Article Text

Abstract
Objectives Diet plays a crucial role in the development of obesity and insulin resistance via multiple mechanisms. Saturated fatty acids can directly trigger tissue specific proinflammatory pathways via Toll-like receptor-4 (TLR4)-dependent mechanisms. Moreover, diet can change the gut microbiome and increase gut permeability. However, very few studies have addressed the obesity-independent role of diet. Dissecting the effects of diet from those of obesity per se will enhance our understanding of the underlying pathogenesis, and, at the translational level, advance our treatment approaches for obesity and its co-morbidities.
Methods Melanin-concentrating hormone (MCH) is an important regulator of appetite and energy balance. MCH-deficient mice are resistant to diet-induced obesity, primarily due to increased locomotor activity. We took advantage of the unique phenotype of these mice to examine the metabolic and inflammatory consequences of a 15-week consumption of a diet high in saturated fat.
Results MCH-deficient mice chronically exposed to a high-fat diet gain less weight compared to their wild-type littermates, despite similar food intake, and are protected from hepatosteatosis. They also lack obesity-associated upregulation of serum leptin and insulin levels and have improved total body insulin sensitivity. Nevertheless, we found indistinguishable liver-specific innate immune responses in both genotypes associated with high-fat feeding, which involved activation of TLR4 and its downstream effectors, MyD88, p38 MAP kinase and STAT-3.
Conclusions Our findings indicate that high-fat feeding is deleterious to the liver, independently of the obesity status. They also suggest that MCH is not necessary for the TLR4-dependent immune response triggered by the high-fat diet.
- Melanin-concentrating hormone
- TLR4
- high-fat diet
- obesity
- liver inflammation
- SOCS3
- fatty liver
- glucose metabolism
- inflammatory mediators
- neuropeptides
- obesity
Statistics from Altmetric.com
- Melanin-concentrating hormone
- TLR4
- high-fat diet
- obesity
- liver inflammation
- SOCS3
- fatty liver
- glucose metabolism
- inflammatory mediators
- neuropeptides
- obesity
Significance of this study
What is already known about this subject?
High-fat diet leads to obesity, insulin resistance and chronic sub-acute inflammation.
Fatty acids directly activate TLR4 thus providing a link between innate immunity and metabolism.
Activation of inflammatory pathways results in insulin resistance.
What are the new findings?
MCH-deficient mice resist liver fat accumulation in response to chronic high-fat feeding.
Despite their leanness, MCH-deficient mice are prone to high-fat diet induced inflammatory responses, which are liver-confined and involve TLR4- and SOCS3-dependent signalling pathways.
In contrast to its proinflammatory role in models of intestinal inflammation, MCH is not necessary for TLR4-dependent innate immune responses.
How might it impact on clinical practice in the foreseeable future?
Diets high in saturated fat might have deleterious effects on the liver, even when consumed by lean individuals.
Exercise alone is not sufficient to prevent diet-induced insulin resistance, unless it is coupled with appropriate dietary modifications.
Modification of dietary habits might be beneficial in patients with liver injury of various aetiologies.
Introduction
Obesity results from an imbalance between energy intake and expenditure and has taken on a pandemic proportion worldwide, with one-third of Americans being overweight or obese.1 The current view is that obesity is a state of chronic low-grade inflammation which originates in the white adipose tissue and ultimately involves the liver and the cardiovascular system, leading to diabetes, atherosclerosis and non-alcoholic fatty liver disease (NAFLD). Nevertheless, the molecular pathways that link obesity and inflammation have just started to emerge.
Toll-like receptors (TLRs) represent a family of microbial pattern recognition molecules that play a crucial role in host defence.2 Several lines of evidence implicate TLRs, and TLR4 in particular, in metabolic processes as well. For example, TLR4 expression is increased in the adipose tissue of both genetically and diet-induced obese mice as well as in the muscle of obese and diabetic patients.3 4 Furthermore, mice with compromised TLR4 signalling develop attenuated inflammatory responses in adipose tissue, liver, muscle and the aorta, along with improved insulin sensitivity in response to high-fat feeding or lipid infusion.3 5–8 Most importantly, it has been clearly demonstrated that certain classes of fatty acids can act as natural ligands for TLR4, in addition to lipopolysaccharide (LPS), hyalouronic acid and high mobility group box 1(HMGB1) and trigger downstream proinflammatory cascades which appear to be NFkB- and JNK-dependent.2 9–11 At the cellular level, activation of TLR4 directly interferes with the insulin signalling pathways resulting in insulin resistance.3 5
Despite solid epidemiological evidence linking consumption of a Western-type diet to the development of obesity and its complications, the underlying mechanisms of such interaction are more complex than originally appreciated. For instance, diet is one of the critical determinants of the composition of the gut flora and an obesity associated microbiome has increased metabolic capacity to harvest energy from the food12 13 Moreover, high-fat feeding directly compromises the epithelial barrier and increases intestinal permeability to endotoxins, most likely due to a downregulation of the tight junction proteins.14 15 Hence, diet-derived fatty acids and diet-induced metabolic endotoxinaemia, both potent TLR4 activators, have been implicated in the inflammatory responses linked to obesity. However, very few studies have addressed the obesity-independent metabolic consequences of the diet.16–18 Disentangling the disease burden of obesity from that of dietary habits will enhance our understanding of the underlying pathogenic mechanisms and, at the translational level, refine our approaches to combat the current epidemic.
We have previously described the phenotype of mice deficient in melanin-concentrating hormone (MCH), an important regulator of energy balance.19 20 MCH is a cyclic 19 amino acid neuropeptide derived from proteolytic cleavage of a preprohormone (ppMCH) and has been identified as a central player in the development of obesity and insulin resistance via multiple mechanisms. The study of the MCH-knockout mice also revealed that MCH is an important regulator of energy expenditure. Thus, MCH genetic ablation was associated with increased energy expenditure, lower weight gain and adiposity in mice with a pure genetic C57BL/6 background particularly on a high fat diet.20 Enhanced energy expenditure in the MCH−/− mice on high-fat diet was attributed primarily to twofold higher locomotor activity.20 In addition, under certain conditions, it played a role in appetite regulation. Intracerebroventricular (ICV) administration of MCH acutely increased chow intake in mice fed a chow diet.21 Similarly, transgenic mice over-expressing MCH showed hyperphagia, obesity and insulin resistance on a high-fat diet.22 This phenotype was recapitulated in wild-type mice receiving chronic ICV infusion of MCH.23 Interestingly, the effects of MCH on appetite appeared to be modulated by the genetic background and MCH-deficient mice in a pure C57BL6 or 129CJ genetic background, consumed the same amount of food as wild-type mice.20
In the present study, we utilised the obesity-resistant phenotype of the MCH knockout mice to examine the potential effects of chronic consumption of a high-fat diet on metabolic and inflammatory parameters that have been previously attributed to obesity. We focused our attention on the liver considering its anatomical proximity to the gut and its critical role in glucose and lipid homeostasis. Furthermore, NAFLD, which encompasses liver pathologies ranging from benign fat accumulation (steatosis) to inflammation (non-alcoholic steatohepatitis (NASH)) and fibrosis, has become a leading form of chronic liver disease worldwide and is one of the most common indications for liver transplantation in the United States.24
Our experiments uncovered significant and TLR4-linked inflammatory responses in the liver of the MCH-knockout mice fed a high fat diet, despite their leanness and lack of hepatosteatosis. These findings show that dietary fats can evoke inflammatory responses in the liver independently of obesity and the accumulation of hepatic lipids.
