Article Text

Abstract
Objective Intravenous iron—a common treatment for anaemia and iron deficiency due to inflammatory bowel disease (IBD)—can cause hypophosphataemia. This trial compared the incidence of hypophosphataemia after treatment with ferric carboxymaltose (FCM) or ferric derisomaltose (FDI).
Design This randomised, double-blind, clinical trial was conducted at 20 outpatient hospital clinics in Europe (Austria, Denmark, Germany, Sweden, UK). Adults with IBD and iron deficiency anaemia (IDA) were randomised 1:1 to receive FCM or FDI at baseline and at Day 35 using identical haemoglobin- and weight-based dosing regimens. The primary outcome was the incidence of hypophosphataemia (serum phosphate <2.0 mg/dL) at any time from baseline to Day 35 in the safety analysis set (all patients who received ≥1 dose of study drug). Markers of mineral and bone homeostasis, and patient-reported fatigue scores, were measured.
Results A total of 156 patients were screened; 97 (49 FDI, 48 FCM) were included and treated. Incident hypophosphataemia occurred in 8.3% (4/48) FDI-treated patients and in 51.0% (25/49) FCM-treated patients (adjusted risk difference: −42.8% (95% CI –57.1% to –24.6%) p<0.0001). Both iron formulations corrected IDA. Patient-reported fatigue scores improved in both groups, but more slowly and to a lesser extent with FCM than FDI; slower improvement in fatigue was associated with greater decrease in phosphate concentration.
Conclusion Despite comparably effective treatment of IDA, FCM caused a significantly higher rate of hypophosphataemia than FDI. Further studies are needed to address the longer-term clinical consequences of hypophosphataemia and to investigate mechanisms underpinning the differential effects of FCM and FDI on patient-reported fatigue.
- iron deficiency
- IBD
- anemia
Data availability statement
No data are available. No individual participant data will be shared, and no trial related information (data, analytical methods, study materials) will be available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Iron deficiency anaemia (IDA) is the most common extraintestinal complication affecting patients with inflammatory bowel disease (IBD), and intravenous iron is a common treatment for IDA in patients with IBD. In a recent systematic literature search covering 42 clinical studies between 2005 and 2020, the pooled incidence of hypophosphataemia after intravenous infusion of ferric carboxymaltose (FCM) was 47% (95% CI 36% to 58%) versus 4% (95% CI 2% to 5%) after intravenous infusion of ferric derisomaltose (FDI). Hypophosphataemia of variable severity and duration is caused by differential effects of FCM and FDI on the phosphate regulating hormone, fibroblast growth factor 23 (FGF23), which induces renal phosphate excretion and downstream effects on 1,25-dihydroxyvitamin D and parathyroid hormone (PTH) that maintain hypophosphataemia. Case series report clinical complications of hypophosphataemia, such as osteomalacia, fractures and severe muscle weakness after FCM, but not after FDI.
WHAT THIS STUDY ADDS
The present trial is the first randomised, double-blind, head-to-head comparison of the risk of hypophosphataemia following equivalent dosing of intravenous FCM or FDI among patients with IDA due to IBD. In the primary outcome, hypophosphataemia (serum phosphate <2.0 mg/dL) occurred in 8.3% of patients treated with FDI versus 51.0% of patients treated with FCM, despite comparable effects on haemoglobin (Hb) and iron stores. Compared with FDI, hypophosphataemia after FCM was associated with more pronounced increases in intact FGF23, renal phosphate excretion, and PTH, and more pronounced decreases in 1,25-dihydroxyvitamin D. Patient-reported fatigue scores improved in both treatment groups, but improvement in patients treated with FDI was faster and greater than in those receiving FCM. Slower improvement in fatigue was associated with more severe hypophosphataemia.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
In patients with IDA due to IBD, the risk of hypophosphataemia is lower in those treated with FDI than those receiving FCM. As both drugs robustly increased Hb over time, the difference in improvement of fatigue may be related to hypophosphataemia. Potential adverse effects of longer-term changes in bone and mineral metabolism warrant further investigation.
Introduction
Anaemia is the most common extraintestinal manifestation of inflammatory bowel disease (IBD),1–3 and iron deficiency and inflammation are its leading causes.2–4 IBD-associated anaemia (iron deficiency and anaemia of chronic disease) is associated with impaired physical and cognitive functioning, increased hospitalisation rates, and reduced quality of life.5–7 The European Crohn’s and Colitis Organisation (ECCO) recommends high-dose intravenous iron as a first-line treatment for iron deficiency anaemia (IDA) in patients with IBD, because the bioavailability and effect of oral iron is limited in patients with inflammation.7 Based on their proven efficacy to treat IDA,8–10 ferric derisomaltose (FDI; previously known as iron isomaltoside) and ferric carboxymaltose (FCM) are among the most widely used intravenous iron formulations in Europe.
Hypophosphataemia is increasingly recognised as an important adverse effect of certain intravenous iron formulations. Intravenous iron-induced hypophosphataemia occurs most commonly in the first 2 weeks following infusion and can be mild and asymptomatic, but in some cases can be severe, symptomatic, persistent, and associated with alterations in mineral and bone metabolism that can culminate in osteomalacia and fractures.11–14 In recent head-to-head randomised clinical trials of patients with IDA predominantly due to heavy uterine bleeding, FCM demonstrated a significantly higher incidence of hypophosphataemia versus FDI.15 Hypophosphataemia due to FCM was driven by acute increases in concentration of the phosphate-regulating and vitamin D-regulating hormone, fibroblast growth factor 23 (FGF23), which triggers a pathophysiological cascade involving renal phosphate wasting, reduced concentration of 1,25-dihydroxyvitamin D, hypocalcaemia, and secondary hyperparathyroidism.11 15 16 These effects are reminiscent of tumour-induced osteomalacia that is caused by excessive ectopic production of FGF23.17
We conducted the PHOSPHARE-IBD randomised, double-blind, clinical trial to compare the incidence of hypophosphataemia after treatment with equivalent doses of FDI versus FCM in patients with IDA due to IBD. We tested for differential effects of the two iron preparations on bone and mineral metabolism. Also, by enrolling patients who required repeated dosing of intravenous iron, we investigated the effects on bone and mineral metabolism in patients who received a second dose of intravenous iron approximately 1 month after their initial infusion. Finally, since the symptoms of hypophosphataemia are relatively non-specific and difficult to discern from similar constitutional symptoms of IDA and IBD,18 we formally studied fatigue scores using a validated patient-reported outcome assessment.
Materials and methods
Trial design
PHOSPHARE-IBD was a randomised, double-blind, comparative clinical trial conducted at 20 outpatient hospital clinics across Europe (Austria, Denmark, Germany, Sweden, and the UK). The trial protocol (EudraCT Number: 2017-002452-87) was approved by local ethics committees and competent authorities.
