
Abstract
Aims/hypothesis
Obesity increases the risk of developing major diseases such as diabetes and cardiovascular disease. Adipose tissue, particularly adipocytes, may play a major role in the development of obesity and its comorbidities. The aim of this study was to characterise, in adipocytes from obese people, the most differentially expressed genes that might be relevant to the development of obesity.
Methods
We carried out microarray gene profiling of isolated abdominal subcutaneous adipocytes from 20 non-obese (BMI 25±3 kg/m2) and 19 obese (BMI 55±8 kg/m2) non-diabetic Pima Indians using Affymetrix HG-U95 GeneChip arrays. After data analyses, we measured the transcript levels of selected genes based on their biological functions and chromosomal positions using quantitative real-time PCR.
Results
The most differentially expressed genes in adipocytes of obese individuals consisted of 433 upregulated and 244 downregulated genes. Of these, 410 genes could be classified into 20 functional Gene Ontology categories. The analyses indicated that the inflammation/immune response category was over-represented, and that most inflammation-related genes were upregulated in adipocytes of obese subjects. Quantitative real-time PCR confirmed the transcriptional upregulation of representative inflammation-related genes (CCL2 and CCL3) encoding the chemokines monocyte chemoattractant protein-1 and macrophage inflammatory protein 1α. The differential expression levels of eight positional candidate genes, including inflammation-related THY1 and C1QTNF5, were also confirmed. These genes are located on chromosome 11q22–q24, a region with linkage to obesity in the Pima Indians.
Conclusions/interpretation
This study provides evidence supporting the active role of mature adipocytes in obesity-related inflammation. It also provides potential candidate genes for susceptibility to obesity.
Similar content being viewed by others
Introduction
Obesity is characterised by increased adipose tissue mass resulting from a chronic energy imbalance between energy intake and energy expenditure [1]. It is widely accepted that both genetic and environmental factors (such as diet) can predispose individuals to the development of obesity [2]. In the United States, more than 30% of the adult population are categorised as obese, defined as having a BMI of over 30 kg/m2 [3]. The increasing prevalence of obesity in recent decades as a public health problem in both developed and developing countries is of concern given the comorbidities of this condition. Obesity is recognised as a risk factor for insulin resistance, which can lead to major diseases, such as type 2 diabetes [4] and cardiovascular disease [5].
A higher risk of obesity-associated metabolic diseases is associated with increased adipose tissue mass in the abdominal region [6]. The role of adipose tissue in the aetiology of obesity [7], as well as its pathophysiological consequences [8], is increasingly being recognised. A few studies in rodents and humans have used microarray gene expression profiling of whole adipose tissue to investigate its role in obesity [9, 10]. Unfortunately, the heterogeneous cell composition of adipose tissue does not provide information on the potentially diverse functions of the specific cell types of the tissue. Adipose tissue contains a mixture of mature, lipid-filled adipocytes and stromal vascular cells, including adipocyte precursor cells (preadipocytes), macrophages, fibroblasts, mast cells and vascular cells [11]. The degree of macrophage infiltration of adipose tissue has been shown to be associated with obesity [12, 13], and macrophage infiltration may play a role in adipose tissue inflammation [14]. Higher expression levels and/or concentrations of inflammatory factors in adipose tissue are associated with increased systemic inflammation in obesity, which may explain, at least in part, the increased risk of obesity-related comorbidities [14].
The function of mature adipocytes as a major cell type in adipose tissue extends beyond lipid storage. These cells actively participate in energy metabolism by secreting bioactive molecules called adipokines, which can act in an autocrine, paracrine, intracrine and/or endocrine fashion [15]. Leptin, a well-known adipokine, can cause obesity by its absence in the ob/ob mouse [16]; this has also been reported in humans [17]. Circulating levels of adiponectin, another well-studied adipokine, are positively correlated with the expression of mRNA encoding adiponectin in adipose tissue, and negatively correlated with obesity-associated insulin resistance [18]. Inflammatory adipokines, such as TNF-α, may contribute to adipose tissue inflammation [14]. Adipocytes may express other genes that contribute to the aetiology or pathophysiology of obesity.