Materials and methods
Animals
The generation of mice with global deletion of prepro-MCH has been described previously.19 MCH−/− mice were back-crossed onto the C57BL/6J genetic background for at least 11 generations and genotyping was performed by PCR.20 Breeding pairs were a gift from Dr E. Maratos-Flier, Boston, Massachusetts, USA. In the current study, MCH−/− mice and their wild-type littermates were generated by heterozygous breeding and maintained single housed after weaning. 6-week-old male wild-type (WT, n=7) and MCH−/− (KO, n=7) mice were fed a high-fat diet (HFD, 45% calories from animal fat (lard), Research Diets D12451) for 15 weeks. A third group of wild-type mice were fed rodent standard chow diet (16.7% calories from animal fat, LabDiet #5008) and served as controls (CD, n=6). Body weight and food intake were monitored on a weekly basis. Body composition was assessed at week 8 of the study in anaesthetised mice by DEXA scan (Lunar PIXImus2; GE Medical Systems, Fitchburg, WI, USA). All animal work was performed in accordance with the rules and guidelines of the Animal Experiment Committee at Beth Israel Deaconess Medical Center.
Biochemical and hormonal analysis
Liver triglyceride content was measured, following a chloroform–methanol (2:1, v/v) extraction using a commercial kit (Cayman Chemical Inc, Ann Arbor, MI, USA). Mouse tail blood was collected at the end of this experiment and serum insulin and leptin levels were measured at the Joslin Diabetes Center Special Assays Core Facility, Boston, Massachusetts, USA.
Glucose tolerance test and insulin tolerance test
At week 12 of the study, overnight fasted mice were injected i.p. with dextrose (1 g/kg of body weight) and tail vein blood glucose levels were monitored using a glucometer (Elite; Bayer, Pittsburgh, PA, USA). A week later, mice were injected i.p. with 1 U/kg insulin (Humulin; Eli Lilly, Indianapolis, IN, USA) and blood glucose levels were measured likewise.
RNA extraction and real-time PCR
Total RNA was prepared from liver and epididymal fat using the RNeasy mini-kit (Qiagen, Valencia, CA, USA). One μg of RNA was reverse transcribed to cDNA using the Advantage RT-PCR kit (Clontech Inc., Mountain view, CA, USA). For the real-time PCR reactions SYBR Green Supermix (Applied Biosystems, Carlsbad, CA, USA) was used and each sample was run in duplicate in an ABI PRISM 7000 Sequence Detection System. Gene-specific primers were designed via Primer Express 2.0 software (Applied Biosystems) and optimised prior to use in the experiment (sequences available upon request), or purchased as pre-developed assays from Applied Biosystems. TATA-binding protein was used as a housekeeping gene to control for input. Results were expressed in arbitrary units (AU) relative to control which was set to 100.
Immunoblotting and immunohistochemical analysis
The following primary antibodies were used: phospho-p38 (1:1000), phospho-JNK (1:1000) (Cell Signaling, Danvers, MA, USA); phospho-STAT -3 (1:200), TLR-4 (1:100) and MyD-88 (1:100) (Santa Cruz Biotechnology, Santa Cruz, California, USA). The indicated blots were stripped by using Restore Western Blot Stripping Buffer (Pierce, Rockford, IL, USA) and reprobed with p38 (1:1000), JNK (1:1000), STAT-3 (1:100) or β-actin (1:5000). The signal intensity was analysed by computerised densitometry (GS-710; Bio-Rad, Hercules, CA, USA). Macrophage infiltration was assessed in paraffin-embedded sections of liver and adipose tissue stained with an antibody against F4/80 (Specialised Histopathology Services, Dana-Farber/Harvard Cancer Center in Boston, Massachusetts, USA).
Statistical analysis
Results are reported as mean±SEM. All statistical comparisons (two-tailed Student t test; Statview 2.0 software) were between MCH−/− and WT mice, both fed a high fat diet. A p value of <0.05 was considered statistically significant.
Results
Absence of hepatic lipid accumulation in MCH−/− mice fed a high-fat diet
At baseline, all groups had similar body weights: 21.8±0.17 g in WT mice fed CD (WT-CD), 21.6±0.45 g in WT mice fed HFD (WT-HFD) and 21.5±0.46 g in MCH−/− mice fed HFD (KO-HFD). At the end of the study period, and consistent with our previous report,20 MCH−/− mice fed HFD weighed significantly less than the WT mice fed the same diet (30.1±1.1 g vs 37.6±2.2 g, respectively; p<0.005) and had indistinguishable body weight to WT mice fed normal diet (31.8±0.5 g) (figure 1A). Accordingly, the percentage of body weight gain throughout the course of this experiment was also found to be significantly higher in WT mice fed HFD than that of MCH−/− mice (65.0±6.3% vs 42.1±3.1%, respectively; p<0.01) (figure 1B). Despite the differences in body weight gain, daily food intake was not significantly different between MCH−/− and WT mice on high-fat diet (2.69±0.65 g vs 2.91±0.87 g, respectively; p=0.87) (figure 1C). Morphologically, WT mice fed HFD exhibited marked heterogeneity in adipocyte size, with significantly increased presence of large adipocytes and immune cell infiltrates. This phenotype was absent in WT fed control diet and MCH−/− fed HFD (figure 1D). The lower body weight in MCH−/− mice was associated with a significant reduction in their total fat mass as compared with the WT mice fed the same diet, and assessed by DEXA scan. At week 8 in the study on high-fat diet, fat mass represented 17.4±1.3% of total body weight in MCH−/− mice versus 30.6±3.3% in WT mice, respectively; p<0.01) (figure 1E). At the end of the experiment, serum leptin levels at the fed state in the MCH-/-mice were significantly lower compared to those in WT mice fed HFD (18.4±3.5 vs 65.1±13.1 ng/ml, respectively; p<0.01) (figure 1F).