Patients
Study personnel at participating trial centres recruited adults aged ≥18 years with IBD and IDA, which was defined as haemoglobin (Hb) <130 g/L and serum ferritin ≤100 ng/mL, who had a history of intolerance or unresponsiveness to oral iron, or in whom there was a clinical need to administer iron rapidly. Inclusion criteria also included body weight ≥50 kg, estimated glomerular filtration rate ≥65 mL/min/1.73 m2 (using the Modification of Diet in Renal Disease formula),19 and serum phosphate >2.5 mg/dL. Exclusion criteria included anaemia predominantly due to factors other than IDA, haemochromatosis or other iron-storage disorder, or intravenous iron use within 30 days prior to screening. Patients were also excluded if they had Hb ≥100 g/L with a body weight <70 kg, if they had a known hypersensitivity to any component in FDI or FCM or had experienced a previous serious hypersensitivity reaction to any intravenous iron compound. The Hb level and weight limitations were necessary to ensure that all patients would be eligible for repeat treatment on Day 35. Severity of IBD was not an exclusion criterion and the trial protocol did not affect the treatment of patients’ underlying IBD, although any patients who were expected to have surgery during the trial period were excluded. All patients provided written informed consent to participate.
Randomisation and masking
Patients were randomised 1:1 by unblinded staff members, using an interactive web response system (eClinicalOS eCRF system randomisation module), which blinded investigators, healthcare staff, and patients to treatment assignment. Blinding was maintained by shielding investigators and patients from observing the preparation of the trial drug, which was performed by unblinded trial personnel not involved in the trial assessments of efficacy or safety. Randomisation was stratified to balance the two treatment groups with regard to screening serum phosphate (<3.5 or ≥3.5 mg/dL).
Procedures
Patients received either FDI (Pharmacosmos A/S, Denmark) or FCM (Vifor Pharma, Switzerland). In each treatment group, the total iron requirement at baseline was calculated from body weight and Hb concentration, as recommended in the ECCO guidelines.7 Patients with baseline Hb <100 g/L received a total iron dose of either 1500 mg or 2000 mg if their body weight was <70 kg or ≥70 kg, respectively; the total iron dose was 1500 mg if baseline Hb was ≥100 g/L. Although FDI can be administered at doses of up to 20 mg/kg body weight,20 both formulations were administered as split doses with a maximum of 1000 mg on each dosing occasion in accordance with the European approved dosing schedule for FCM (maximal single FCM dose is 1000 mg).21 To allow direct comparison, patients received FDI or FCM as a single 20 min intravenous infusion of 1000 mg at baseline (Day 0) and, depending on the a priori calculated iron dose, either 500 mg or 1000 mg at Day 35. During the trial, treatment with erythropoiesis-stimulating agents, blood transfusion, radiotherapy, and chemotherapy were prohibited.
A screening period of up to 28 days was followed by a baseline randomisation visit on Day 0 and follow-up visits on Days 1, 7, 14, 35, 42, 49, and 70 to assess serum phosphate. Additional non-fasting blood and spot urine samples were collected at each visit; blood samples were analysed at a central laboratory and the results were reported to the physician investigators. The Day 1 assessments were included to capture physiological responses 24 hours after the initial intravenous iron administration. Fatigue was assessed on Days 0, 14, 35, 49, and 70 using the Functional Assessment of Chronic Illness Therapy (FACIT) Fatigue Scale, a patient-reported outcome. The FACIT scale has been validated in patients with IBD.22 Adverse events (AEs) were recorded at each visit.
Outcomes
The primary endpoint was the incidence of hypophosphataemia (defined as serum phosphate <2.0 mg/dL) at any time from baseline to Day 35, after all patients had received 1000 mg of either iron formulation, regardless of the degree of underlying iron deficiency. Among several secondary safety and efficacy endpoints, and exploratory endpoints (online supplemental table S1), this report focuses on the incidence of hypophosphataemia at any time through Day 70, changes from baseline to each postrandomisation visit in Hb, ferritin, transferrin saturation (TSAT), and biomarkers of mineral and bone homeostasis, including serum phosphate, urinary fractional excretion of phosphate, intact FGF23, 1,25-dihydroxyvitamin D, 24,25-dihydroxyvitamin D, ionised calcium, parathyroid hormone (PTH) and bone-specific alkaline phosphatase (ALP), as well as FACIT Fatigue Scale score. AEs were evaluated for severity and seriousness.
Supplemental material
Statistical analysis
Based on an assumed incidence of hypophosphataemia of 15% in the FDI treatment arm and 40% in the FCM arm,11 49 patients in each treatment group would be required to detect a significant difference between the groups with 5% alpha and 80% power. To account for potential imprecision in the estimated incidence rates at the time of trial design, and to gain more safety information, we planned to randomise 60 patients per treatment group. However, recruitment was ended early due to the COVID-19 pandemic.
The primary endpoint, secondary safety endpoints, and bone-specific ALP (pre-specified exploratory safety endpoint), were analysed using the safety analysis set, which included all patients who received at least one dose of study drug. The secondary efficacy endpoints, and FACIT Fatigue Scale score (prespecified exploratory patient-reported outcome), were analysed using the intention-to-treat (ITT) analysis set, which included all randomised patients. For the primary endpoint, the difference between the incidence of hypophosphataemia in the FDI versus the FCM group was calculated using the Cochran–Mantel–Haenszel method with 95% CIs, adjusting for randomised strata (screening serum phosphate <3.5 or ≥3.5 mg/dL). For the primary endpoint, and for the secondary safety endpoint of hypophosphataemia at any time through Day 70, patients without postbaseline observations were imputed as having hypophosphataemia, as prespecified in the trial’s statistical analysis plan. Post hoc sensitivity analyses either imputed these patients as remaining free of hypophosphataemia or excluded them from analyses of the primary endpoint. To assess whether a diagnosis of Crohn’s disease (CD) versus ulcerative colitis (UC) modified the effect of the different intravenous iron formulations on the incidence of hypophosphataemia, a test of interaction was conducted using logistic regression, and stratified analyses by IBD diagnosis were also performed. Longitudinal changes in anaemia and iron parameters, biomarkers of bone and mineral homeostasis, and the FACIT Fatigue Scale score were analysed using a restricted maximum likelihood-based mixed model for repeated measures (MMRM) approach. For patients with no postbaseline measurements, the change from baseline was set to zero at the first postbaseline visit. Otherwise, no imputation of missing values was performed. The MMRM model included the categorical fixed effects of treatment (FDI and FCM), stratum, day, and treatment-by-day interaction, as well as the continuous mixed covariates of baseline value and baseline value-by-day interaction. As a sensitivity analysis, terms for CD, UC, and their interactions with treatment, were included in the MMRM analysis of change in FACIT Fatigue Scale score. AE data were summarised descriptively. A post hoc analysis was conducted to assess whether decreases in phosphate concentration were associated with changes in FACIT Fatigue Scale scores or Hb. The average change from baseline to each subsequent time point was calculated for serum phosphate for the overall trial population (across both treatment groups). The quartiles of the average phosphate concentration change were used as a group variable (instead of treatment) in a MMRM model analysing changes in FACIT Fatigue Scale score or Hb over time.
