
Abstract
The gut microbiota has been hailed as an accessory organ, with functions critical to the host including dietary metabolic activities and assistance in the development of a proper functioning immune system. However, an aberrant microbiota (dysbiosis) may influence disease processes such as colorectal cancer. In this review, we discuss recent advances in our understanding of the contributions of the microbiota to prevention, initiation/progression, and treatment of colorectal cancer, with a major focus on biofilms and the antimicrobial and antitumoural immune response.
Similar content being viewed by others
Main
Colorectal cancer (CRC) is one of the leading causes of cancer deaths among both men and women in the world. The framework for our understanding of CRC is that a series of mutations and epigenetic changes accumulate slowly over the span of decades, leading to a gain of function in oncogenes and a loss of function in tumour-suppressor genes as healthy tissue is transformed along the adenoma–carcinoma sequence (Fearon and Vogelstein, 1990; Wood et al, 2007). Although inherited genetic susceptibility has a key role in a subset of CRC cases, the vast majority of CRC cases are sporadic and non-inherited. For example, a Western diet, smoking, obesity, diabetes, alcohol consumption, and exposure to carcinogens have been validated as important risk factors for the development of sporadic CRC. However, despite long-standing associations between diet, the microbiome, and CRC (Aries et al, 1969; Hill et al, 1971), only recently have we begun to appreciate the specific mechanisms by which the gut microbiome may influence not only the initiating events of carcinogenesis but also its progression. This review covers recent advances in our understanding of the various roles in which bacteria may initiate or alter the course of CRC and discusses some of the pressing questions in the field.
Introduction to colon structure, biology, and immunology
As our scientific understanding and interest in how the gut microbiota influences complex disease processes such as CRC expands, it is important to first highlight biological differences along the colorectal axis with regards to the composition and function of the microbiota and the cognate inflammatory response.
Biological differences along the colorectal axis in CRC
The entire gastrointestinal tract spans a distance of 25 feet, although the largest burden of microbes resides in the most distal portion of the gut, namely, the colorectum. Features of CRC that differ along the axis of the colon are outlined in Figure 1. Traditionally, the conceptual division between the right and left colon has been located near the splenic flexure, based on developmental biology. However, recent findings by Dejea et al (2014) proposed redefining the right vs left colon based on the overwhelming burden of microbial biofilms proximal to the hepatic flexure. Thus, for the purposes of this review on CRC, we term the right colon to be the region from the caecum through the ascending colon and hepatic flexure and the left colon to contain the remaining distal portions consisting of the transverse colon, splenic flexure, descending and sigmoid colon, and rectum.

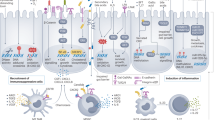
Molecular features of the colon during homeostasis and carcinogenesis. (Top) General biological differences among healthy tissues along the colorectal axis are depicted in blue. (Bottom) Differences in the prevalence of genetic features and biofilms among tumours along the colorectal axis are summarised in orange and red. The dashed line represents the transition between right and left colon based on the overwhelming burden of biofilms in tumours proximal to the hepatic flexure as reported in Dejea et al (2014).
Genetic differences in CRC have been noted along the colon as well, including a high frequency of KRAS mutations specifically in the caecum and a gradient in the prevalence of CpG island methylator phenotypes, microsatellite instability (MSI), and BRAF mutations that is highest in the ascending colon but then gradually decreases as one moves distally along the colonic axis (Yamauchi et al, 2012). Given these major differences in the microbial organisation (e.g., biofilms) and genetic features of CRC as one moves along the colonic axis, one might expect to see similarly divergent changes in the microbiome along this axis. Surprisingly, the composition of the mucosal microbiome is largely consistent within a healthy adult (Eckburg et al, 2005). The faecal microbiome, however, is much richer in diversity than the mucosal microbiome, but, for reasons that yet remain unclear, not all of the mucosal species in a given patient are necessarily detected in stool (Eckburg et al, 2005; Mira-Pascual et al, 2015). Thus stool samples thus far have been poor indicators of the mucosal microbiome. Furthermore, adherent microbes are more likely to be key players in CRC pathogenesis than those passing fleetingly through the gut via the stool, given that adherent microbes are the only organisms that have direct access to the colonic epithelial cells (CECs) for any extended period of time. Thus the prognostic use of stool samples for CRC detection may be limited.