The aim of this study was to investigate the gene expression profiles of isolated adipocytes from abdominal subcutaneous adipose depots in obese and non-obese non-diabetic Pima Indians, a population with one of the highest prevalence rates of obesity and type 2 diabetes [19]. This study was carried out to characterise the most differentially expressed genes in adipocytes from the two subject groups. As a complementary approach to our ongoing positional cloning effort, this study may also help to identify candidate susceptibility genes for obesity on chromosome 11q22–q24, the peak of linkage to obesity in this population [20].
Subjects and methods
Subjects
All subjects provided written informed consent prior to participation. This study was approved by the Tribal Council of the Gila River Indian Community and by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The anthropometric and metabolic characteristics of the subjects are described in Table 1. All subjects were in good health, as determined by medical history, physical examination and routine blood and urine tests, and none were taking medication at the time of the study.
Metabolic measurements
Volunteers were admitted to the Clinical Research Unit, NIDDK, Phoenix, AZ, USA, where they consumed a weight-maintaining diet (containing 50% of calories as carbohydrates, 30% as fat and 20% as protein) for 2–3 days prior to clinical testing. BMI was calculated as weight (in kg) divided by the square of height (in m2). Body composition was measured by dual energy X-ray absorptiometry (DXA) using a total body scanner (DPX-L; Lunar Radiation, Madison, WI, USA), as described previously [21]. A 3-h 75-g OGTT was performed after a 12-h overnight fast. Diabetes was defined according to the criteria of the World Health Organization.
Fat biopsy and extraction of total RNA from isolated adipocytes and adipose tissue
Subcutaneous abdominal fat biopsies were obtained from 58 non-diabetic Pima Indians. Samples from 39 subjects were used for microarray hybridisation, and those from 19 individuals for real-time PCR (RT-PCR) of specific inflammation-related genes. After an overnight fast, subjects underwent subcutaneous abdominal fat needle biopsies under local anaesthesia with 1% lidocaine. Collagenase digestion of the adipose tissue biopsy was carried out as described previously [22], and the floating adipocyte fraction was collected. Total RNA was extracted from adipose tissue and isolated mature adipocytes using an RNeasy Lipid Mini Kit and an RNeasy Mini Kit from Qiagen (Valencia, CA, USA), respectively. During the extraction, RNA was treated with DNase using the RNase-free DNAse Set (Qiagen) according to the manufacturer’s instructions.
Microarray
Target preparation, array hybridisation and scanning
Double-stranded cDNA for each subject was synthesised using the Superscript Choice System (Invitrogen, Carlsbad, CA, USA). A 10-μg aliquot of adipocyte total RNA from each subject was first used to synthesise single-stranded cDNA and then biotin-labelled cRNA, as recommended by the manufacturer of the microarray (Affymetrix; Santa Clara, CA, USA). Labelled cRNA from each sample was individually hybridised to the five microarray chips included in the Affymetrix Human Genome U95 (HG-U95A, B, C, D, and E) Set, which contains a total of approximately 60,000 probe sets. Following hybridisation, the washing and staining procedure was performed in the Affymetrix GeneChip Fluidics Station. To enhance the signals, an antibody amplification step was added using normal goat IgG and biotinylated goat anti-streptavidin antibody (Vector Laboratories, Burlingame, CA, USA). The arrays were scanned twice using a Hewlett Packard GeneArray scanner (Palo Alto, CA, USA). Raw data were analysed using Microarray Suite, Version 5.0 software (Affymetrix). To allow comparison of microarray data across different arrays and individual samples, we normalised the data by global scaling to an arbitrary target intensity value of 2,500 using Microarray Suite 5.0 software. The data for absolute analysis have been deposited in NCBI Gene Expression Omnibus (GEO; available at http://www.ncbi.nlm.nih.gov/geo/, last accessed in June 2005) and may be accessed through GEO Series accession number GSE2508.