Body weight, food intake and adiposity in mice fed a high fat diet (WT, wild type; KO, MCH−/−; HFD, high fat diet; CD, control diet). (A) Body weight in MCH−/− mice (KO-HFD) was significantly lower than that in WT mice fed the same diet (WT-HFD). (B) Body weight gain during the study expressed as % of initial body weight. (C) Average daily food intake in the three groups of mice. (D) Representative sections of epididymal fat tissue from the three different groups of mice. (E) MCH−/− mice fed HFD exhibited significantly lower % fat mass compared to WT mice fed a high fat diet. (F) Under high fat feeding, MCH−/− mice had significantly lower serum leptin levels compared to WT mice fed high fat diet. **p<0.01; ns, not significant.
Consistent with their increased adiposity, WT mice fed HFD showed abundant fat droplet accumulation in the liver (figure 2A). In contrast, in MCH−/− mice on the same diet the liver had a normal appearance at the microscopic level. Similarly, hepatic triglyceride (TG) levels were more than 2.5-fold higher in WT mice fed HFD compared to that in MCH−/− mice (35.4±5.1 vs 13.5±1.4 mg/g tissue, respectively; p<0.001) (figure 2B).
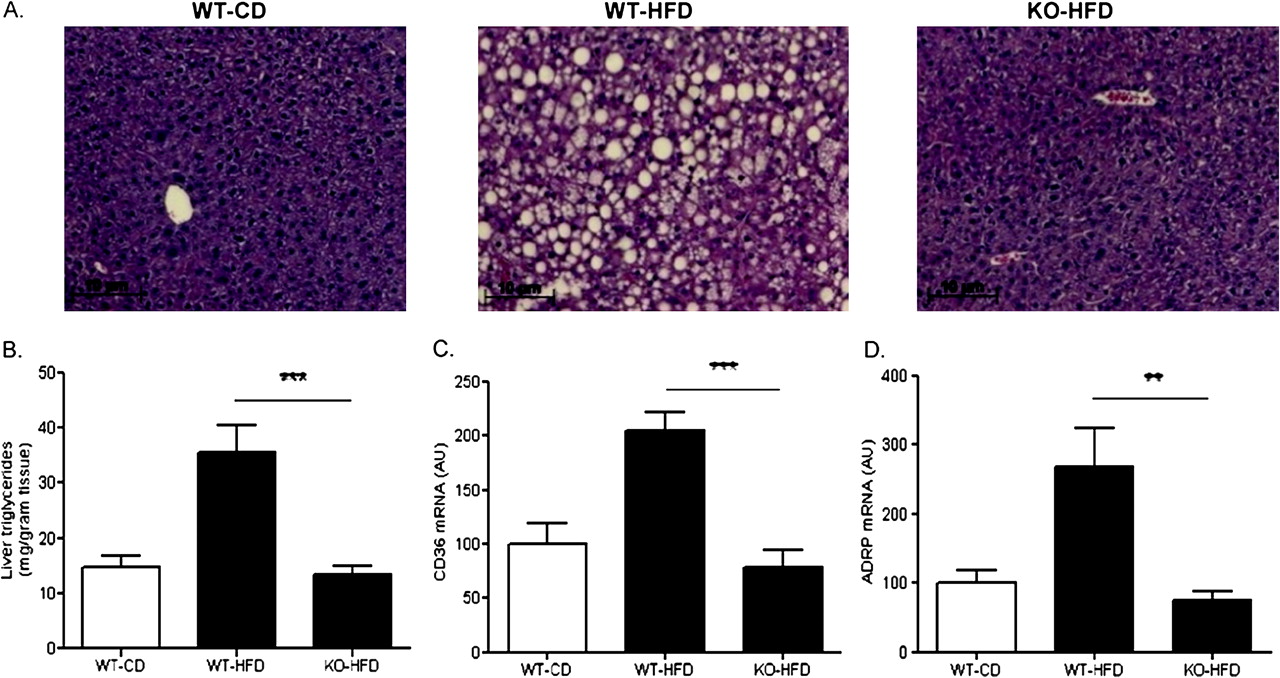
Lack of hepatic lipid accumulation in MCH−/− mice fed a high-fat diet. (A) Representative H&E stained liver sections from mice in each group. High-fat feeding for 15 weeks resulted in abundant fat droplet accumulation in the liver of WT mice but not in the liver of MCH−/− mice. (B) MCH−/− mice fed a high fat diet had significantly lower hepatic total triglyceride content as compared with WT mice fed a high fat diet. (C,D) Chronic high fat feeding significantly upregulated hepatic gene expression of the lipid transporters CD36 and adipose differentiation-related protein (ADRP) in WT mice but not in MCH−/− mice. **p<0.01; ***p<0.001; AU, arbitrary units.
Hepatic gene expression of CD36, one of the major fatty acid transporters upregulated in mouse models of liver steatosis and also in humans with NAFLD,25 was significantly induced by high-fat diet in wild type mice but not in MCH−/− mice (204.5±17.8 vs 78.5±15.4, respectively; p<0.001) (figure 2C). Similarly, adipose differentiation-related protein (ADRP), a lipid coating protein recently indicated to be involved in long chain fatty acid uptake,26 was also induced by HFD in WT mice but not in MCH−/− mice (268.9±55.6 vs 75.9±12.1, respectively; p<0.01) (figure 2D).
Lipogenic gene expression in wild type and MCH−/− mice fed a high-fat diet
In WT mice, feeding of a HFD for 15 weeks caused significant upregulation in the gene expressions of several key and rate-limiting enzymes in lipid metabolism. Specifically, in mice fed a high fat diet, hepatic expression of acetyl-CoA carboxylase (ACC)-1 which converts acetyl-CoA to malonyl-CoA was significantly lower in MCH−/− mice compared to WT (121.7±17.9 vs 221.3±34.8, respectively; p<0.05) (figure 3A). Similarly, the liver expression of fatty acid synthase (FAS) which catalyses malonyl-CoA to palmitic acid (C16:0), and stearol-CoA desaturase (SCD)-1 which desaturates palmitic acid for diacylglyceride and TG synthesis, were also significantly lower in MCH−/− mice (133.3 ± 31.9 vs 438.1 ± 76.2 vs for FAS, p<0.01; and 206.7±28.5 vs 83.1±13.8 for SCD-1, p<0.01, KO-HFD vs WT-HFD respectively) (figure 3B,C).