Symbiotic and antimicrobial immune defenses in the colon
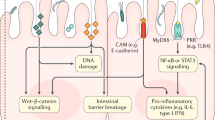
Because all CRCs exhibit excess, subclinical inflammation, it is also important to note the unique characteristics of the gut-associated lymphoid tissue (GALT), as the interplay between the GALT and the gut microbiome likely impacts the inflammation involved in CRC initiation, progression, and treatment. Whereas interaction with a pathogen in the spleen or other central lymphoid organs is rare and triggers an immediate and robust inflammatory response, the gut has evolved to coexist with commensal bacteria as it depends on these microbes for not only digestive health but also for the development of a properly functioning mucosal and systemic immune system. In the colon, a thick, mucus bi-layer serves as the primary innate defense barrier, with some bacteria colonising the thin outer layer but the inner, Muc-2-dominated layer being largely sterile (Johansson et al, 2008). Tonic stimulation of several pattern recognition receptors (PRRs) on hematopoietic cells and intestinal epithelial cells by commensals appears to be instrumental in the maintenance of this innate mucus barrier, as disruption of these PRRs results in a dramatic thinning of the mucus layer and increases susceptibility to colitis in mouse models (Philpott et al, 2014; Wlodarska et al, 2014). Early mutagenic events, including loss of APC and/or activation of β-catenin signalling, may also alter Muc-2 and tight junctional proteins and allow for infiltration of protumorigenic microbial products (Grivennikov et al, 2012). Secreted antimicrobial peptides (AMPs) and IgA serve as additional innate immunity factors in the colon but can also be manipulated by microbial players (Levy et al, 2015; Moon et al, 2015).
Underneath this innate defense barrier lies an adaptive immune compartment dominated by the anti-inflammatory cytokines TGFβ and IL-10, which are largely secreted by epithelial cells, regulatory T cells (Tregs), and stromal cells (Smythies et al, 2005; Smythies et al, 2006). TGFβ, in particular, is critical for a number of different functions, including the induction of class switch recombination in B cells from IgM to secretory IgA (by both T-cell-dependent and -independent pathways), for the induction of and maintenance of a tolerogenic phenotype in dendritic cells, macrophages, and Tregs, and for the induction of specific immunosuppressive pathways, such as indoleamine 2,3-dioxygenase (Smythies et al, 2005; Contractor et al, 2007; Belladonna et al, 2009; Pallotta et al, 2011). Importantly, this suppressive milieu can be overcome by IL-6 in the gut, which in combination with TGFβ triggers differentiation of naive CD4+ T cells into proinflammatory Th17 cells (Hu et al, 2011) that in turn lead the adaptive immune response against invading microbial pathogens.
Despite the critical role of Th17 cells in antimicrobial defense, these cells are thought to be causally related to the chronic, subclinical, unbalanced mucosal inflammation revealed to be of importance to the pathogenesis of colon carcinogenesis in recent years. A Th1-dominated inflammatory state seems to be protective in CRC patients (Tosolini et al, 2011) and boosts antitumoural T-cell responses via the production of IFNγ. In contrast, Th17 (or IL-17 produced by other cell types) has been shown to be a key driver of CRC in multiple intestinal neoplasia (Min) mouse models, including the spontaneous tumorigenesis ApcMin/+ mouse model (Chae et al, 2010) and in ApcMin/+ mice colonised with enterotoxigenic Bacteroides fragilis (ETBF) (Wu et al, 2009). Furthermore, a Th17 signature is associated with a worse prognosis in CRC patients (Tosolini et al, 2011). Thus the balance between Th1/Th17 inflammatory responses may have a key role in patient outcomes.
The Treg/Th17 balance may also be critically important to intestinal homeostasis, as Tregs are one of the pivotal cell types responsible for keeping colon inflammation in check. Paradoxically, two recent reports have shown that Tregs are necessary for early Th17 responses to pathogens, including ETBF in the distal mouse colon of Min mice (Geis et al, 2015) and Candida albicans in the mouse oropharyngeal tract (Pandiyan et al, 2011). Mechanistically, Tregs, which are highly IL-2 dependent but incapable of producing their own, were shown to deplete exogenous IL-2, creating an IL-2-deprived environment that favoured Th17 development at the expense of Th1 (Chen et al, 2011; Geis et al, 2015). In the ETBF model, the depletion of Tregs reduced early microadenoma formation in the colon by limiting Th17 cell differentiation despite the persistence of marked Th1-skewed inflammation. The dependence of Th17 responses on Tregs was only required during early events, showing that not only is the type of inflammation important (Th1 vs Th17) but also the timing (early vs late infection) following challenge with specific pathogens.
Microbial initiators and progressors of tumorigenesis
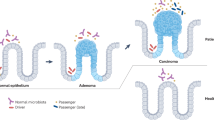
Theories on how the microbiota may contribute to the initiation and progression of CRC can be divided according to evidence for individual bacteria, evidence for aggregate changes in the microbiota as a whole largely through metabolism and diet, and finally the effects of differential microbial organisation as in the case of biofilms. These theories are summarised in Figure 2.