The associations between the average change in FACIT Fatigue Scale score and the average change in phosphate were examined using linear regression.
All statistical analyses were performed using SAS release V.9.4 (SAS Institute), and two-tailed p-values <0.05 were considered statistically significant.
No interim analyses took place. There was no data monitoring committee for this trial.
Patient and public involvement
Patients or the public were not involved in the design, or conduct, or reporting of this study. Patients will be consulted about plans for dissemination of the study results through specific advisory boards, including patients and relevant patient organisations.
Results
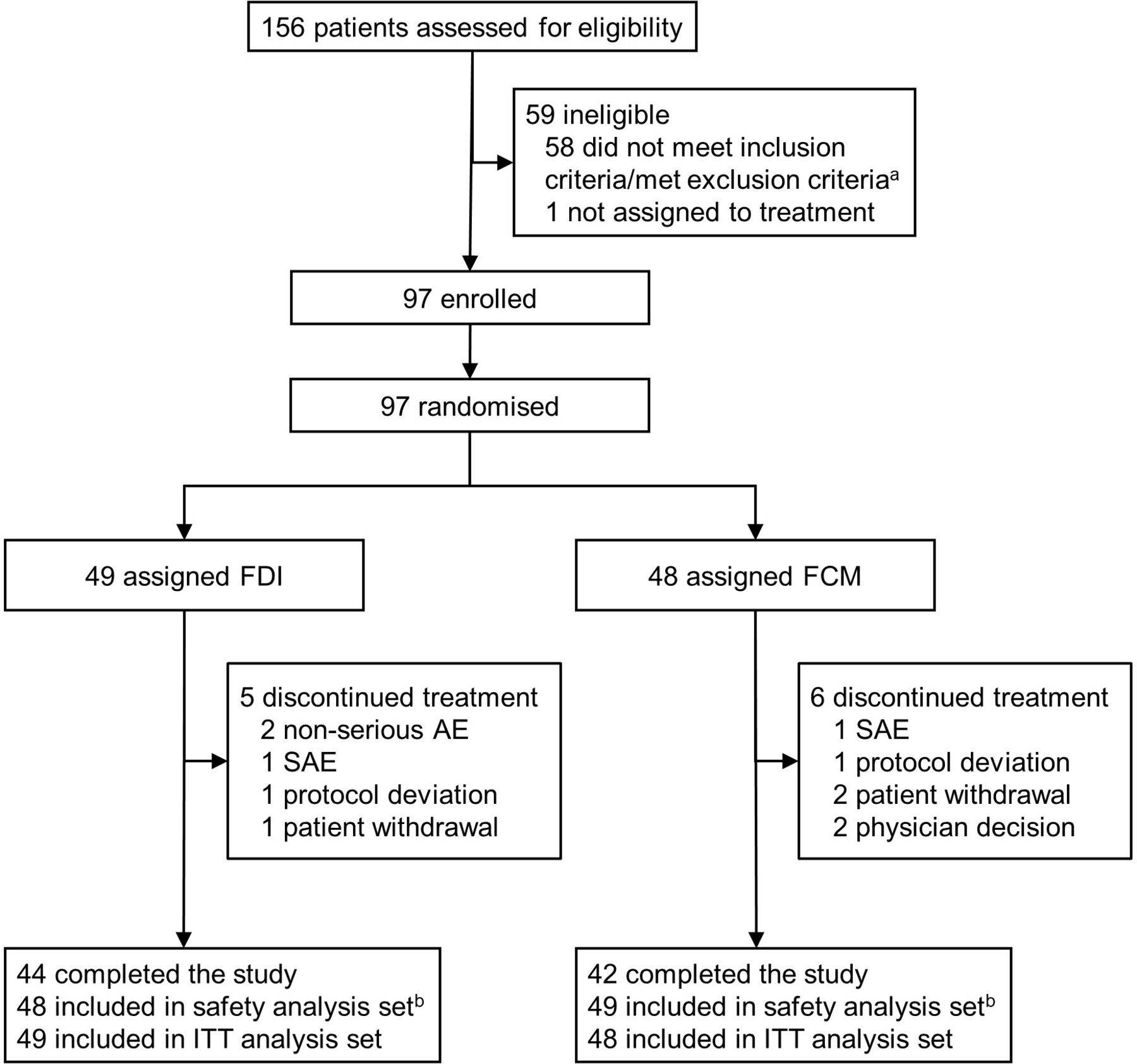
Between 23 May 2018 and 25 May 2020, 156 patients were screened and 97 were included and treated in the trial; 48 patients received FDI and 49 patients received FCM (figure 1). Patients’ baseline characteristics are summarised in table 1. More patients with UC than CD were included, and the proportion of UC was higher in the FDI treatment group. Otherwise, patients’ characteristics were well balanced between treatment groups. On average, clinical disease activity status was mild. At baseline, mean Hb was 105 g/L. Mean 25-hydroxyvitamin D concentrations were low, indicating a high prevalence of vitamin D insufficiency at baseline. At inclusion, other bone and mineral metabolism parameters were within the normal range and were well balanced between study groups.
Baseline demographics, disease characteristics and comorbidities, concomitant medication, and laboratory parameters

Trial profile. aFive patients were not eligible due to low serum phosphate at screening, that is, did not meet the inclusion criteria of serum phosphate >2.5 mg/dL. Two of the five also met additional exclusion criteria. bOne patient was randomised to FDI, but received FCM. AE, adverse event; FCM, ferric carboxymaltose; FDI, ferric derisomaltose; ITT, intention-to-treat; SAE, serious adverse event.
