünemann, Agnes Hartemann, Bolette Hartmann, Gerard Helft, Malene Hornbak, Lesley Hoyles, Jean-Sebastien Hulot, Richard Isnard, Sophie Jaqueminet, Niklas Rye Jørgensen, Hanna Julienne, Johanne Justesen, Judith Kammer, Nikolaj Karup, Mathieu Kerneis, Jean Khemis, Lars Køber, Ruby Kozlowski, Michael Kuhn, Aurelie Lampure, Véronique Lejard, Ivica Letunic, Florence Levenez, Lajos Marko, Lea Lucas-Martini, Laura Martinez-Gili Robin Massey, Nicolas Maziers, Jonathan Medina-Stamminger, Lucas Moitinho-Silva, Gilles Montalescot, Sandrine Moutel, Ana Luisa Neves, Michael Olanipekun, Laetitia Pasero Le Pavin, Luis Pedro Coelho, Christine Poitou, Francoise Pousset, Laurence Pouzoulet, Andrea Rodriguez-Martinez, Sebastien Schmidt, Tatjana Schütz, Lucas Silva, Johanne Silvain, Mathilde Svendstrup, Timothy D Swartz, Thierry Vanduyvenboden, Camille Vatier, Eric O Verger, Stefanie Walther
Contributors EB, GF, JR and KC conceived and designed the project. EB and GF provided principal metagenomic analyses linked with patient phenotypes and adjusted for microbial cell loads. VT, LV, JD, TLR, CA and LM-H performed in vivo murine studies, provided murine biological samples, and their analyses. EB provided metagenomic analyses for in vivo murine studies. KEA and SA determined dietary analysis and patterns and associations with patient clinical phenotypes and inflammatory profiles, BH provided interpretations for dietary FFQ data, SA and FM performed analyses for intestinal inflammation and interpreted patient inflammatory data, VP provided molecular analyses for gene expression in human tissues, FA, MB, RC, JE-S, J-MO, JA-W, TN, KC and MS recruited patients and RC, JA-W and TN contributed to patient investigation and data management. SH and PG provided healthy volunteers from the NutriNet-Santé Study, EB, SF, ELC, GF, NP, EP, IL, JN, SV-S, BJ and PD developed databases, analytical pipelines and performed metagenomics and functional analysis. BQ and HR contributed to stool sample processing and sequencing all patient stool samples, CR, SAn and FM performed all measurements of inflammatory cells and systemic markers, and SFe performed the centralised measures of metabolic variables. JC and AM performed metabolomic measurements on patients, NBS provided network analyses to assist in interpreting data, J-DZ, SH, M-ED, JR, J-MO, OBP, PB, MS and SDE contributed to results’ discussion. EB, LV, VT, GF, SA, TLR, KEA, FB, JR and KC wrote the paper. All authors commented and edited the manuscript. KC is the guarantor who accepts full responsibility for the work and/or the conduct of the study, had access to all data, and controlled the decision to publish.
Funding This work was supported by European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement HEALTH-F4-2012-305312 (METACARDIS). Funding supports were also obtained from Leducq Foundation(17CVD01), JPI-Microdiet study (2017-01996_3). Part of the work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG): SFB 1052 (project B1), the Fondation pour la Recherche Médicale (FDT201904008276, FDT202106012793), and the French Agency of Research (ANR-CAPTOR, ANR-DeepIntegromics).
Competing interests KC is a consultant for Danone Research, Ysopia and CONFO therapeutics for work not associated with this study. KC held a collaborative research contract with Danone Research in the context of MetaCardis project. FB is a shareholder of Implexion pharma AB. MB received lecture and/or consultancy fees from AstraZeneca, Boehringer-Ingelheim, Lilly, Novo Nordisk, Novartis and Sanofi.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
these sentences have to be deleted because related experiments are not shown in this version