Roles of the microbiota in CRC prevention, initiation, progression, and therapy. Colonic epithelial cells are depicted with the mucus layer (dark yellow) facing the gut lumen. (Left) Bacteria have multiple protective roles against CRC, including production of anti-inflammatory metabolites and regulation of crypt epithelial cell proliferation. Most notably, bacteria have integral roles in the maintenance and repair of the colonic epithelial barrier, by triggering controlled innate immune responses through PRRs on host cells. Disruption of PRR signalling leads to breaches of the epithelial barrier and excessive inflammation that may instead promote tumorigenesis. (Middle) Multiple avenues by which bacteria may initiate or promote CRC tumorigenesis are also depicted, including the direct genotoxicity of specific bacteria, as well as the proinflammatory effects triggered by either specific microbes, a dysbiotic microbiota as a whole, and/or colonic biofilms. (Right) Immune responses to commensals have also been proposed to be essential for the efficacy of multiple chemotherapies and immunotherapies.
The roles of individual microbes in CRC initiation
The carcinogenic potential of individual bacterial species has recently been covered in depth by others, with two major theories emerging for how these bacteria may initiate CRC (Irrazabal et al, 2014; Sears and Garrett, 2014). The first and perhaps most direct is that certain bacteria have direct DNA mutagenesis capabilities and/or interfere with the host DNA repair machinery, as seen with ETBF that express the B. fragilis toxin (BFT), superoxide-producing E. faecalis, and the polyketide synthase (pks)-expressing clade of E. coli. The second is that many of the implicated bacteria share a common ability to enhance Wnt-mediated signalling pathways or other specific proinflammatory pathways that are commonly mutated and/or overexpressed in CRC, as seen in the above three species as well as F. nucleatum. Importantly, however, none of the bacteria have consistently been demonstrated to be enriched in all tumour specimens compared with healthy controls, suggesting that no single organism is likely responsible for all CRC cases. Additionally, all of these mouse models rely on genetically predisposed mice with mutations in tumour-suppressor genes such as Apc or mice that have been administered chemicals to help drive carcinogenesis such as dextran sodium sulphate (DSS), which induces inflammation and disruption of colonic mucosa, and azoxymethane (AOM), a DNA alkylater. Likewise, while germ-free mice have proven invaluable in our understanding of specific pathogens, the absence of commensals hinders development of the immune system and further complicates their relevance to understanding human disease. Thus, while murine studies have demonstrated strong evidence for the procarcinogenic activity of certain bacteria, they have not proven that bacteria can provide the first hit to initiate cancer nor has clear relevance to human disease been established.
The roles of individual microbes in CRC progression
All of the specific microbial mechanisms above that may have a role in CRC initiation likely also have a role in CRC progression. Additionally, observational studies on patient outcomes have given us clues as to which microbes may be associated with CRC progression. For example, a trend towards increased prevalence of BFT toxin (particularly BFT-2) was recently demonstrated in more advanced CRC cases (100%) vs early-stage CRC (72.7%) (Boleij et al, 2015). There is also evidence for an association of fusobacteria with worse prognoses, although the data are more complex. For example, a high level of F. nucleatum DNA in the tumour tissue was associated with increased lymph node metastases (Castellarin et al, 2012) and a worse outcome in CRC patients (Mima et al, 2015a) but was not associated with worse mortality in a separate study (Mima et al, 2015b). Likewise, while the presence of F. nucleatum DNA in tumours was associated with MSI status (Mima et al, 2015a), which is typically associated with better patient outcomes, in another study, F. nucleatum was associated with a decrease in CD3+ T cells within the tumour in CRC patients (Mima et al, 2015b), a feature that is typically associated with MSS status and worse outcomes in patients (Kawakami et al, 2015). These observational studies clearly need to be validated by prospective studies in patients to clarify the role of these bacteria in CRC progression, as these microbes may merely be taking advantage of a compromised host defense/mucus layer, rather than contributing to actual tumorigenesis.
The aggregate microbiota and microbial metabolites in CRC initiation
The gut microbiota as a whole may also alter cancer susceptibility, largely via interactions with host diet and metabolism. Louis et al (2014) recently reviewed in depth the potential procarcinogenic roles of bacterial products, including toxins, hydrogen sulphide (H2S), polyamines, secondary bile acids, and reactive oxygen species (ROS). Elevations in many of these factors have been linked to diets rich in saturated fats and sugars and low in fibre. For example, high levels of saturated fats leads to increased production of bile acids by the liver, and these, in turn, are metabolised by bacteria into secondary bile acids, such as lithocholic and deoxycholic acid, which have proinflammatory and procarcinogenic properties (Sears and Garrett, 2014). In addition, these high fat/high sugar/low fibre diets may also result in a less diverse microbiota overall, which is also associated with an increased risk of CRC.
Conversely, the vast metabolomic potential of the gut microbiota can also be harnessed to prevent CRC. For example, short chain fatty acids (SCFA) have potent anti-inflammatory properties, including the promotion of colonic Treg expansion (Arpaia et al, 2013; Smith et al, 2013), and several studies have shown a protective effect of alternative bacterially produced bile acid products such as urodeoxycholic acid, as reviewed by Louis et al (2014). Most recently, the microbial metabolites taurine, histamine, and spermine were found to alter host inflammasome processes in mice, either positively or negatively, and may represent other easily modifiable targets (Levy et al, 2015).