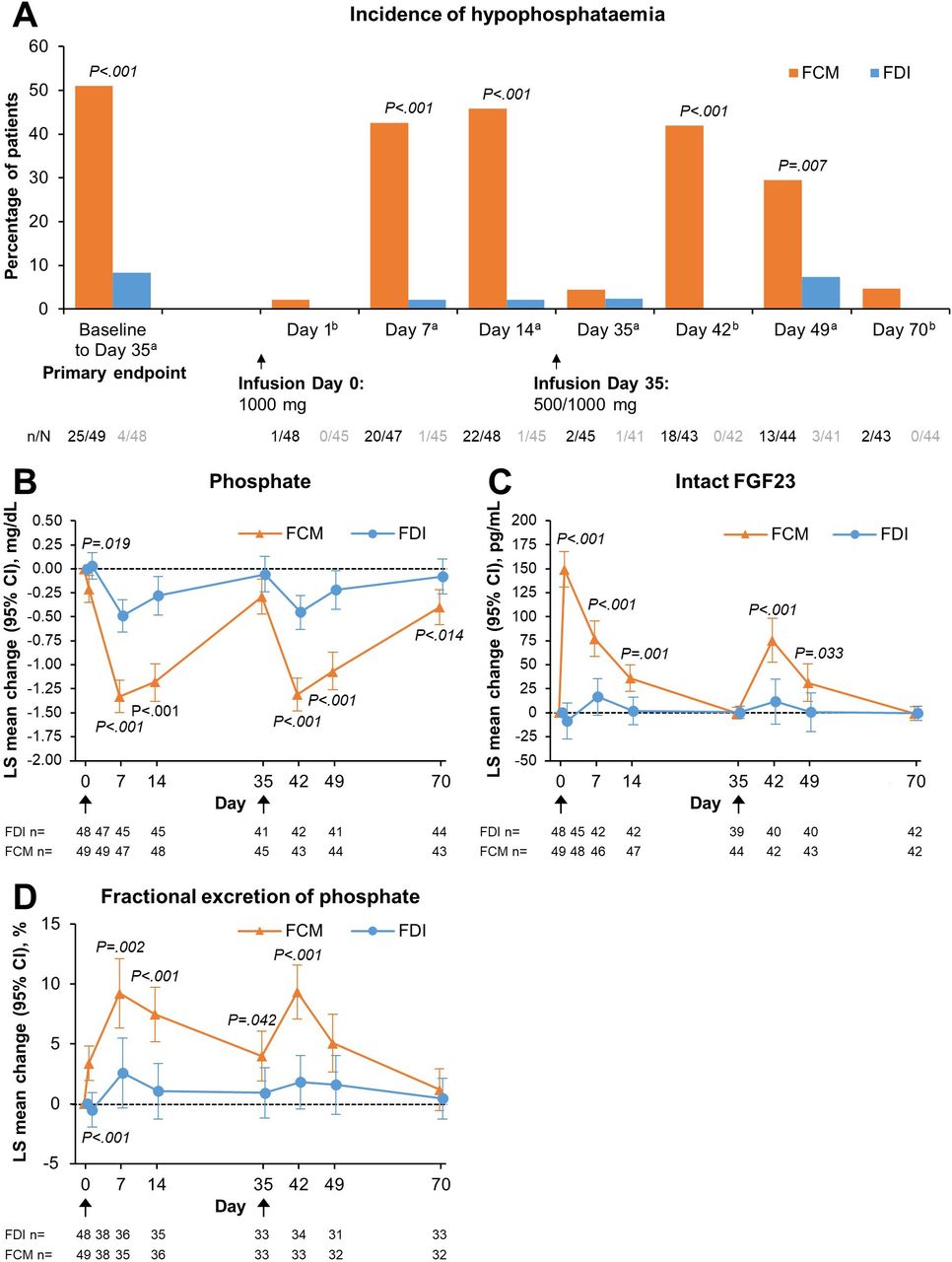
All patients received the first infusion of 1000 mg of either FDI or FCM on Day 0, except for two patients in the FDI group for whom the infusion had to be stopped due to infusion reactions (non-serious; drop in blood pressure in both patients) and they subsequently did not receive the total dose. The primary endpoint—the incidence of hypophosphataemia (defined as serum phosphate <2.0 mg/dL) at any time after the first dose to Day 35—was 8.3% (n=4/48) in the FDI group and 51.0% (n=25/49) in the FCM group (adjusted risk difference: −42.8% (95% CI –57.1 to –24.6); p<0.0001) (figure 2A; online supplemental table S2). In both arms, the highest incidence of hypophosphataemia occurred within 2 weeks of treatment. Two patients in the FDI group and one patient in the FCM group did not have a postbaseline observation and were imputed as having hypophosphataemia in the primary prespecified analysis. The results were qualitatively unchanged in post hoc sensitivity analyses, in which these patients were imputed as remaining free of hypophosphataemia or excluded from the analysis (online supplemental table S3). The higher risk of hypophosphataemia among FCM-treated versus FDI-treated patients was not modified by diagnosis of IBD (p=0.1948), and the absolute risk difference between the FCM and FDI groups was similar within each diagnosis of IBD (for CD, the absolute risk difference was 45.5% higher in the FCM arm; for UC, the absolute risk difference was 43.1% higher in the FCM arm).

(A) Incidence of hypophosphataemia from baseline at each study visit, and changes from baseline in key phosphaturic hormone biochemical parameters—(B) Phosphate; (C) Intact FGF23; (D) Fractional excretion of phosphate—according to intravenous iron treatment. (A) Data are presented for the safety analysis set. For baseline to Day 35 (primary safety endpoint), two patients in the FDI group and one patient in the FCM group did not have a postbaseline observation and were, therefore, set as having hypophosphataemia. Black arrows indicate infusion of intravenous iron (FDI or FCM). aP-values are for risk difference with 95% Newcombe CI adjusted for stratum using Cochran–Mantel–Haenszel method. bP-values for unadjusted risk difference and 95% Wald CI. (B–D) Data are presented for the safety analysis set. Black arrows indicate infusion of intravenous iron (FDI or FCM). Due to lack of space, the x-axis Day one labels and tick marks are not shown. (A–D) P-values are for between-group differences. FCM, ferric carboxymaltose; FDI, ferric derisomaltose; FGF23, fibroblast growth factor 23; LS, least squares.
A second infusion was administered on Day 35 to correct the total iron deficit. For FDI, 18.8% (n=9/48) of patients received 1000 mg, and 66.7% (n=32/48) received 500 mg as a second infusion; 14.6% (n=7/48) did not receive a second dose. For FCM, 22.4% (n=11/49) received 1000 mg, and 69.4% (n=34/49) received 500 mg as a second infusion; 8.2% (n=4/49) did not receive a second dose. Incidence rates of hypophosphataemia at any time from baseline to Day 70 (secondary safety endpoint) were 12.5% (n=6/48) for FDI and 59.2% (n=29/49) for FCM (adjusted risk difference: −46.6% (95% CI −60.9% to –28.1%); p<0.0001) (online supplemental table S2). The incidence of hypophosphataemia was higher in FCM-treated versus FDI-treated patients at all postbaseline visits, reaching a peak incidence of 45.8% at 2 weeks after the first FCM treatment (figure 2A; online supplemental table S2). Serum phosphate changes after treatment are shown in figure 2B. Despite lower overall iron doses being administered on Day 35 vs Day 0, the mean decreases in phosphate concentration from baseline were comparable after the first and second doses, and were significantly greater after FCM versus FDI treatment (figure 2B). The majority of patients recovered from hypophosphataemia by Day 70, but 4.7% (n=2/43) FCM-treated patients remained hypophosphataemic more than 1 month after the second infusion, and overall mean serum phosphate remained significantly lower on Day 70 in FCM-treated versus FDI-treated patients (figure 2B).