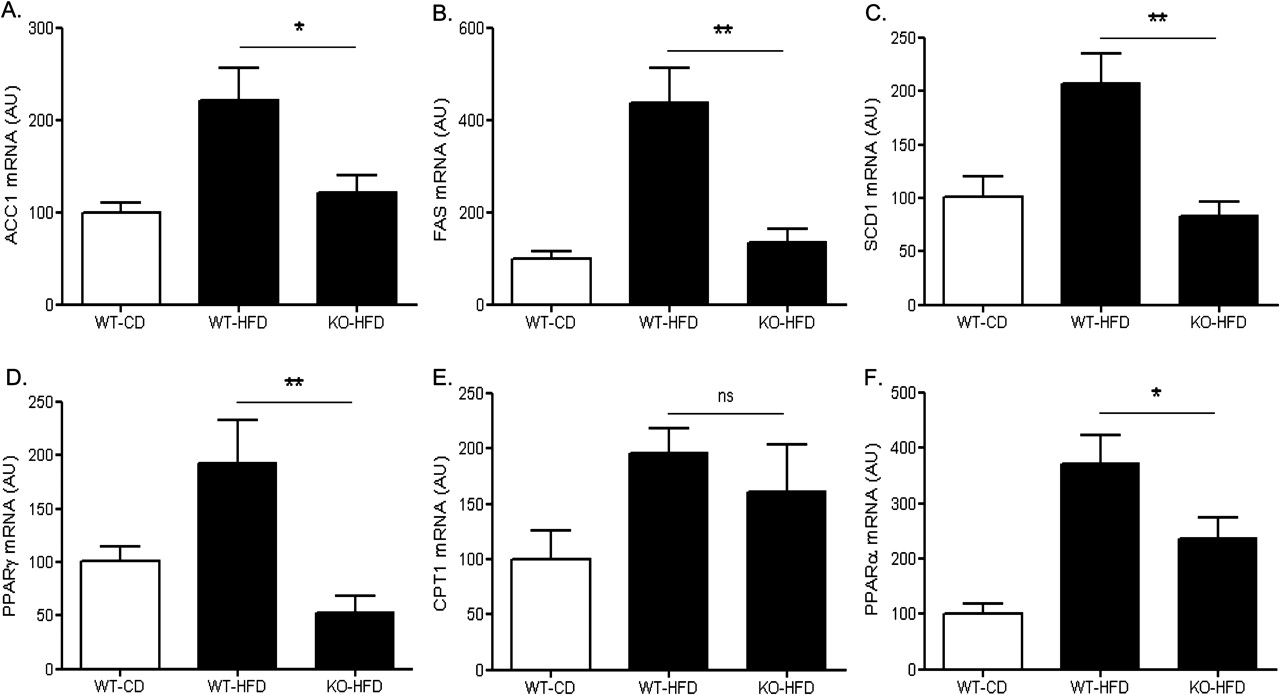
Effects of high-fat feeding on hepatic expression of key metabolic regulators. Three groups of mice were included in the study (WT, wild-type; KO, MCH−/−; HFD, high fat diet; CD, control diet) (A) acetyl-CoA carboxylase (ACC)-1; (B) fatty-acid synthase (FAS); (C) stearol-CoA desaturase (SCD)-1; (D) peroxisome proliferator-activated receptor (PPAR)-γ; (E) carnitine palmitoyltransferase (CPT)-1α; (F) peroxisome proliferator-activated receptor (PPAR)-α *p<0.05; **p<0.01; ns, not significant; AU, arbitrary units.
Peroxisome proliferator-activated receptor (PPAR) γ over-expression, in both in vivo and in vitro settings, increases triglyceride synthesis and upregulates lipogenic enzymes as well as CD36.26 27 Consistent with our findings on CD36, ACC-1, FAS and SCD-1, hepatic gene expression of PPARγ was lower in MCH−/− mice (52.7±11.6 vs193.0±39.6 KO-HFD vs WT-HFD respectively; p<0.01) (figure 3D).
Hepatic lipid oxidation in MCH−/− and WT mice fed a high-fat diet
Hepatic mRNA expression of carnitine palmitoyltransferase (CPT)-1a, the essential regulatory enzyme that transfers fatty acids from the cytosol to the mitochondria for β-oxidation,28 was similar in MCH−/− and WT mice fed a high fat diet (figure 3E) (161.2±42.2 vs 195.6±22.4, respectively; p>0.05). Liver transcripts for PPARα, a transcriptional regulator of genes involved in lipid oxidation,28 were lower in the MCH−/− mice compared to WT (235.5 ± 37.9 vs 371.3 ± 51.9, respectively, p<0.05) (figure 3F).
An inflammatory response in the liver, but not in the white adipose tissue, of MCH−/− mice fed a high-fat diet
Immunostaining of liver biopsies with a macrophage-specific F4/80 antibody revealed the presence of an increased number of monocytic cells in both WT and MCH−/− mice fed a high fat diet (figure 4A, upper panel). In accordance with this observation, hepatic mRNA expression of the macrophage marker CD68 was indistinguishable in KO-HFD and WT-HFD mice (181.7±20.6 vs 180.7±36.5, respectively; p>0.05) (figure 4B, left panel). However, monocytic infiltration in the perigonadal adipose tissue was significantly lower in MCH−/− mice compared to wild type mice fed a high fat diet (126.7±62.00 vs 380.7±87.06, respectively, p<0.01) (Figure 4B, right panel), a feature consistent with their overall reduced adiposity (figure 1E).

Inflammatory responses in the liver and adipose tissue of MCH−/− and WT mice fed a high fat diet for 15 weeks. F4/80 immunostaining of liver (upper panel) and white adipose tissue (epididymal fat) (lower panel) in the three groups of mice (WT, wild type; KO, MCH−/−; HFD, high fat diet; CD, control diet). MCH−/− mice fed a high fat diet had hepatic but not adipose tissue accumulation of F4/80 positive cells. (B) mRNA expression of the monocytic marker CD68 in the liver (left panel) and white adipose tissue (right panel) in the three groups of mice. **p<0.01; ns, not significant; AU, arbitrary units.