Quantitative RT-PCR
We used quantitative RT-PCR to verify the differential expression levels of several genes based on either their biological functions or chromosomal locations. Genes in the biological function category were verified using the cDNA samples from adipocytes isolated from nine non-obese and ten obese Pima Indians who were different from those who provided the microarray data (Table 1). Confirmation of the biologically relevant genes in this separate group of subjects should help control for stochastic biological sampling error exacerbated by multiple testing.
As a preliminary step in our effort to complement the ongoing positional cloning of obesity susceptibility genes in Pima Indians, we also verified the differential expression of 12 positional candidate genes. These genes are located on chromosome 11q22–q24, which encompasses the linkage peak to BMI in Pima Indians [20]. Quantitative RT-PCR of these genes was carried out using cDNA samples from the subjects used to obtain the microarray data, as differential expression of these genes might be associated with genetic variations of the subjects.
Quantitative RT-PCR was carried out as previously described [22], using TaqMan Assays-on-Demand Gene Expression Products (Applied Biosystems, Foster City, CA, USA) to measure the level of transcription of all tested genes except for two positional candidate genes. The two genes, THY1 and SNF1LK2 (formerly known as SIK2 or KIAA0781), were analysed using Assays-by-Design products (Applied Biosystems). The mRNA expression of each gene was normalised to that of the gene encoding human TATA-box binding protein (TBP) (Applied Biosystems), a normalising gene that was invariable in isolated human adipocyte samples, as assessed by both microarray and quantitative RT-PCR methods (data not shown).
Data analyses
The raw microarray data obtained from Microarray Suite, Version 5.0 software were analysed using the Mann–Whitney U-test, as well as the two-sided t-test corrected for unequal variances (Welch test) to compare the mean expression level for each gene between the non-obese and obese individuals. The Bayesian posterior probability of being a false discovery (expressed as false discovery rate [FDR]) was estimated for each probe set individually, based on the Welch p values, using a mixture model described elsewhere [23, 24]. We focused on the genes among those most differentially expressed that had corresponding probe sets with a less than 0.1% FDR, i.e. with a posterior probability of being differentially expressed of 99.9%. These genes were annotated with their chromosomal locations, UniGene number, LocusLink ID and Gene Ontology (GO) information (according to biological process, cellular component and molecular function) using NetAffx resources [25]. Genes that could not be annotated using NetAffx were classified using the Human Proteome Survey Database [26]. We used Onto-Express (available at http://vortex.cs.wayne.edu:8080/index.jsp, last accessed in June 2005) [27] to calculate whether any of the GO terms were significantly over-represented among the most differentially expressed genes, as determined by hypergeometric p values. A two-sided binomial test was used to calculate whether there was a significant difference between the numbers of upregulated and downregulated genes within each GO class. The p value calculation is only valid if the expression levels of the genes are independent; thus, the p values reported for these analyses should only be considered as heuristic ranking statistics.
The quantitative RT-PCR data on those genes that were differentially expressed between the obese and the non-obese groups were analysed using a two-tailed Student’s t-test, with a p value of less than 0.05 considered significant.
Results
The most differentially expressed genes
The distribution of p values for all probe sets hybridised with adipocyte samples from non-obese and obese subjects showed that there were more than 3.5 times as many genes with significant differential expression (i.e. a p value of between 0 and 0.05) than would be expected at random. The plot of p values for all (approximately 60,000) probe sets can be seen in Fig. 1 of the Electronic supplementary material (ESM).
A subset of 703 probe sets with a less than 0.1% FDR (an arbitrary cut-off level that indicated the highest probability of being truly differentially expressed between the two groups) was selected for further analysis. These probe sets had p values less than 0.00002 by the Welch test and less than 0.0001 according to the Mann–Whitney U-test. At these p values, practically none of the genes identified as being significantly different between the obese and non-obese subjects would be false positives, whereas approximately 75% of the genes called non-significant would truly not be different between the two groups (data not shown).