Changes from baseline in intact FGF23 (which controls urinary phosphate excretion) were highest on the day after the first infusion of FCM and the levels returned to baseline by Day 35 (figure 2C). Intact FGF23 concentrations rose significantly after both FCM infusions compared with FDI (figure 2C). This was reflected by higher urinary excretion of phosphate in the FCM-treated patients (figure 2D). In contrast to the serum phosphate concentration, fractional excretion of phosphate returned towards baseline in both treatment groups by Day 70.
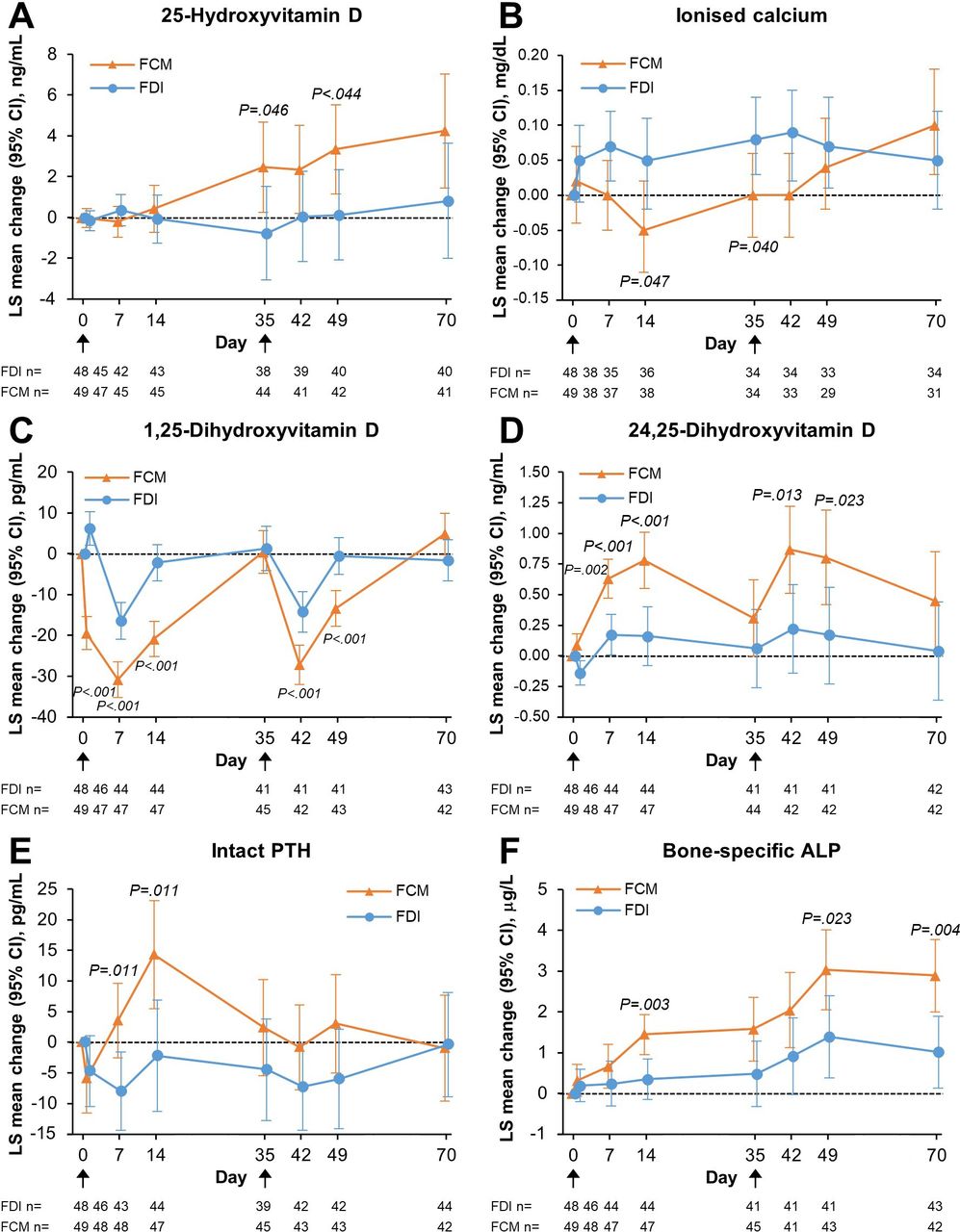
25-hydroxyvitamin D concentrations increased from baseline in FCM-treated patients (figure 3A). Ionised calcium also increased by the end of the trial (figure 3B). In accordance with these changes, the proportion of patients receiving vitamin D supplements increased during the study period from 12.5% (n=6/48) at baseline to 29.5% (n=13/44) at Day 70 in FDI-treated and from 20.4% (n=10/49) at baseline to 37.2% (n=16/43) at Day 70 in FCM-treated patients. Calcium supplement use changed from 35.4% (n=17/48) at baseline to 34.1% (n=15/44) at Day 70 in FDI-treated patients and from 18.4% (n=9/49) at baseline to 27.9% (n=12/43) at Day 70 in FCM-treated patients.

Changes from baseline in mineral and bone parameters, according to intravenous iron treatment—(A) 25-Hydroxyvitamin D; (B) Ionised calcium; (C) 1,25-Dihydroxyvitamin D; (D) 24,25-Dihydroxyvitamin D; (E) Intact PTH; (F) Bone-specific ALP. Data are presented for the safety analysis set. P-values are for between-group differences. Black arrows indicate infusion of intravenous iron (FDI or FCM). Due to lack of space, the x-axis Day one labels and tick marks are not shown. ALP, alkaline phosphatase; FCM, ferric carboxymaltose; FDI, ferric derisomaltose; LS, least squares; PTH, parathyroid hormone.