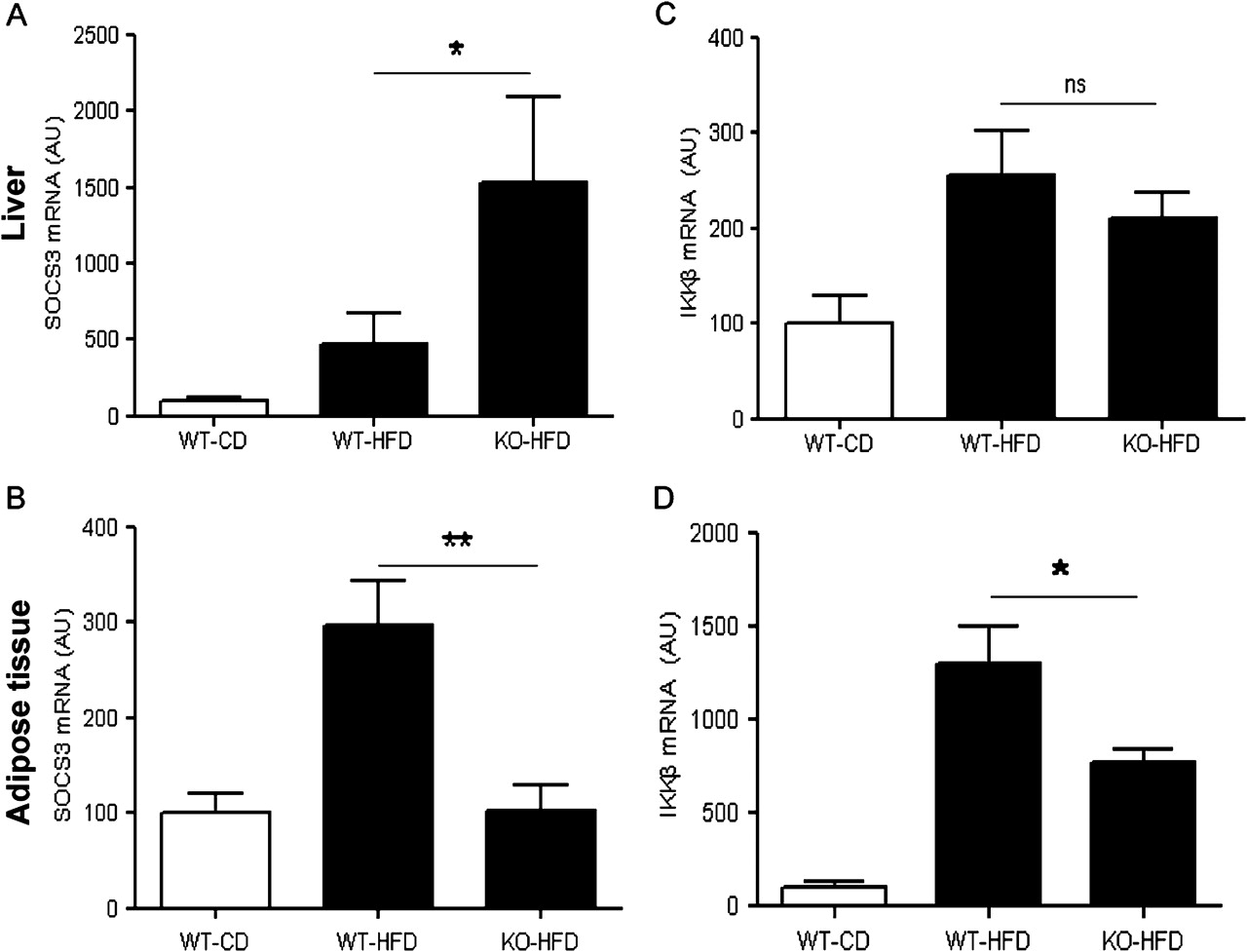
Moreover, high-fat feeding was associated with an increase in hepatic SOCS-3 gene expression in WT mice which was even higher in MCH−/− mice (467.1±203.0 vs 1528.0±562.4, respectively; p<0.05) (figure 5A). The inflammatory response of the MCH−/− mice appeared to be liver specific, since SOCS-3 expression in epididymal fat was about threefold lower in the MCH−/− mice compared to wild-type (102.9±27.1 vs 296.5±47.0, respectively; p<0.01) (figure 5B). Likewise, hepatic expression of IKK-β, an upstream IkB kinase, was found significantly upregulated in both MCH−/− and wild-type mice fed a high-fat diet (210.5±26.9 vs 256.1±45.9, respectively, p>0.05) (figure 5C), while in adipose tissue, IKK-β mRNA levels were significantly lower in MCH−/− mice fed a high fat diet compared to WT mice (768.3±69.5 vs 1297.8±202.2, respectively, p<0.05) (figure 5D).

Inflammatory responses in the liver (upper panel) and adipose tissue (lower panel) of MCH−/− and WT mice fed a high fat diet for 15 weeks. (A,B) mRNA expression of suppressor of cytokine signalling (SOCS) -3 (C,D) mRNA expression of IKB-kinase β (IKKβ) as measured by real time PCR. (WT, wild type; KO, MCH−/−; HFD, high fat diet; CD, control diet). *p<0.05; **p<0.01; ns, not significant; AU, arbitrary units.
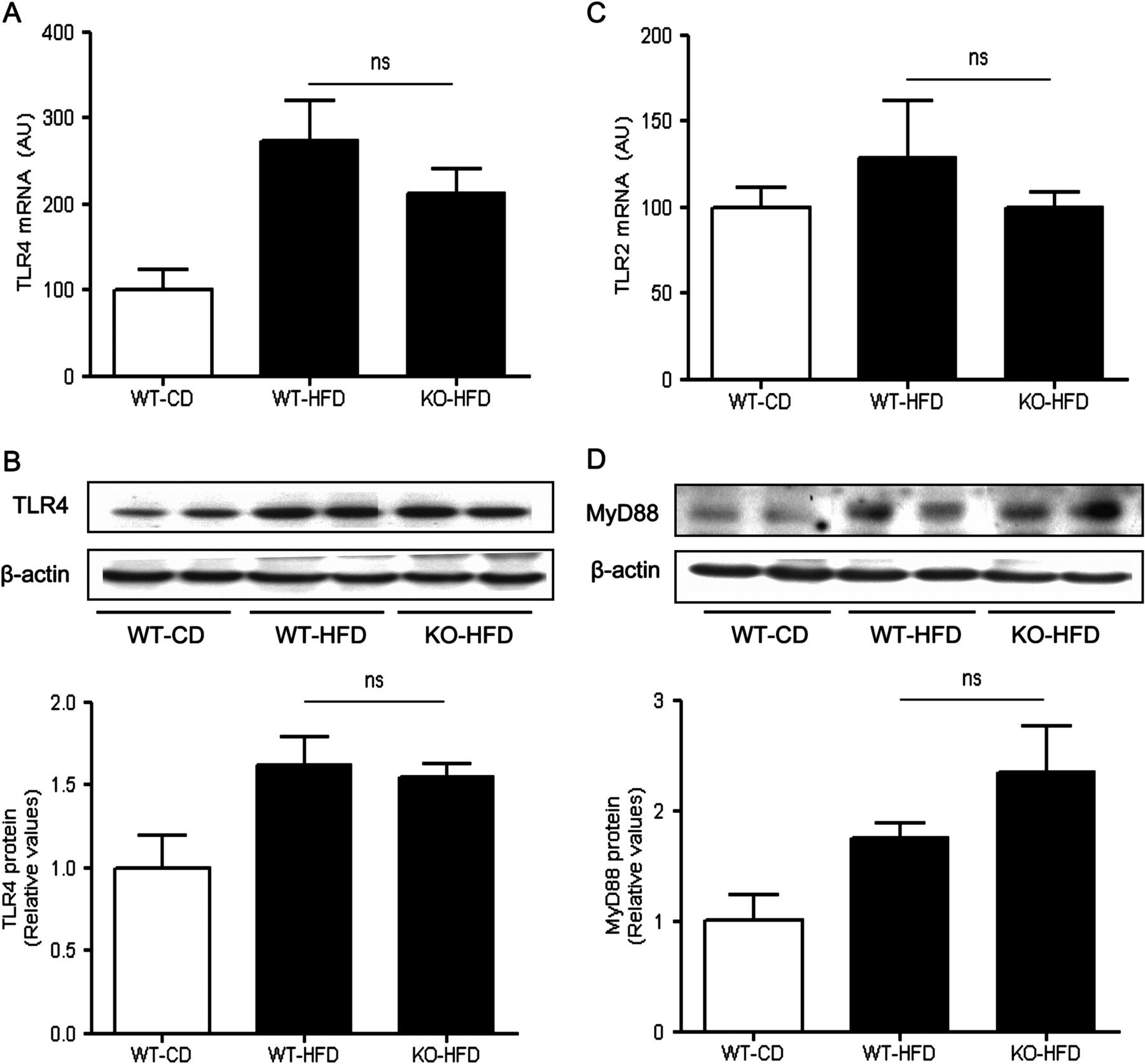
Activation of TLR-4 signalling in the liver of wild-type and MCH−/− mice fed a high-fat diet
TLRs constitute a family of pattern-recognition receptors critical for the body's antimicrobial defence mechanisms. Among them, TLR4 is activated by LPS, a component of the cell wall of Gram-negative bacteria, and, to a lesser extent, by non-bacterial products, such as saturated fatty acids.3 4 29 Hepatic gene expression of TLR4 was significantly induced in both WT and MCH−/− mice fed a high-fat diet (213.2±28.1 vs 273.6±46.7, KO-HFD vs WT-HFD; p>0.05) (figure 6A), as well as TLR4 protein expression (1.5±0.1 vs 1.6±0.2, KO-HFD vs WT-HFD; p>0.05) (figure 6B). While TLR2 