Because some of the transcripts were represented on multiple probe sets, the selected 703 probe sets corresponded to 677 unique transcripts. Of these 677 transcripts, 433 were upregulated and 244 were downregulated in mature adipocytes of obese individuals. The 651 genes corresponding to the 677 transcripts were localised to specific chromosomal regions, and 410 genes were classified into 20 GO Biological Process categories (Table 2). The identification of each gene, its gene symbol, chromosomal location, representative probe set and the relative expression of the gene in adipocytes of obese subjects (upregulated or downregulated compared with non-obese control subjects) are presented in Table 1 of the ESM. These 410 most differentially expressed genes will be the focus of the remainder of this manuscript, unless otherwise noted.
Over-represented GO Biological Process categories
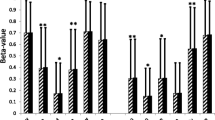
The GO Biological Process category with the highest number of differentially expressed genes was inflammation/immune responses (54 genes). Furthermore, there were significantly more genes in this category (52 out of 54) showing increased expression in adipocytes of obese subjects. The upregulated inflammation-related genes included those encoding chemotactic factors for monocytes/macrophages (chemokines) and those encoding members of the complement system, immunoglobulin superfamily and interferon pathway (ESM Table 1). The chemokine genes consisted of those encoding monocyte chemoattractant protein-1 (MCP-1; gene symbol CCL2), macrophage inflammatory protein 1α (MIP-1α; CCL3), MIP-1β (CCL4), chemokine (C-X-C motif) ligand 1 (CXCL1), macrophage inflammatory protein 2α (MIP-2α; CXCL2), MIP-2β (CXCL3), stromal cell-derived factor 1 (SDF-1; CXCL12), and interleukin-8 (IL8, formerly known as CXCL8). Macrophage migration inhibitory factor (MIF), a lymphokine that regulates macrophage function in inflammation, and chemokine-like factor super family 3 (CKLFSF3), a potential chemokine, were also upregulated in adipocytes of obese subjects. The increased expression of representative inflammatory genes (CCL2 and CCL3) in a separate group of nine non-obese subjects, as compared with ten obese subjects, was confirmed by quantitative RT-PCR (Fig. 1).

Confirmation of the increased mRNA expression levels of CCL2 (MCP-1) and CCL3 (MIP-1α) in adipocyte samples from obese subjects by quantitative RT-PCR. Adipocyte samples were isolated from abdominal subcutaneous biopsies from a group of nine non-obese (white bars) and ten obese (black bars) Pima Indians who were separate from those who provided the microarray data. The expression of CCL2 (MCP-1) (a) and CCL3 (MIP-1α) (b) was normalised to that of TBP. The data are presented as means±SEM. *p=0.02 vs lean subjects; **p=0.04 vs lean subjects
To assess whether the inflammation/immune response genes in the adipocytes of obese subjects were actually genes expressed by potentially contaminating adipose tissue macrophages, we examined the expression of several macrophage genes (ADAM8, CD163, CD68, EMR1, FCGR1A, ITGAM, MARCO). All probe sets, apart from one coding for CD163, were assigned an absent call for all subjects, as determined by the Microarray Suite software, and none were differentially expressed between the non-obese and obese subjects. The expression levels of CD68 and EMR1 were further determined using quantitative RT-PCR. The lack of differential CD68 expression between the two groups was confirmed, despite the presence of CD68 mRNA transcripts in adipocyte samples from all subjects (data not shown). The expression of EMR1 in adipocytes was not detectable by quantitative RT-PCR (data not shown).
The other two over-represented GO Biological Process categories were cellular signalling and transcription regulation. The former category included the genes adrenomedullin; phosphoinositide-3-kinase, regulatory subunit 1; regulator of G-protein signalling 2; adrenergic, beta, receptor kinase 2; phosphodiesterase 3B, cGMP-inhibited; protein phosphatase 1, regulatory (inhibitor) subunit 1B; and protein phosphatase 2, regulatory subunit B (B56), alpha isoform. The transcription regulation category included the genes nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor); forkhead box C1; E2F transcription factor 4, p107/p130-binding; and Sp1 transcription factor. In contrast to the category of inflammation/immune response, described above, the numbers of upregulated and downregulated genes within these two broad categories were not significantly different.