Despite increased use of vitamin D and calcium supplements in the FCM group, 1,25-dihydroxyvitamin D concentrations decreased from baseline after each iron administration, significantly more so after FCM than FDI, and comparably after the first and second dosing (figure 3C). In contrast, concentrations of 24,25-dihydroxyvitamin D (a measure of vitamin D degradation that is stimulated by FGF23) increased significantly after each infusion of FCM compared with FDI (figure 3D). Mean changes from baseline in PTH were significantly higher in the FCM compared with the FDI group on Day 7 and Day 14 (figure 3E). Bone-specific ALP increased after both treatments, but to significantly higher concentrations after FCM versus FDI; the changes were more pronounced after the second infusion of each study drug (figure 3F). Mean decreases in the anabolic bone marker N-terminal propeptide of type 1 collagen (serum P1NP) were greater at all time points from Day 1 to Day 49 in FCM-treated patients compared with FDI-treated patients (online supplemental figure S1), reaching statistical significance between groups on Day 14. By comparison, changes in the bone resorption marker carboxy-terminal collagen crosslinks (CTx) were less pronounced than P1NP in both groups and did not significantly differ between groups throughout the study period (online supplemental figure S1).
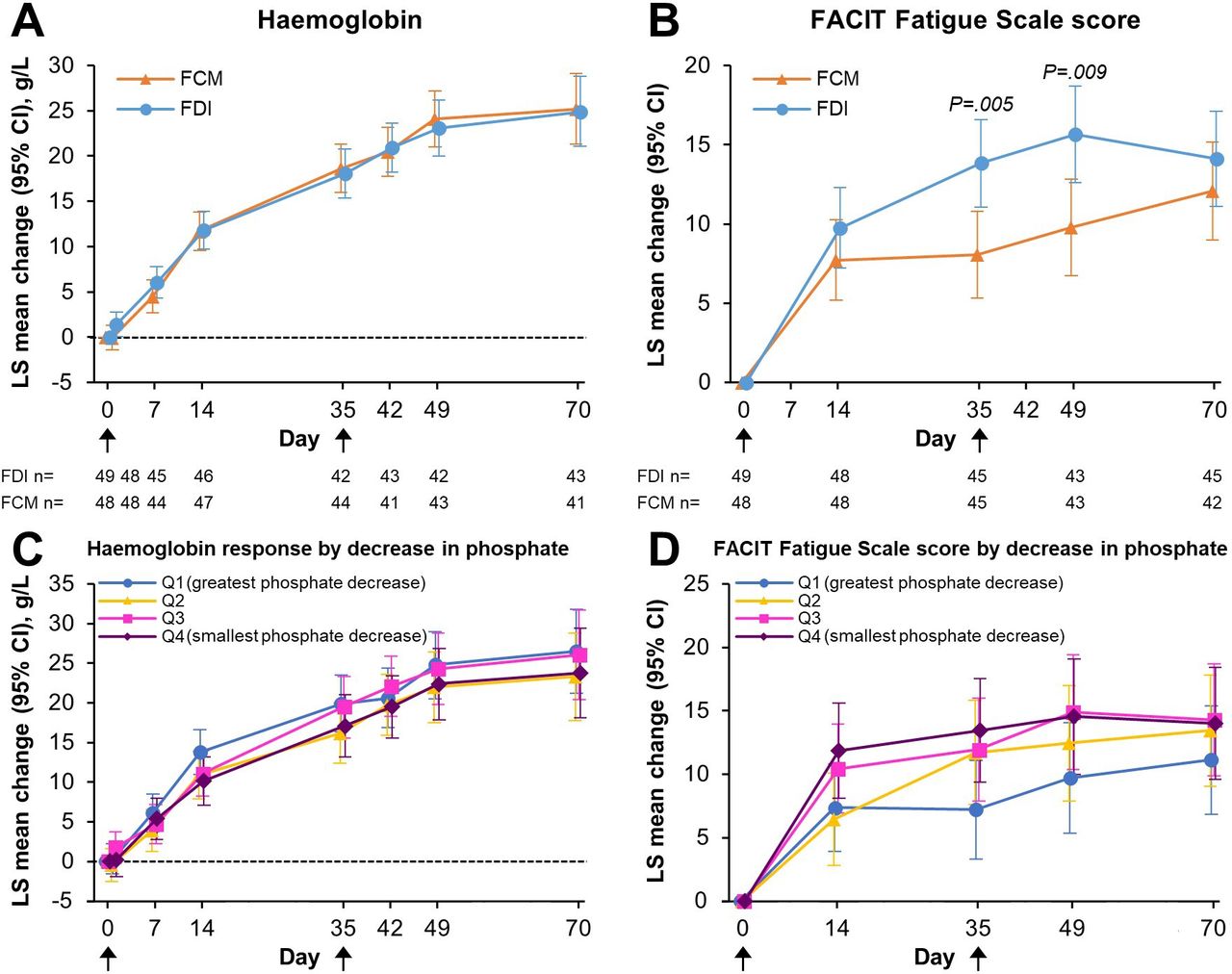
By the end of the trial (Day 70), ferritin and TSAT had increased comparably in the FCM and the FDI treatment groups (online supplemental figure S2), and both iron formulations resulted in robust Hb increases by Day 70 (FDI: 24.9 g/L (95% CI 21.1 to 28.8); FCM: 25.2 g/L (95% CI 21.3 to 29.1); figure 4A). Concomitantly, both study drugs promoted improvement in fatigue symptoms, marked by increased FACIT Fatigue Scale scores; the specific IBD diagnosis did not modify the overall effect of the different intravenous iron formulations on change in FACIT Fatigue Scale score (online supplemental table S4). Interestingly, the improvement in FACIT Fatigue Scale scores over time was significantly greater for FDI versus FCM at Days 35 and 49 (figure 4B). The magnitude of improvement in FACIT Fatigue Scale scores was inversely associated with the magnitude of decrease in phosphate concentration (p=0.0063); more severe hypophosphataemia was associated with slower improvement in fatigue (figure 4D). In contrast, Hb responses were similar across quartiles of hypophosphataemia severity (figure 4C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes from baseline in haemoglobin and FACIT Fatigue Scale score, according to intravenous iron treatment (A, B), and by serum phosphate quartiles (C, D). Data are presented for the ITT analysis set (A, B), and for the safety analysis set with FDI and FCM pooled (C, D). P-values are for between-group differences. Black arrows indicate infusion of intravenous iron (FDI or FCM). Due to lack of space, the x-axis Day 1 labels and tick marks are not shown. FACIT, Functional Assessment of Chronic Illness Therapy; FCM, ferric carboxymaltose; FDI, ferric derisomaltose; ITT, intention-to-treat; LS, least squares; Q, quartile.
Overall, AEs and serious AEs (SAEs) occurred with comparable frequency in the FDI and FCM groups (table 2). Hypophosphataemia and vitamin D deficiency were reported as an AE more often in the FCM group than in the FDI group. Headache and nausea were reported as an AE more often in the FDI group than in the FCM group. Equivalent proportions of treatment-emergent AEs were considered related to trial drug and the majority were mild or moderate in severity (table 2). There were no deaths during the trial.