is also involved in pathogen recognition in many disease paradigms, feeding a high-fat diet for 15 weeks did not lead to any significantly changes on hepatic TLR2 gene expression in any group in our study (figure 6C). This result is consistent with previous reports specifically implicating TLR4, but not TLR2, in the pathogenesis of non-alcoholic steatohepatitis.30

Toll-like receptor (TLR) involvement in the liver inflammatory responses associated with high fat feeding. (A) TLR4 mRNA expression as measured by real time PCR in the liver of three groups of mice (WT, wild type; KO, MCH−/−; HFD, high fat diet; CD, control diet). (B) Western blot analysis of TLR4 expression in the liver of MCH−/− and WT mice fed a high fat diet. (C) Hepatic TLR2 mRNA expression. (D) Western blot analysis of MyD88 expression in the liver of MCH−/− and WT mice fed a high fat diet. ns, not significant; AU, arbitrary units.
Along with the observed diet-induced hepatic activation of the TLR4 signalling in the MCH−/− mice, expression of MyD88, the essential cytosolic TLR4 adaptor protein, was increased in both MCH−/− and WT mice fed a high fat diet (2.3±0.4 vs 1.7±0.1, respectively; p>0.05) (figure 6D).
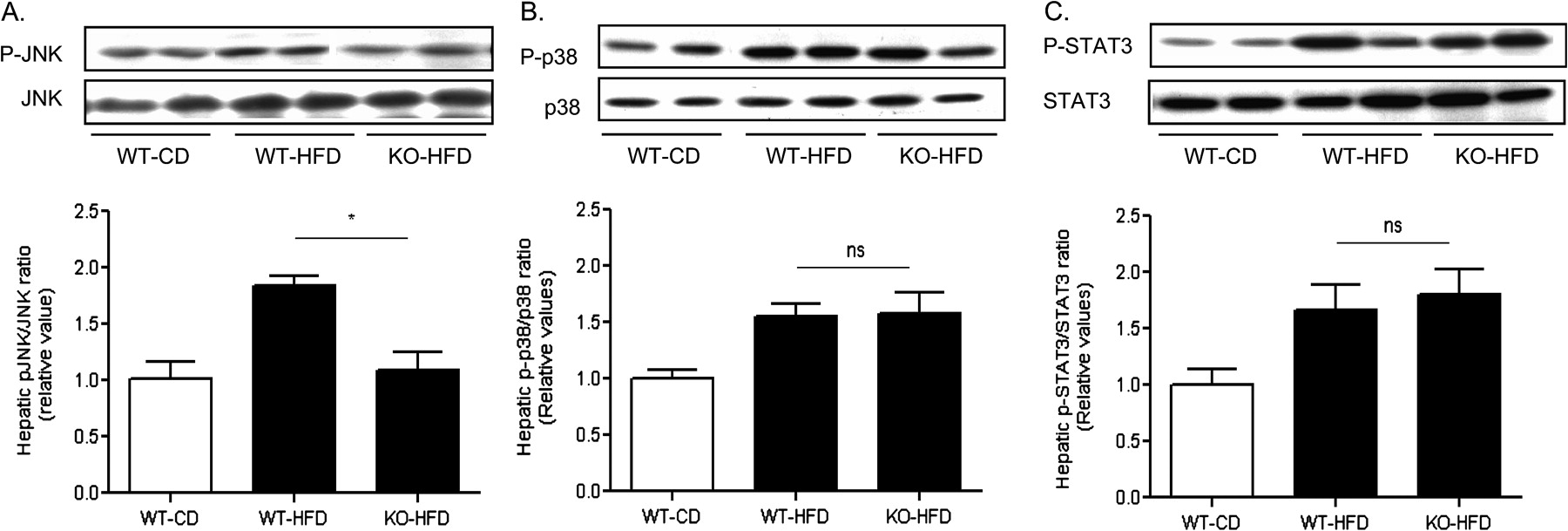
TLR4/MyD88 signalling downstream effectors in the liver of MCH−/− mice fed a high-fat diet
Stress kinase responses (Erk1/2, JNK and p38 MAP kinases) and signal transducer and activator of the transcription (STAT)-3 activation represent major components of the TLR4- and/or MyD88-induced proinflammatory responses.2 11 31 While MCH−/− fed a high fat diet were protected from increased JNK phosphorylation in their livers, compared to WT mice treated likewise (1.1±0.1 vs 1.8 ± 0.1, KO-HFD vs WT-HFD; p<0.05) (figure 7A), they exhibited similar to the wild type mice levels of p38 activation (1.6±0.2 vs 1.5±0.1, KO-HFD vs WT-HFD; p>0.05) (figure 7B). Likewise, MCH−/− mice had similar to WT mice hepatic activation of STAT-3 (1.8±0.2 vs 1.7±0.2, KO-HFD vs. WT-HFD; p>0.05) (figure 7C).