The remaining categories were not significantly over-represented, but a few categories had a significantly greater number of upregulated genes (cell cycle control/cell proliferation, cell adhesion, structural protein/cytoskeleton organisation, apoptosis, proteolysis/peptidolysis, cell growth and maintenance, blood coagulation), and one category had a significantly greater number of downregulated genes (energy pathway/electron transport).
Quantitative RT-PCR confirmation of the differential expression of positional candidate genes for obesity
There were 12 differentially expressed genes that mapped to chromosome 11q22–q24, the region with linkage to BMI in the Pima Indians [20]. Six of these positional candidate genes were upregulated in adipocytes of obese subjects (C1q and tumor necrosis factor related protein 5 [C1QTNF5]; Thy-1 cell surface antigen [THY1]; fasciculation and elongation protein zeta 1 [FEZ1]; nicotinamide N-methyltransferase [NNMT]; hypoxia up-regulated 1 [HYOU1]; and transgelin [TAGLN]), and six were downregulated (immunoglobulin superfamily, member 4 [IGSF4]; vacuolar protein sorting 11 [VPS11]; acetyl-Coenzyme A acetyltransferase 1 [ACAT1]; SNF1-like kinase 2 [SNF1LK2]; sterol-C5-desaturase-like protein [SC5DL]; and roundabout axon guidance receptor, homolog 3 [ROBO3]). Notably, C1QTNF5 and THY1 are classed as inflammation/immune response genes (ESM Table 1).
The quantitative RT-PCR results for the 12 positional candidate genes confirmed the differential expression of all six upregulated genes and two of the six downregulated genes (Table 3)].
Discussion
The results of this study indicate that the genes in isolated abdominal subcutaneous adipocytes that are most differentially expressed between non-diabetic obese subjects and non-diabetic non-obese subjects are those classified as involving inflammation/immune response. The fact that a larger proportion of inflammation-related genes were upregulated (rather than downregulated) in adipocytes of the obese individuals further implies an active role for these cells in adipose tissue inflammation. Increased levels of transcripts for these inflammation-related genes will presumably result in increased secretion of these factors from adipocytes. Some of these inflammation-related genes encode chemokines, such as MCP-1 and MIP-1α, that can attract monocytes/macrophages to infiltrate adipose tissue. Other upregulated genes encode adhesion molecules, such as fibronectin and integrin, that may help retain any infiltrating monocytes/macrophages in the tissue. These data are in line with recent studies indicating that the percentage of adipose tissue macrophages is positively correlated with obesity [12, 13]. The resident macrophages may be an important source of inflammation in the tissue [12, 13]. Further investigation of the contribution of augmented adipose tissue inflammation to the pathophysiology of obesity is warranted.
The increased expression of chemokines and cell adhesion molecules in adipocytes of obese individuals might have been induced, at least in part, by proinflammatory factors derived from the adipocytes themselves, such as TNF-α [28]. Consistent with previous studies (for review see [29]), adipocyte TNF-α expression was upregulated in obese subjects, as assessed by microarray and quantitative RT-PCR (p=0.05 and p=0.01, respectively; data not shown); however, TNF-α was excluded from our list of the most differentially expressed genes because the FDR of the gene was higher than the arbitrary cut-off value of 0.1%.
We investigated whether the observed upregulation of inflammation-related genes in adipocytes of obese subjects was due to contamination of the adipocyte preparation by macrophages. The lack of differential expression of macrophage markers between obese and non-obese subjects indicates that the increased levels of expression of chemokines in adipocytes of obese subjects were not directly derived from the potentially higher number of macrophages that might have contaminated the adipocyte fraction during isolation.