Adverse events and serious adverse events, according to intravenous iron treatment
Discussion
Randomised clinical trials included in systematic reviews and meta-analyses have shown that FCM is the intravenous iron formulation associated with the highest rates of hypophosphataemia of approximately 50%.14 18 23 In contrast, the hypophosphataemia rate after FDI treatment has been reported as <5%.18 Significant heterogeneity in reported incidence rates for FCM was, in part, attributed to differences in kidney function, varying aetiologies and severity of underlying iron deficiency, and to different doses of intravenous iron.14 A prospective observational study of patients with IBD showed that 71.2% of patients developed hypophosphataemia 2 weeks after FCM infusion as compared with 11.1% after FDI.8 The PHOSPHARE-IDA trials (conducted in the USA), which were the first randomised comparisons of the two drugs, reported hypophosphataemia rates of 74.4% and 8.0% in FCM-treated and FDI-treated patients, respectively, but the dose and administration schedules of the drugs differed in accordance with the recommended dosing regimens in their respective US Prescribing Information.15
The current trial—PHOSPHARE-IBD—was the first double-blind, randomised, controlled trial to use an identical dosing regimen to directly compare the effect of FCM and FDI on hypophosphataemia, specifically in an IBD population. The trial included anaemic patients with mild to moderately severe IBD (both UC and CD), which is a representative cohort of patients with IBD presenting for iron supplementation in clinical gastroenterology practice.7 24 The rationale for the split dosing was mandated by the approved dosing of FCM in Europe, where the maximal single dose is 1000 mg.21 Considering the trial design, it can be concluded that the observed differences in hypophosphataemia rates are caused solely by differences in iron formulations, and are not attributable to differences in total iron dose, administration schedule, CD versus UC, or other patient-related factors such as kidney function, severity of inflammation, degree of iron deficiency, or vitamin D deficiency. As in prior clinical trials of intravenous iron,25 site investigators underreported hypophosphataemia as an AE, which could be attributed to poor awareness of hypophosphataemia as an AE of intravenous iron.
Although previous trials have reported a significantly increased risk of hypophosphataemia in patients treated with FCM compared with FDI in other patient populations,14 15 the current trial is noteworthy because it advances the field in several important ways. First, it focuses exclusively on patients with IBD, who constitute one of the largest populations that receive intravenous iron and are the population that has borne some of the most devastating consequences of hypophosphataemia reported in the literature.26 Second, this is one of only a few randomised clinical trials (if not the first) to formally test the effects of two doses of intravenous iron in patients who were determined to have an iron requirement >1000 mg. Third, this randomised clinical trial was positioned to collect novel, double-blinded data on patient-reported outcomes, in contrast to prior, unblinded trials, where patient knowledge of the iron formulation they received could have biased assessments of patient-reported outcomes. Thus, the finding of significant differences in patient-reported fatigue (based on FACIT scores), and its association with severity of hypophosphataemia, is an important new finding that advances the field and will help inform clinicians and patients about the short-term effects of hypophosphataemia on patient well-being.
Previous studies with shorter observation periods have shown that a considerable proportion of patients remained hypophosphataemic on Day 35–50 after FCM treatment.14 A long-term observational study demonstrated that some patients required several months to recover from hypophosphataemia after a single infusion of FCM.8 In the present trial, the majority of patients recovered from hypophosphataemia by Day 70; however, a small proportion of FCM-treated patients remained hypophosphataemic more than 1 month after the second infusion.
The present trial also shows that, despite recovery from hypophosphataemia, the time course of the cascade of biochemical changes associated with high FGF23 expression (termed the ‘6H syndrome’)27 is protracted in FCM-treated patients. In particular, bone-specific ALP remained elevated on Day 70 in the FCM group relative to the FDI group, implying that there are effects of FCM on bone turnover during the entire trial period and beyond. This finding could link hypophosphataemia with osteomalacia, which is an increasingly reported complication of FCM that has been recently included as a specific warning in the FCM Patient Information in Europe, as requested by the Pharmacovigilance Risk Assessment Committee from the European Medicines Agency and the UK’s Medicines and Healthcare products Regulatory Agency.13 18 28 29 The latter specifies IBD as a particular risk factor in this context.29
Despite the dramatically different effects on plasma phosphate homeostasis,14 15 23 FDI and FCM treatment have both been consistently shown to improve quality of life in patients with iron deficiency.30–32 The present trial confirms that both formulations effectively corrected IDA, as evidenced by almost identical Hb responses leading to a rise of 20–30 g/L in both groups as well as correction of serum iron parameters. This observation argues against the possibility that the high hypophosphataemia rate in FCM-treated patients is caused by high erythropoietic efficacy. It is important to note this because regeneration of red blood cells during recovery from haemolytic or pernicious anaemia, or after peripheral blood stem cell transplantation, can cause redistribution of phosphate from plasma to nascent blood cell membranes, which could result in hypophosphataemia.33–35
Patient-reported outcomes are of particular importance in clinical trials. In the current double-blind trial, we used the FACIT Fatigue Scale to assess fatigue, which improved in both treatment groups and was not attributable to the specific IBD diagnosis; the improvement was faster and greater in the FDI-treated group, although this difference was not significant by Day 70. While this difference is unexplained by the haematological response to the treatments administered, our data suggest that reduction in serum phosphate partially undermines the beneficial effect of intravenous iron on fatigue. It is challenging to disentangle fatigue that is due to IDA from fatigue that is due to hypophosphataemia, especially after treatment with intravenous iron, which improves fatigue overall. However, we found that reduction in serum phosphate correlated with reduced improvement in fatigue, suggesting that use of an iron formulation that avoids significant hypophosphataemia could lead to more pronounced improvements in fatigue symptoms.
In comparison with another study assessing hypophosphataemia in patients with IBD, FCM-treated patients in our trial experienced less frequent hypophosphataemia overall, and they recovered more quickly from hypophosphataemia.8 A potential explanation for these differences is the increased use during the current trial of vitamin D and calcium as concomitant medications. Baseline 25-hydroxyvitamin D (the main circulating storage form of vitamin D) was low in both groups, with an overall mean of 23 ng/mL, which could be caused by reduced intestinal absorption of vitamin D seen in IBD.36 During the course of the trial, investigators continuously received unblinded data with regard to patients’ calcium and vitamin D status, and they increased vitamin D and calcium supplementation, which were marked by the longitudinal increase of 25-hydroxyvitamin D concentrations in FCM-treated patients and the lack of reduced serum ionised calcium after the second dose of FCM. It is conceivable that the excess vitamin D and calcium supplementation in the FCM group during the course of this trial negated the effects of FCM on calcium and PTH levels after the second, compared with the first, dosing and could have mitigated the negative effects of FCM on phosphate homeostasis.