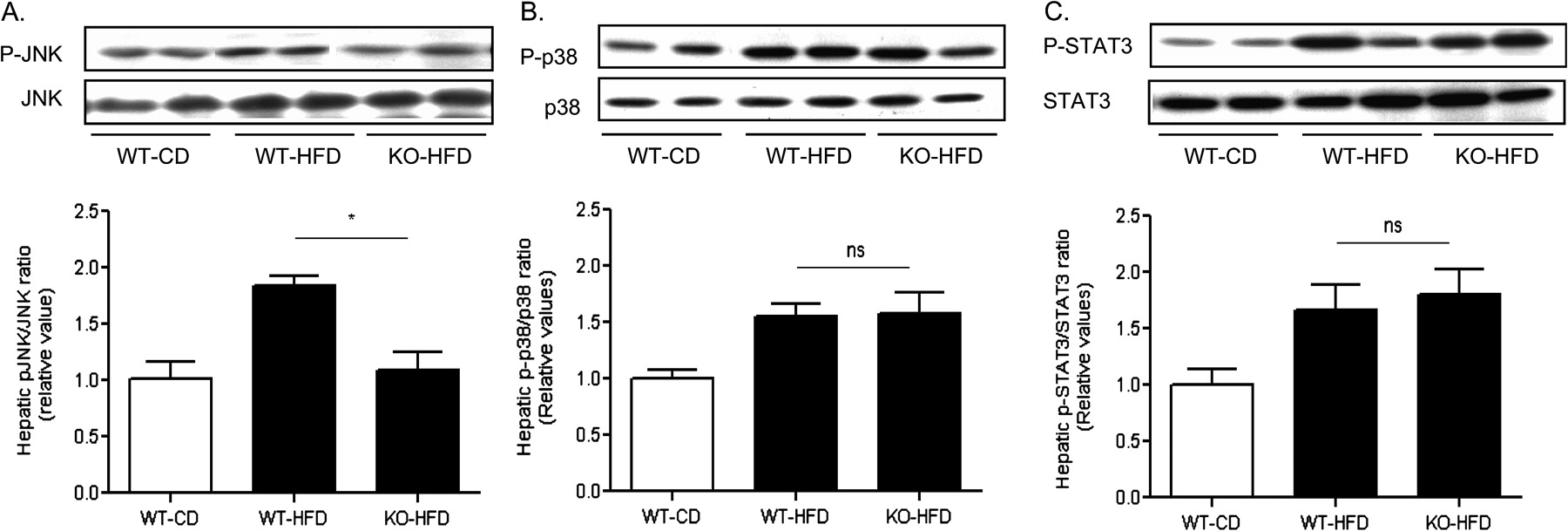
Proinflammatory signalling in the liver of MCH−/− and WT mice fed a high fat diet. Liver tissue homogenates were analysed by western blot in three groups of mice (WT, wild type; KO, MCH−/−; HFD, high fat diet; CD, control diet) for the activated and total protein expression of (A) JNK; (B) p38; and (C) STAT-3. The lower panels represent quantification of the specific western blot signals. *p<0.05; ns, not significant.
Global insulin sensitivity in the wild type and MCH−/− mice fed a high-fat diet
Blood glucose levels at the random fed state were similar in MCH−/− and WT mice fed a high fat diet (151.9±12.6 mg/dl vs 155.1±12.4 mg/dl respectively; p>0.05 (figure 8A), as well as the area under the curve (AUC) in a glucose tolerance test (34623±3288 vs 36499±3460, KO-HFD vs WT-HFD; p>0.05) (figure 8B). However, significantly lower serum insulin levels at the random fed state were observed in MCH−/− mice compared to WT mice fed a high fat diet (3.7±0.6 ng/ml vs 6.2±1.1 ng/ml, respectively; p<0.05) (figure 8C), which was consistent with their lower body weight gain and reduced adiposity as seen in figure 1. Likewise, MCH−/− mice fed a high fat diet exhibited attenuated blood glucose levels throughout a 2-h insulin tolerance test as shown by a significantly lower AUC compared to WT mice (6341±475 vs 9664±378, KO-HFD vs WT-HFD; p<0.05) (figure 8D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Indices of insulin sensitivity of MCH−/− and wild-type mice fed a high fat diet. (A) Blood glucose levels at the random fed state in three groups of mice (WT, wild type; KO, MCH−/−; HFD, high fat diet; CD, control diet). (B) Blood glucose levels overtime (left panel) during a glucose tolerance test (GTT) and calculation of the area under the curve (AUC) right panel. (C) Serum insulin levels at the random fed state. (D) Percentage drop in blood glucose levels overtime (left panel) in response to an insulin challenge (ITT) and calculation of the area under the curve (AUC) right panel. (E) Liver mRNA expression of PEPCK, a key enzyme in gluconeogenesis. (F) Liver mRNA expression of PGC1α and (G) SREBP1c, as measured by real time PCR. Enzyme in liver tissues measured by real-time *p<0.05; ns, not significant; AU, arbitrary units.
Hepatic gluconeogenesis in the wild type and MCH−/− mice fed a high-fat diet
Gene expression of phosphoenol-pyruvate carboxykinase (PEPCK), one of the key enzymes involved in hepatic gluconeogenesis, was similar in MCH−/− and WT mice fed a high fat diet (199.4±31.1 vs 173.1±49.5, respectively; p>0.05) (figure 8E). Likewise, hepatic mRNA levels of peroxisome proliferator-activated receptor-α coactivator 1(PGC-1α), a positive regulator of gluconeogenesis,32 were indistinguishable between MCH−/− and WT mice fed a high-fat diet (200.8±15.7 vs 247.3±49.6, KO-HFD vs WT-HFD; p>0.05) (figure 8F). Sterol regulatory element binding protein (SREBP)-1c mRNA levels were also comparable between MCH−/− and WT mice fed a high fat diet (238.8±75.4 vs 218.8 ± 41.5, KO-HFD vs WT-HFD; p>0.05) (figure 8G).
Discussion
In this study, we report hepatic inflammatory responses in mice fed a high fat diet, which appear to be liver specific and independent of the obesity status and lipid accumulation in the liver. Specifically, we have shown that despite the fact that MCH−/− mice fed a high fat diet have less hepatic fat accumulation compared to wild type mice, they have similar activation of key inflammatory cascades in their livers.