The expression of other inflammation-related genes, such as those induced by interferon, were also increased in the adipocytes of obese subjects. The elevated levels of these interferon-responsive genes might constitute a reaction to interferon secreted by lymphocytes, thus implicating the potential role of lymphocytes in adipose tissue inflammation in obese subjects. This possibility warrants further investigation, since the presence of lymphocytes in adipose tissue remains a controversial topic.
The standard adipocyte isolation procedure using collagenase digestion has been reported to induce the expression of many inflammation-related genes [30]. Nevertheless, the differential expression of inflammation-related genes in this study was unlikely to be caused by the isolation procedure, since all samples were subject to similar isolation procedures. A similar upregulation of inflammation-related genes was observed in preadipocytes/stromal vascular cells of adipose tissue from obese Pima Indians that had been cultured for a couple of weeks [31]. The cultured cells had presumably recovered from acute inflammatory responses induced by collagenase digestion, similar to unpublished observations that the expression of pro-inflammatory genes, including TNF and IL1B, by stromal vascular cells is reduced to levels comparable to those measured prior to collagenase digestion (X. Xu and A. Ferrante, Naomi Berrie Diabetes Center, Columbia University, NY, USA). Furthermore, a recent study by Clement et al. [32] reported that the expression of inflammation-related genes in adipose tissue of obese subjects decreased following weight loss. Taken together, the results imply that the upregulation of inflammation-related genes in the adipocytes of obese subjects in our study was not simply due to the adipocyte isolation procedure itself. Nevertheless, we cannot exclude the possibility that compared with adipose tissue samples from non-obese people, those from obese individuals are predisposed to increased expression of inflammatory-related genes when subjected to the adipocyte isolation procedure.
With respect to the most differentially expressed genes, the other over-represented GO Biological Process categories were cellular signalling and transcription regulation. The numbers of upregulated vs downregulated genes in these categories were not different between non-obese and obese subjects; furthermore, the genes did not indicate any specific molecular pathways.
The general upregulation of genes in the categories of cell cycle control/cell proliferation, structural protein/cytoskeleton organisation, and cell growth and maintenance might reflect the ability of adipocytes from obese subjects to accommodate increased cell size. On the other hand, the adipocytes of obese subjects had decreased expression of genes encoding energy pathway/electron transport. The overall gene expression profile of these isolated human adipocytes is similar to that of the adipose tissue of diet-induced obese C57BL/6J mice [14] and that of genetically obese (ob/ob) mice with a mutation in the gene encoding leptin on different strain backgrounds [9, 12, 13]. Further studies are required to investigate the relative contributions of genetic background and dietary intake to the differential adipocyte gene expression in obese people.
As a complementary approach to linkage analysis in identifying candidate susceptibility genes for obesity in this population, we used quantitative RT-PCR to confirm the differential expression levels of genes on chromosome 11q22–q24. This chromosome region encompasses the linkage peak to BMI in Pima Indians [20]. The region contains two inflammation-related genes, THY1 and C1QTNF5, together with a further ten of the most differentially expressed genes. THY1 is a major cell surface glycoprotein; its expression in activated endothelial cells facilitates the adhesion and transendothelial migration of monocytes [33]. C1QTNF5 is a member of the C1q tumor necrosis factor superfamily, which is likely to be involved in inflammation [34]. The upregulated expression of THY-1 and C1QTNF5 in adipocytes from obese subjects was confirmed, as well as four other upregulated genes (HYOU1, NNMT, TAGLN, FEZ1), and two other downregulated genes (IGSF4 and SC5DL). The differential expression of these genes in adipocytes may contribute to, or result from, obesity. Notably, HYOU1 was identified as the gene encoding oxygen regulated protein (150 kDa), a member of heat shock protein 70 family, which may play a role in protein folding and secretion in the endoplasmic reticulum [35]. This gene is also upregulated in skeletal muscle of obese insulin-resistant Pima Indians, as compared with equally obese but insulin-sensitive control subjects, and polymorphisms in this gene are associated with insulin resistance, but not with BMI [36]. The other genes with confirmed differential expression, including THY1 and C1QTNF5, serve as good positional candidate genes for obesity that are transcriptionally dysregulated in adipocytes. Further screening of genetic polymorphisms that may be associated with obesity in/surrounding these genes in Pima Indians is underway.