This trial was limited by the relatively short follow-up period, which did not allow sufficient time to assess for clinical consequences of the observed biochemical derangements. This is particularly relevant in relation to the persistent elevation of serum bone-specific ALP seen in the FCM group at the end of the trial. Future large cohort studies or registries would be helpful in assessing long-term clinical consequences of FCM use on the musculoskeletal system.
In conclusion, the PHOSPHARE-IBD trial confirmed that hypophosphataemia is not a class or dose effect of intravenous iron, but a particularly common adverse effect of FCM that is driven by marked increases in FGF23. Despite equivalent dosing schedules, the effects of FDI on FGF23 and, thus, serum phosphate are much smaller. This study shows that hypophosphataemia is a common complication of FCM use in patients with IBD.
Data availability statement
No data are available. No individual participant data will be shared, and no trial related information (data, analytical methods, study materials) will be available.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Austria (Reference no: EK Nr: 1243/2017) Centre No. 4301 - Ethikkommission der Medizinischen Universität Innsbruck Centre No. 4302 - Ethikkommission der Medizinischen Universität Wien Centre No. 4303 - Ethikkommission f.d. Bundesland Salzburg Centre No. 4304 - Ethikkommission des Landes Niederösterreich Centre No. 4305 - Ethikkommission des Landes Oberösterreich, Kepler Universitätsklinikum, Denmark (Reference no: Sagsnr. 1-10-72-205-17) De Videnskabsetiske Komitéer for Region Midtjylland Germany (Reference no: FF114/2018) Centre No. 4901 - Ethik-Kommission bei der Landesärztekammer Hessen Centre No. 4902 - Ethik-Kommission der Ärztekammer Hamburg Centre No. 4903 - Ethik-Kommission des Fachbereichs Medizin der Johann Wolfgang Goethe-Universität Centre No. 4904 - Ethikkommission der Universität Ulm Centre No. 4905 - Ethikkommission der TU Dresden Centre No. 4906 - Sächsische Landesärztekammer - Ethikkommission Sweden Regionala etikprövningsnämnden i Lund UK (IRAS Project ID: 236810) West Midlands - Edgbaston Research Ethics Committee. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank all the investigators, trial personnel, and participating patients for their contributions to the trial. Data management and statistical analyses were provided by Biostata ApS, Birkeroed, Denmark, and Jens-Kristian Slott Jensen, Slott Stat, Denmark, and data analysis input was provided by Jakob Bue Bjørner, QualityMetric Incorporated LLC, Johnston, Rhode Island, USA. Editorial support was provided by ‘Cambridge—a Prime Global Agency’, Cambridge, UK, funded by Pharmacosmos A/S.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SL, HZ, MW, TI, and LLT conceived the trial. SL, HZ, MW, TI, LLT, and WR designed the trial. SL, WR, HZ, TI, IB, and CP were trial site investigators and collected data. HZ, MW, TI, and LLT performed statistical analysis. All authors contributed to the preparation of the manuscript, including critical review and approval. All authors had full access to all the data in the trial and had final responsibility for the decision to submit for publication. HZ was chief investigator and acts as the guarantor for this study and publication.
Funding Pharmacosmos A/S funded the trial and was the Good Clinical Practice (GCP) sponsor. The sponsor participated in the design of the trial, commissioned contract research organisations and was responsible for and coordinated protocol writing, preparation of the statistical analysis plan, data collection, writing of the clinical study report, project management, and data analysis. The sponsor and the academic authors interpreted the data; the sponsor reviewed and commented on the manuscript that was developed by the academic authors. Journal selection was made by the academic author group and agreed on by the sponsor. The sponsor did not have the right to veto submission, in general, or to any particular journal.
Competing interests HZ has received honoraria for lecturing, for consulting and grant support for research from Vifor, the manufacturer of ferric carboxymaltose, and from Pharmacosmos, the manufacturer of ferric derisomaltose, as well as from Medice and Pierre Fabre, the distributors of ferric derisomaltose in Austria and Switzerland. HZ has also received payments for lecturing and consulting from Abbvie, Bayer, Falk, Gilead, Merz, and Sanofi during the 36 months prior to submission of this manuscript. MW reports personal fees from Pharmacosmos, during the conduct of the study; personal fees from Amgen, personal fees from Bayer, personal fees and other from Akebia, personal fees from Jnana, personal fees from Walden, personal fees from Reata, personal fees from Enyo, personal fees from Unicycive, outside the submitted work. IB has no competing interests to declare. CP has served as a speaker for Abbvie, MSD, Falk, Janssen, Takeda, Merck, Ferring, and Astro Pharma, and at an advisory board meeting/as a consultant for Abbvie, MSD, Takeda, and Janssen. SL has no competing interests to declare. LLT is a full-time employee of Pharmacosmos A/S, which funded and was the GCP sponsor of the trial. LLT is co-inventor of patents related to parenteral iron products. WR has served as a speaker for Abbvie, Boehringer Ingelheim, Celltrion, Falk Pharma GmbH, Ferring, Galapagos, Janssen, Medice, Mitsubishi Tanabe Pharma Corporation, Pfizer, Pharmacosmos, PLS Education, Roche, Takeda, Therakos, at an advisory board meeting/as a consultant for Abbvie, Algernon, Amgen, AM Pharma, AMT, AOP Orphan, Arena Pharmaceuticals, Astellas, AstraZeneca, Roland Berger GmbH, Bioclinica, Boehringer-Ingelheim, Bristol-Myers Squibb, Calyx, Celgene, Celltrion, Covance, Eli Lilly, Ernest & Young, Falk Pharma GmbH, Ferring, Fresenius, Galapagos, Gatehouse Bio Inc., Genentech, Gilead, Grünenthal, ICON, Index Pharma, Inova, Intrinsic Imaging, Janssen, Johnson & Johnson, Kyowa Hakko Kirin Pharma, Landos Biopharma, Lipid Therapeutics, LivaNova, Mallinckrodt, Medahead, MedImmune, Millenium, Mitsubishi Tanabe Pharma Corporation, MSD, Nash Pharmaceuticals, Nippon Kayaku, Novartis, Ocera, OMass, Otsuka, Parexel, Periconsulting, Pharmacosmos, Pfizer, Prometheus, Protagonist, Provention, Quell Therapeutics, Sandoz, Seres Therapeutics, Setpointmedical, Sublimity, Takeda, Teva Pharma, Therakos, Theravance, Tigenix, Zealand, and has received research funding from Abbvie, Amgen, Janssen, MSD, Sandoz, and Takeda. TI has attended paid advisory boards for Pharmacosmos and has received fees for presenting at academic meetings.
Patient and public involvement Patients or the public were not involved in the design, or conduct, or reporting of this study. Patients will be consulted about plans for dissemination of the study results through specific advisory boards, including patients and relevant patient organisations.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.