The aggregate microbiota in CRC progression
It is also becoming increasingly clear that an aberrant microbiota, also known as dysbiosis, may have an important role in tumour progression. Although dysbiosis may take many forms and a consistent signature in all CRC patients is unlikely to be unveiled, the protumorigenic mechanism of dysbiosis appears to involve enhanced microbial stimulation of the Toll-like receptor (TLR)/MyD88 pathway, as knockout of MyD88 in both the ApcMin/+ and AOM-IL10−/− colitis-associated cancer models abrogates tumorigenesis, as does treatment with antibiotics or the use of germ-free mice (Rakoff-Nahoum and Medzhitov, 2007; Uronis et al, 2009; Song et al, 2014). Most recently, this elevated TLR/MyD88 signalling was found to enhance calcineurin-mediated activation of NFAT signalling that, in turn, enhanced intestinal stem cell proliferation (Peuker et al, 2016), providing a probable mechanism of action.
Biofilms as CRC initiators
An emerging concept in the role of the microbiota in CRC initiation is that it is not just the composition of the microbiota that is important but that complex community structures such as bacterial biofilms can also dramatically alter both host and microbial function in CRC. Bacterial biofilms along the colorectal axis have been shown to be present in approximately 15% of healthy patients upon colonoscopy (Swidsinski et al, 2009) but were recently shown to be a feature of nearly 100% of right-sided CRC (Dejea et al, 2014). Biofilms are massive bacterial invasions of the mucus layer, are encased in a polymeric matrix, and often consist of many different types of bacteria and even fungi. They are commonly found in the environment but have also been reported to be a feature of dozens of problematic medical infections, particularly those involving artificial medical devices such as catheters as well as with living tissue as in the case of endocarditis (Costerton et al, 1999). How and why they form in the colon, where they involve invasion of the protective mucus layer adjacent to the CECs, is largely unknown. However, one hypothesis is that biofilm formation is, in part, a defense mechanism of the microbiota against the host. For example, biofilms may provide a degree of stability to the gut microbiota in the face of the rapid turnover of the gut mucus layer. Biofilms may also protect luminal microbiota from host factors such as IgA, AMPs, and even antibiotics, which may be inactivated by or do not diffuse well through the biofilm matrix (Costerton et al, 1999).
The bacterial species that make up colorectal mucosal biofilms are clearly a small subset of what is found in the faecal microbiota, with only approximately 100 species present in colon biofilms compared with the nearly 1000 species detected in stool. The class Bacteroidetes (encompassing Bacteroides and Prevotella) is typically the predominant constituent of the biofilms, although proteobacteria (including enterobacteria such as E. coli), Clostridium spp. (e.g., eubacterium), actinobacteria (e.g., bifidobacteria), bacilli (including the putatively protective lactobacilli), and fusobacteria are also commonly detected (Macfarlane et al, 2004; Swidsinski et al, 2005; Dejea et al, 2014). Although colon biofilms are observed sporadically in healthy controls (∼15%), those biofilms tend to be much thinner and more disjointed as compared with the thick, continuous biofilms seen in inflammatory bowel disease and CRC patients (Swidsinski et al, 2005; Dejea et al, 2014). Importantly, 100% of the CRC patients examined to date who had biofilm-positive tumours also had biofilms on their paired, normal tissue. Tissues underlying the biofilms in CRC patients and in healthy individuals were demonstrated to have decreased or altered E-cadherin, enhanced IL-6 and Ki67 expression (a measure of CEC proliferation) alongside in the tumour hosts, phospho-Stat3, suggesting that the biofilms were eliciting a procarcinogenic effect (Dejea et al, 2014). This is perhaps not surprising, given that a feature of biofilms is the invasion of the mucus layer by bacteria, which allows for bacteria to directly interact with colonocytes and potentially trigger inflammatory responses as well as oncogenic mechanisms in the CEC layer. Conversely, CECs and/or leukocytes can be seen invading the biofilms, again suggesting that the biofilms are immunogenic and involve highly dynamic bacterial:host interactions (Dejea et al, 2014).