In terms of candidate genes for obesity in other populations, it is interesting to note that one such candidate gene—NR3C1 on chromosome 5q31, which encodes glucocorticoid receptor—was downregulated in adipocytes of obese subjects in this study. The expression of this gene was also reduced in cultured preadipocytes/stromal vascular fraction of obese Pima Indians [22]. NR3C1 belongs to the category of transcription regulation, and a single nucleotide polymorphism in exon 2 of the gene (N363S) has been shown to be associated with obesity in two different study populations [37, 38].
In summary, the expression of genes involving inflammation/immune response was increased in isolated abdominal subcutaneous adipocytes from obese people, indicating the active role of mature adipocytes in obesity-related inflammation. Two inflammation-related genes were localised to chromosome 11q22–q24 where there is a linkage to BMI in Pima Indians; together with six other genes in this region with confirmed differential expression, these genes serve as good candidate genes for obesity.
Abbreviations
- DXA:
-
dual energy X-ray absorptiometry
- ESM:
-
Electronic supplementary material
- FDR:
-
false discovery rate
- GO:
-
Gene Ontology
- MCP-1:
-
monocyte chemoattractant protein-1
- MIP-1α:
-
macrophage inflammatory protein 1α
- RT-PCR:
-
real-time PCR
References
Tataranni PA, Ravussin E (2002) Energy metabolism and obesity. In: Wadden TA, Stunkard AJ (eds) Handbook of obesity treatment. The Guilford, New York, pp 42–72
Pi-Sunyer FX (2002) The obesity epidemic: pathophysiology and consequences of obesity. Obes Res 10[Suppl 2]:97S–104S
Seidell JC, Flegal KM (1997) Assessing obesity: classification and epidemiology. Br Med Bull 53:238–252
Tataranni PA (2002) Pathophysiology of obesity-induced insulin resistance and type 2 diabetes mellitus. Eur Rev Med Pharmacol Sci 6:27–32
Nielsen S, Jensen MD (1997) Obesity and cardiovascular disease: is body structure a factor? Curr Opin Lipidol 8:200–204
Chan DC, Watts GF, Sussekov AV et al (2004) Adipose tissue compartments and insulin resistance in overweight-obese Caucasian men. Diabetes Res Clin Pract 63:77–85
Arner P (2000) Hunting for human obesity genes? Look in the adipose tissue! Int J Obes Relat Metab Disord 24[Suppl 4]:S57–S62
Trayhurn P, Beattie JH (2001) Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 60:329–339
Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS, Attie AD (2000) The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci U S A 97:11371–11376
Gomez-Ambrosi J, Catalan V, Diez-Caballero A et al (2004) Gene expression profile of omental adipose tissue in human obesity. FASEB J 18:215–217
Klaus S, Keijer J (2004) Gene expression profiling of adipose tissue: individual, depot-dependent, and sex-dependent variabilities. Nutrition 20:115–120
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Moraes RC, Blondet A, Birkenkamp-Demtroeder K et al (2003) Study of the alteration of gene expression in adipose tissue of diet-induced obese mice by microarray and reverse transcription-polymerase chain reaction analyses. Endocrinology 144:4773–4782
Hauner H, Hochberg Z (2002) Endocrinology of adipose tissue. Horm Metab Res 34:605–606
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Montague CT, Farooqi IS, Whitehead JP et al (1997) Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387:903–908
Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G (2003) Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 52:1779–1785
Lillioja S, Bogardus C (1988) Obesity and insulin resistance: lessons learned from the Pima Indians. Diabetes Metab Rev 4:517–540
Hanson RL, Ehm MG, Pettitt DJ et al (1998) An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet 63:1130–1138