Overall, our experimental approach allowed us to distinguish inflammatory mediators triggered by high-fat diet from those associated with diet-induced obesity. In the first group belong molecules such as TLR4, and p38 MAPkinase, IKK-β, STAT-3 and SOCS-3, which have been shown to represent downstream targets of TLR4 signalling.2 9 Most likely, this TLR4 upregulation occurs in Kupffer cells, which are primarily involved in innate immune responses2 3 33 and were also expanded in our study in both wild-type and MCH−/− mice fed a high-fat diet. Interestingly, each one of the above mediators has also been demonstrated to independently compromise insulin sensitivity by converging inflammatory and metabolic processes. For example, infusion of bacterial LPS, a potent TLR4 activator, in mice leads to liver-specific insulin resistance while chronic consumption of a high-fat diet results in a two- to threefold increase in plasma LPS, a condition described as metabolic endotoxinemia.34 Moreover, increased expression of SOCS1 and/or SOCS3 can impair insulin-induced tyrosine phosphorylation of insulin receptor substrate (IRS)-1 and -2, a hallmark of insulin resistance while hepatic insulin resistance in obese mice could be alleviated by inhibition of the SOCS proteins.35 Profound hepatic insulin resistance and development of type 2 diabetes also characterise transgenic mice with hepatocyte specific expression of constitutively active IKK-β.36 MCH−/− mice chronically exposed to a high fat diet have lower serum insulin levels compared to wild type mice fed a high fat diet and improved total body insulin sensitivity as measured by an insulin tolerance test, consistent with their lean phenotype. Whether these mice exhibit a certain degree of hepatic insulin resistance needs to be further investigated in functional experiments including insulin signalling in various tissues and clamp studies.32 Upregulation of SREBP1c expression in the liver of MCH−/− mice is also puzzling, given the lack of activation of lipogenic pathways in those mice. Moreover, increased levels of SREBP1c, as in the MCH−/− mice fed a high fat diet, might contribute to liver insulin resistance, since it has been previously demonstrated that SREBP directly binds to the promoter of IRS-2 and suppresses its expression, leading to impaired insulin sensitivity.37 Taken together, these findings further underscore the complexity of the hepatic regulation of glucose and fat metabolism.
MyD88, a common TLR adaptor,9 11 was found to be upregulated in the liver of MCH−/− fed a high fat diet and likewise in wild-type mice, consistent with TLR4 activation in those mice.29 In contrast, the effects of TLR4 activation in alcoholic steatohepatitis and in liver inflammation associated with ischaemia/reperfusion are rather MyD88-independent and involve the IFN regulatory factor 3.38 39 It has been previously shown that saturated fatty acids can activate TLR2/4 and JNK dependent proinflammatory pathways in macrophages.10 27 Still, in the liver of MCH−/− mice fed a high fat diet we detected a selective TLR4 upregulation that did not involve activation of JNK. Instead, the phenotype of MCH−/− mice is associated with increased p38 phosphorylation, another downstream target of TLR4 signalling.31 Thus, it seems likely that different ligands of TLR4 utilise distinct downstream signalling pathways to mediate their effects, a possibility that warrants further investigation. Notably, phosphorylation of p38 in response to TLR4 activation by LPS was also described in alcohol-induced liver injury.2
Another interesting observation in the current study is a twofold upregulation of CD68 expression, one of the markers of activated Kupffer cells, in the liver of MCH−/− mice fed a high fat diet. CD68 expression was similar in both mouse genotypes exposed to high-fat diet, suggesting a direct effect of diet instead of obesity status. The pivotal role of Kupffer cells in non-alcoholic fatty liver disease has been well established11 40 and the number of CD68 positive cells in the liver of patients with NAFLD appears to be a positive predictor of disease progression.41 Beyond the propagation of inflammatory responses, recent studies have also implicated macrophages in the mechanisms of TLR4-mediated insulin resistance.10 33 42 On the other hand, and of particular relevance to the present study, mice with haematopoietic lineage-specific TLR4 inactivation become obese when fed a high-fat diet, while sustaining insulin sensitivity in their liver and adipose tissue.17 To our knowledge, our study and that by Saberi et al17 are unique in their design because they use complementary approaches to uncouple diet-induced TLR4 activation and insulin resistance from obesity. Previous studies from our group have shown that MCH, in addition to its role in energy balance, is a mediator of inflammation, at least in the intestine.43 44 Hence, MCH−/− were expected to be protected from liver inflammation, not only because their lack of obesity, but also lack of MCH itself. From a different perspective, it becomes apparent from this study that MCH is dispensable for TLR4-mediated immune responses triggered by the high-fat diet.
In the case of MCH−/− mice fed a high fat diet, which lack hepatosteatosis, experimental evidence suggests additional TLR4 ligands as inducers of their hepatic inflammatory responses. A strong candidate for such effects could be gut-derived LPS. It is well documented that consumption of certain types of diet, for example diets high in saturated fats or fructose, results in portal endotoxinaemia, by at least two mechanisms: compromising the integrity of the gut epithelial barrier and altering the gut microbiome.13 14 42 45 Moreover, a recent study of RELMβ knock-out mice, which resist diet-induced obesity like the MCH−/− mice presented here, clearly demonstrated that high-fat diet per se determines the composition of the gut microbiome, independently of the obesity status.16 Nevertheless, the phenomenon is not confined to diets rich in saturated fats and cholesterol,8 and activation of TLR4 signalling also occurs in mice and humans with fructose-induced NAFLD.46 47 Most importantly, our results offer a paradigm of development of diet-induced liver inflammation in the absence of hepatic fat accumulation. In corroboration of these findings, combined LDL-receptor null and ApoE2 knock-in mice, which are hyperlipidaemic, develop severe hepatic inflammation in the absence of steatosis in response to feeding a high fat/high cholesterol diet.18
In summary, the unexpected finding in the present study is the dissociation between hepatic fat accumulation and activation of liver-specific innate immune responses in MCH−/− mice fed a high fat diet. From a translational perspective, given the lack of adiposity in those mice due to increased locomotor activity,20 our results suggest that unless appropriate modification of dietary habits is achieved, chronic consumption of a diet enriched with saturated fat could lead to liver damage, even in lean individuals. In fact, occurrence of liver inflammation has been reported in lean subjects and rodent models in the absence of obesity, as well as obesity in the absence of liver inflammation,16 17 clearly indicating that mechanisms additional to body fat accumulation are implicated in the pathogenesis of hepatic insulin resistance and NAFLD.
Acknowledgments
We thank Dr E. Maratos-Flier for kindly providing us with the MCH−/− breeding pairs. We thank Dana-Farber/Harvard Cancer Center in Boston, Massachusetts, for the use of the Specialized Histopathology Core, which provided the immunohistochemical staining service. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516.
References
Footnotes
Funding This work was supported by NIH grant RO1DK080058 (EK).
Competing interests None.
Ethics approval The mice used in this study were cared for and handled according to animal protocols approved by the Beth Israel Deaconess Medical Center Animal Care Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Digest