Although studies on the specific bacterial mechanisms rendering biofilms pro-oncogenic have only just begun, the polymicrobial make-up of the biofilms suggests a community mechanism is at play rather than one driven by individual bacteria. For example, biofilm-positive tissues from both the tumour region and histologically normal colon tissue display enrichment of bacterially synthesised acetylated polyamine N1N12-diacetylspermine compared with biofilm-negative tumours and healthy controls (Johnson et al, 2015). Polyamine synthesis is a near-universal feature of both bacterial and host cells and is a critical modulator of a multitude of both bacterial and host processes. Furthermore, polyamine synthesis is required for biofilm formation in at least one bacteria, Yersinia pestis (Di Martino et al, 2013). In CRC patients, N1N12-diacetylspermine was found to be highly localised to the luminal edge of the mucosa, consistent with bacterial production of this polyamine. Additional data suggested that both the host colon mucosa and the bacterial biofilms contributed to the production of N1N12-diacetylspermine. Whether this polyamine is required for the formation of biofilms in CRC patients remains to be examined, but it may be an important contributor to the increased cell proliferation and pro-oncogenic state seen in the biofilm-positive tissues.
Protective roles of the microbiota in CRC
PRRs, inflammasomes, and the aggregate microbiota in CRC
Another emerging hypothesis is that loss-of-function mutations in PRRs may enhance CRC tumorigenesis by affecting the detection of pathogen- or cellular damage-associated molecular patterns (DAMPs). There are five major classes of PRRs: TLRs, C-type lectins (CTLs; e.g., DC-SIGN), RIG-I-like receptors, Absent in Melanoma 2 (AIM2)-like receptors (also known as the HIN-200 family of receptors), and NOD-like receptors (NLRs). These receptors detect diverse foreign antigens and trigger similarly diverse responses, including NFκB activation, MAPK activation, and production of proinflammatory cytokines, that in turn modulate downstream adaptive immune responses. Several PRRs (e.g., AIM2 and the NLR family members NLRP1, NLRP3, NLRC4, and NLRP6) have also been shown to be sensor components of inflammasome complexes, cellular machinery designed to churn out copious amounts of mature caspase-1 that in turn proteolytically activates the proinflammatory cytokines IL-1β and IL-18. Cytosolic PRRs have also been shown to detect DAMPs (e.g., extracellular ATP) (Mariathasan et al, 2006) and may be instrumental in priming antitumoural T-cell responses, highlighting the diverse nature of the PRR family.
Inherited mutations in these PRR families have been linked to a wide variety of autoimmune and inflammatory disorders. Given the key role of inflammation in CRC, it is not surprising that these genes are commonly mutated in CRC as well and in some cases have also been associated with CRC progression (Nonaka et al, 2011; Ungerback et al, 2012; Lu et al, 2013; Luddy et al, 2014; Philpott et al, 2014; Liu et al, 2015; Sharma and Jha, 2015). For example, loss-of-function mutations in the cytoplasmic PRR NOD2 is one of the strongest, heritable genetic links to Crohn’s disease and has also been associated with an increased risk of CRC in patients (Tian et al, 2010). Although NOD1−/− and NOD2−/− deficiencies are not sufficient to induce spontaneous colitis in mice (Natividad et al, 2012), NOD2−/− in combination with DSS resulted in a dysbiosis-mediated susceptibility to tumorigenesis that was transmissible via the microbiota and triggered tumours in wild-type mice also treated with DSS (Couturier-Maillard et al, 2013). Similarly, mutations in NLRP6 and AIM2, which are the sensor PRR molecules of two separate inflammasome complexes, have also been shown to exacerbate chemically induced (DSS/AOM) tumorigenesis in mice via a microbially mediated mechanism that is transmissible to wild-type mice also treated with DSS and AOM (Hu et al, 2013; Man et al, 2015). Mutations in AIM2, similarly to NOD2, are frequently found in CRC and are associated with a worse prognosis (Dihlmann et al, 2014). Mutations in another inflammasome PRR sensor, NLRP3, are associated with an increased risk of developing inflammatory disorders such as Crohn's disease in patients as well as DSS-associated colitis in mice (Schoultz et al, 2009), but direct links between NLRP3 and CRC initiation in patients have yet to be validated nor has the role of the microbiota in NLRP3-mediated attenuation of colitis been demonstrated.
Despite the consistent role of the microbiota in transmitting susceptibility to CRC in many of the PRR knockout studies above, the actual microbial players suggested to be involved in the dysbiosis were quite different. For example, NOD2−/− dysbiosis was associated with an overall loss of diversity as well as increased Bacteroides and a decrease in genera of the phylum Firmicutes (Butyrivibrio and Lachnobacterium); the NLRP6−/− dysbiosis was associated with an increase in Prevotella and TM7 and reductions in Lactobacilli and Firmicutes; finally, the AIM2−/− dysbiosis was associated with a decrease in Prevotella and an increase in Akkermansia mucinophila in one study but was recently associated with an increase in E. coli in another study (Hu et al, 2015). These studies suggest that more than one colitogenic phenotype may exist following PRR mutagenesis, which is perhaps not surprising given the diverse nature of the PRRs and the ligands that they recognise.