Tataranni PA, Ravussin E (1995) Use of dual-energy X-ray absorptiometry in obese individuals. Am J Clin Nutr 62:730–734
Permana PA, Nair S, Lee YH, Luczy-Bachman G, Vozarova De Courten B, Tataranni PA (2004) Subcutaneous abdominal preadipocyte differentiation in vitro inversely correlates with central obesity. Am J Physiol Endocrinol Metab 286:E958–E962
Allison D, Gadbury G, Heo M et al (2002) A mixture model approach for the analysis of microarray gene expression data. Comput Stat Data Anal 39:1–20
Gadbury G, Page G, Edwards J et al (2004) Power and sample size estimation in high dimensional biology. Stat Methods Med Res 13:325–338
Liu G, Loraine AE, Shigeta R et al (2003) NetAffx: affymetrix probesets and annotations. Nucleic Acids Res 31:82–86
Hodges PE, Carrico PM, Hogan JD et al (2002) Annotating the human proteome: the Human Proteome Survey Database (HumanPSD) and an in-depth target database for G protein-coupled receptors (GPCR-PD) from Incyte Genomics. Nucleic Acids Res 30:137–141
Khatri P, Draghici S, Ostermeier GC, Krawetz SA (2002) Profiling gene expression using onto-express. Genomics 79:266–270
Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF (2002) Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 51:1319–1336
Fasshauer M, Paschke R (2003) Regulation of adipocytokines and insulin resistance. Diabetologia 46:1594–1603
Ruan H, Zarnowski MJ, Cushman SW, Lodish HF (2003) Standard isolation of primary adipose cells from mouse epididymal fat pads induces inflammatory mediators and down-regulates adipocyte genes. J Biol Chem 278:47585–47593
Nair S, Lee YH, Rousseau E et al (2005) Increased expression of inflammation-related genes in cultured preadipocytes/stromal vascular cells from obese compared with non-obese Pima Indians. Diabetologia 48:DOI 10.1007/s00125-005-1868-2
Clement K, Viguerie N, Poitou C et al (2004) Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J 18:1657–1669
Wetzel A, Chavakis T, Preissner KT et al (2004) Human Thy-1 (CD90) on activated endothelial cells is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Immunol 172:3850–3859
Shapiro L, Scherer PE (1998) The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr Biol 8:335–338
Ikeda J, Kaneda S, Kuwabara K et al (1997) Cloning and expression of cDNA encoding the human 150 kDa oxygen-regulated protein, ORP150. Biochem Biophys Res Commun 230:94–99
Kovacs P, Yang X, Permana PA, Bogardus C, Baier LJ (2002) Polymorphisms in the oxygen-regulated protein 150 gene (ORP150) are associated with insulin resistance in Pima Indians. Diabetes 51:1618–1621
Roussel R, Reis AF, Dubois-Laforgue D, Bellanne-Chantelot C, Timsit J, Velho G (2003) The N363S polymorphism in the glucocorticoid receptor gene is associated with overweight in subjects with type 2 diabetes mellitus. Clin Endocrinol (Oxf) 59:237–241
Lin RC, Wang XL, Dalziel B, Caterson ID, Morris BJ (2003) Association of obesity, but not diabetes or hypertension, with glucocorticoid receptor N363S variant. Obes Res 11:802–808
Acknowledgements
We would like to thank the members and leaders of the Gila River Indian Community, without whose cooperation these studies would have been impossible. We gratefully acknowledge J. Bunt, the nurses of the clinical research ward, and the metabolic kitchen staff for the care of the research volunteers. We also thank G. Luczy-Bachman for technical help. This work was partly supported by NIH grant award P30DK056336 to D. B. Allison.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Lee, Y., Nair, S., Rousseau, E. et al. Microarray profiling of isolated abdominal subcutaneous adipocytes from obese vs non-obese Pima Indians: increased expression of inflammation-related genes. Diabetologia 48, 1776–1783 (2005). https://doi.org/10.1007/s00125-005-1867-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1867-3