Additionally, our knowledge of how PRRs help regulate gut homeostasis in other ways continues to increase. For example, tonic sensing of commensals by NOD1 and NOD2 may be critical for mucus and AMP generation (Philpott et al, 2014). Similarly, NLRP6 helps promote goblet cell autophagy and mucus secretion (Wlodarska et al, 2014), while also regulating crypt epithelial cell proliferation (Normand et al, 2011). Thus, beyond protection against any individual pathogen, these PRRs have critical roles in maintaining general epithelial barrier integrity, heightening their importance to CRC. Clearly, further studies are needed in order to clarify how and why mutations in particular PRRs trigger dysbiosis, and whether mutations in PRRs in patients increase the risk of initial transforming events in CRC or are merely associated with CRC progression.
Microbial modulation of sensitivity to treatment
Finally, the microbiota may also influence the progression of a wide array of cancers in a positive manner by providing a critical boost to antitumoural T-cell responses following certain types of chemotherapy and immunotherapy. For example, treatment with cyclophosphamide was found to trigger the translocation of several Gram-positive bacterial species to the secondary lymphoid organs; surprisingly, this infiltration was required for the promotion of antitumoural Th1 and Th17 responses via a MyD88-dependent pathway, as germ-free mice and mice co-treated with either broad-spectrum antibiotics or antibiotics specific for Gram-positive bacteria in addition to the cyclophosphamide displayed larger tumours than mice with an intact microbiota (Viaud et al, 2013). Likewise, the antitumoural response following treatment with the ROS-inducing chemotherapeutic agent oxaliplatin was similarly reliant on functional MyD88 signalling triggered by microbes, as germ-free mice, mice treated with antibiotics, and MyD88−/− mice did not demonstrate successful tumour regression (Iida et al, 2013). Most recently, efficacy of the immunotherapy checkpoint blockade inhibitors anti-PDL1 and anti-CTLA-4 were found to be reliant on commensal bacteria (bifidobacteria in the former, and Bacteroides, among others, in the latter) (Sivan et al, 2015; Vetizou et al, 2015), though whether these antitumoural responses also relied on MyD88 signalling remain unclear. A critical point of all of these studies is that antibiotics, which are often life saving for cancer patients whose immune systems are suppressed by chemotherapy, should be administered with care due to the dependence of various cancer drugs on a functioning microbiota as well as to avoid the adverse consequences of antibiotic-resistant bacteria.
Conclusion
Overall, the interactions between the gut microbiota and CRC are complex and involve both positive and negative factors. Pathogenic roles of the microbiota involve aspects as varied as direct genotoxic activity, alterations of dietary metabolism, and triggering of proinflammatory immune responses. Alternatively, the gut microbiota may also have a protective role against CRC through maintenance of the epithelial/mucus barrier (largely through stimulation of PRRs) and production of protective metabolites, such as SCFAs. The potential for the microbiota to enhance antitumoural T-cell responses following chemotherapy and immunotherapy has added yet another exciting dimension to the interplay between the host and the microbiota. Emerging areas of research include delineating how different types of inflammation affect CRC, the role of biofilms and other complex bacterial structures in predicting and/or causing CRC, and the myriad roles of PRR sensors in maintaining innate defenses against not only microbes but also potentially early transformed cells. Prospective studies in patients will be absolutely critical in the validation of the proposed theories. All of these areas may provide novel targets for therapy or provide much-needed biomarkers of disease, as colonoscopy is unlikely attainable for all in resource-limited settings where CRC appears to be increasing in incidence.
References
Aries V, Crowther JS, Drasar BS, Hill MJ, Williams RE (1969) Bacteria and the aetiology of cancer of the large bowel. Gut 10: 334–335.
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504: 451–455.
Belladonna ML, Orabona C, Grohmann U, Puccetti P (2009) TGF-beta and kynurenines as the key to infectious tolerance. Trends Mol Med 15: 41–49.
Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC, Platz EA, Pardoll DM, Sears CL (2015) The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis 60: 208–215.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA (2012) Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22: 299–306.
Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL (2010) Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci USA 107: 5540–5544.
Chen Y, Haines CJ, Gutcher I, Hochweller K, Blumenschein WM, McClanahan T, Hämmerling G, Li MO, Cua DJ, McGeachy MJ (2011) Foxp3(+) regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity 34: 409–421.
Contractor N, Louten J, Kim L, Biron CA, Kelsall BL (2007) Cutting edge: Peyer's patch plasmacytoid dendritic cells (pDCs) produce low levels of type I interferons: possible role for IL-10, TGFbeta, and prostaglandin E2 in conditioning a unique mucosal pDC phenotype. J Immunol 179: 2690–2694.
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284: 1318–1322.
Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, Gaillot O, Schreiber S, Lemoine Y, Ryffel B, Hot D, Nùñez G, Chen G, Rosenstiel P, Chamaillard M (2013) NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123: 700–711.
Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M, Stein E, Vadivelu J, Roslani AC, Malik AA, Wanyiri JW, Goh KL, Thevambiga I, Fu K, Wan F, Llosa N, Housseau F, Romans K, Wu X, McAllister FM, Wu S, Vogelstein B, Kinzler KW, Pardoll DM, Sears CL (2014) Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci USA 111: 18321–18326.
Di Martino ML, Campilongo R, Casalino M, Micheli G, Colonna B, Prosseda G (2013) Polyamines: emerging players in bacteria-host interactions. Int J Med Microbiol 303: 484–491.
Dihlmann S, Tao S, Echterdiek F, Herpel E, Jansen L, Chang-Claude J, Brenner H, Hoffmeister M, Kloor M (2014) Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int J Cancer 135: 2387–2396.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA (2005) Diversity of the human intestinal microbial flora. Science 308: 1635–1638.
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759–767.
Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Housseau F (2015) Regulatory T-cell response to enterotoxigenic Bacteroides fragilis colonization triggers IL17-dependent colon carcinogenesis. Cancer Discov 5: 1098–1109.
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M (2012) Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491: 254–258.
Hill MJ, Drasar BS, Hawksworth G, Aries V, Crowther JS, Williams RE (1971) Bacteria and aetiology of cancer of large bowel. Lancet 1: 95–100.
Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, Jin C, Wunderlich C, Wunderlich T, Eisenbarth SC, Flavell RA (2013) Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci USA 110: 9862–9867.
Hu S, Peng L, Kwak YT, Tekippe EM, Pasare C, Malter JS, Hooper LV, Zaki MH (2015) The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep 13: 1922–1936.
Hu W, Troutman TD, Edukulla R, Pasare C (2011) Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity 35: 1010–1022.
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai RM, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, Trinchieri G, Goldszmid RS (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342: 967–970.
Irrazabal T, Belcheva A, Girardin SE, Martin A, Philpott DJ (2014) The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell 54: 309–320.
Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC (2008) The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105: 15064–15069.
Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, Ivanisevic J, Cho K, Wick EC, Hechenbleikner EM, Uritboonthai W, Goetz L, Casero RA Jr, Pardoll DM, White JR, Patti GJ, Sears CL, Siuzdak G (2015) Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab 21: 891–897.
Kawakami H, Zaanan A, Sinicrope FA (2015) Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol 16: 30.
Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, Pevsner-Fischer M, Shapiro H, Christ A, Harmelin A, Halpern Z, Latz E, Flavell RA, Amit I, Segal E, Elinav E (2015) Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163: 1428–1443.
Liu R, Truax AD, Chen L, Hu P, Li Z, Chen J, Song C, Chen L, Ting JP (2015) Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget 6: 33456–33469.
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12: 661–672.
Lu S, Bevier M, Huhn S, Sainz J, Lascorz J, Pardini B, Naccarati A, Vodickova L, Novotny J, Hemminki K, Vodicka P, Försti A (2013) Genetic variants in C-type lectin genes are associated with colorectal cancer susceptibility and clinical outcome. Int J Cancer 133: 2325–2333.
Luddy KA, Robertson-Tessi M, Tafreshi NK, Soliman H, Morse DL (2014) The role of toll-like receptors in colorectal cancer progression: evidence for epithelial to leucocytic transition. Front Immunol 5: 429.
Macfarlane S, Furrie E, Cummings JH, Macfarlane GT (2004) Chemotaxonomic analysis of bacterial populations colonizing the rectal mucosa in patients with ulcerative colitis. Clin Infect Dis 38: 1690–1699.
Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, Sharma D, Li L, Malireddi RK, Gurung P, Neale G, Olsen SR, Carter RA, McGoldrick DJ, Wu G, Finkelstein D, Vogel P, Gilbertson RJ, Kanneganti TD (2015) Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 162: 45–58.
Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232.
Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song M, Kostic AD, Giannakis M, Bullman S, Milner DA, Baba H, Giovannucci EL, Garraway LA, Freeman GJ, Dranoff G, Garrett WS, Huttenhower C, Meyerson M, Meyerhardt JA, Chan AT, Fuchs CS, Ogino S (2015a) Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut; e-pub ahead of print 26 August 2016; doi:10.1136/gutjnl-2015-310101.
Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, Kim SA, Masuda A, Nowak JA, Nosho K, Kostic AD, Giannakis M, Watanabe H, Bullman S, Milner DA, Harris CC, Giovannucci E, Garraway LA, Freeman GJ, Dranoff G, Chan AT, Garrett WS, Huttenhower C, Fuchs CS, Ogino S (2015b) Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol 1: 653–661.
Mira-Pascual L, Cabrera-Rubio R, Ocon S, Costales P, Parra A, Suarez A, Moris F, Rodrigo L, Mira A, Collado MC (2015) Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J Gastroenterol 50: 167–179.
Moon C, Baldridge MT, Wallace MA, Burnham CA, Virgin HW, Stappenbeck TS (2015) Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521: 90–93.
Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, Philpott D, Garcia Rodenas CL, McCoy KD, Verdu EF (2012) Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−; Nod2−/− mice. Inflamm Bowel Dis 18: 1434–1446.
Nonaka M, Ma BY, Imaeda H, Kawabe K, Kawasaki N, Hodohara K, Kawasaki N, Andoh A, Fujiyama Y, Kawasaki T (2011) Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) recognizes a novel ligand, Mac-2-binding protein, characteristically expressed on human colorectal carcinomas. J Biol Chem 286: 22403–22413.
Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M (2011) Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA 108: 9601–9606.
Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, Grohmann U (2011) Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol 12: 870–878.
Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, Hernandez-Santos N, Edgerton M, Gaffen SL, Lenardo MJ (2011) CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34: 422–434.
Peuker K, Muff S, Wang J, Kunzel S, Bosse E, Zeissig Y, Luzzi G, Basic M, Strigli A, Ulbricht A, Kaser A, Arlt A, Chavakis T, van den Brink GR, Schafmayer C, Egberts JH, Becker T, Bianchi ME, Bleich A, Röcken C, Hampe J, Schreiber S, Baines JF, Blumberg RS, Zeissig S (2016) Epithelial calcineurin controls microbiota-dependent intestinal tumor development. Nat Med.
Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE (2014) NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14: 9–23.
Rakoff-Nahoum S, Medzhitov R (2007) Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317: 124–127.
Schoultz I, Verma D, Halfvarsson J, Torkvist L, Fredrikson M, Sjoqvist U, Lördal M, Tysk C, Lerm M, Söderkvist P, Söderholm JD (2009) Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn's disease in Swedish men. Am J Gastroenterol 104: 1180–1188.
Sears CL, Garrett WS (2014) Microbes, microbiota, and colon cancer. Cell Host Microbe 15: 317–328.
Sharma N, Jha S (2015) NLR-regulated pathways in cancer: opportunities and obstacles for therapeutic interventions. Cell Mol Life Sci 73: 1741–1764.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350: 1084–1089.
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341: 569–573.
Smythies LE, Maheshwari A, Clements R, Eckhoff D, Novak L, Vu HL, Mosteller-Barnum LM, Sellers M, Smith PD (2006) Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol 80: 492–499.
Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD (2005) Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 115: 66–75.
Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N, Shi Y, Iwakura Y, Qian Y (2014) Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40: 140–152.
Swidsinski A, Loening-Baucke V, Herber A (2009) Mucosal flora in Crohn's disease and ulcerative colitis—an overview. J Physiol Pharmacol 60 (Suppl 6): 61–71.
Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H (2005) Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43: 3380–3389.
Tian Y, Li Y, Hu Z, Wang D, Sun X, Ren C (2010) Differential effects of NOD2 polymorphisms on colorectal cancer risk: a meta-analysis. Int J Colorectal Dis 25: 161–168.
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagès F, Galon J (2011) Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 71: 1263–1271.
Ungerback J, Belenki D, Jawad ul-Hassan A, Fredrikson M, Fransen K, Elander N, Verma D, Söderkvist P (2012) Genetic variation and alterations of genes involved in NFkappaB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 33: 2126–2134.
Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C (2009) Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One 4: e6026.
Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Bérard M, Nigou J, Opolon P, Eggermont A, Woerther PL, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350: 1079–1084.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther PL, Eberl G, Bérard M, Ecobichon C, Clermont D, Bizet C, Gaboriau-Routhiau V, Cerf-Bensussan N, Opolon P, Yessaad N, Vivier E, Ryffel B, Elson CO, Doré J, Kroemer G, Lepage P, Boneca IG, Ghiringhelli F, Zitvogel L (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342: 971–976.
Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, Elinav E, Finlay BB, Flavell RA (2014) NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156: 1045–1059.
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B (2007) The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113.
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15: 1016–1022.
Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, Liao X, Waldron L, Hoshida Y, Huttenhower C, Chan AT, Giovannucci E, Fuchs C, Ogino S (2012) Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 61: 847–854.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Drewes, J., Housseau, F. & Sears, C. Sporadic colorectal cancer: microbial contributors to disease prevention, development and therapy. Br J Cancer 115, 273–280 (2016). https://doi.org/10.1038/bjc.2016.189
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.189
Keywords
This article is cited by
-
High expression of CCDC69 is correlated with immunotherapy response and protective effects on breast cancer
BMC Cancer (2023)
-
Early life Lactobacillus rhamnosus GG colonisation inhibits intestinal tumour formation
British Journal of Cancer (2022)
-
Bee pollen in zebrafish diet affects intestinal microbiota composition and skin cutaneous melanoma development
Scientific Reports (2022)
-
Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies
Nature Reviews Gastroenterology & Hepatology (2019)
-
Mutation profiling of cancer drivers in Brazilian colorectal cancer
Scientific Reports (2019)
