
Abstract
Meconium ileus is most often associated with mutations in the CFTR gene; however recently, mutations in GUCY2C in the Bedouin population have also been shown to result in this phenotype. This gene codes for an intestinal transmembrane receptor that generates cyclic GMP, which activates cystic fibrosis transmembrane receptor. We report a third family that supports the association of variants in the GUCY2C gene with meconium ileus (MI). A Lebanese kindred was studied and individuals affected with MI had either homozygous or compound heterozygous variants in GUCY2C. The earliest manifestation of the affected individuals was the presence of second trimester fetal echogenic bowel, thus resulting in the expansion of the differential diagnosis of this ultrasound finding.
Similar content being viewed by others


Introduction
The detection of echogenic bowel during a second trimester fetal ultrasound may be the first indication that the developing fetus has cystic fibrosis due to the abnormal consistency of the meconium in the intestine. However, this ultrasound finding could also indicate that the fetus has trisomy 21 or a congenital infection. Alternatively, this finding may be associated with no sequelae to the child. If the neonate presents with meconium ileus (MI), then the likelihood that the baby has cystic fibrosis is extremely high (~80%).1
In 2012, Romi et al2 reported two unrelated consanguineous Bedouin kindreds, who presented with MI, without pulmonary or pancreatic features of cystic fibrosis. Mutations were identified in the GUCY2C gene, which encodes guanylyl cyclase 2C, a regulator of ion and fluid balance that is predominantly localized at the apical brush border membrane of the intestinal epithelium.3, 4 Binding of ligands to GUCY2C catalyzes the synthesis of cyclic GMP, which in turn causes activation of PKGII that leads to phosphorylation and subsequent opening of the cystic fibrosis transmembrane receptor.5 The first family was found to have a homozygous missense mutation (p.Asp367Gly) in the GUCY2C gene, in a highly conserved area in the extracellular ligand binding domain. Romi et al2 were able to demonstrate experimentally that this amino-acid substitution leads to a reduction in ligand binding and a decrease in the guanylate cyclase activity. Decreased penetrance of the postnatal MI phenotype was evident, as there were only 11 affected individuals with MI out of 15 who were found to have the homozygous genotype. One individual had ultrasound evidence late in pregnancy of MI, but was able to pass stool unassisted at birth. The second reported family had a single affected individual who had severe MI, requiring surgery. This individual had a homozygous truncating mutation (p.Asn757Lysfs*2) that removes the guanylate cyclase catalytic domain.
Here we report a third family, with two different variants in the GUCY2C gene, segregating with an increased risk of MI.
Clinical description
The proband (IV-2) (Figure 1) first came to medical attention at 18 weeks gestational age. His mother had undergone a detailed fetal ultrasound, as her first pregnancy had been associated with multiple congenital anomalies. The parents were of Lebanese background, had unremarkable medical histories and were first cousins. No fetal anomalies were detected, but the fetal bowel was echogenic. The parents underwent screening for cystic fibrosis, and the CFTR gene was analyzed for 37 common mutations, with none being detected. Maternal serology for congenital viral infections was negative. Maternal serum screening for trisomy 21 was likewise unremarkable (1/14 000 risk). The proband was born at 39 weeks gestation. At 3 days of age, the meconium had not passed. He underwent surgical bowel cleansing after the diagnosis of MI. Sequencing of the entire CFTR gene was performed on the proband, with no detection of mutations. A sweat chloride test was likewise normal. He was otherwise well grown, and at 8 years of age is healthy and developmentally normal.

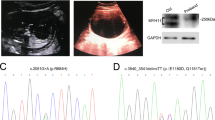
Pedigree of the family showing individuals affected with MI (solid circle/squares) and sequencing results of the proband and his first-degree relatives (WT—wild type/reference sequence). The non-penetrant individual is indicated by the gray circle.
Shortly after the proband’s diagnosis of MI, a paternal cousin (III-3) was also diagnosed with MI that needed surgical assistance to clear the meconium. Both her parents are Lebanese, with no known links of consanguinity with her father’s family to that of the proband’s. Her sweat chloride test and sequencing of the CFTR gene were normal.
The parents of the proband underwent a third pregnancy that was unremarkable. In the fourth pregnancy (IV-4), they came to medical attention again when echogenic bowel was noted at 19 weeks gestational age. Repeat serology for congenital infections was unremarkable, as was the maternal serum screen for trisomy 21. After delivery, MI was diagnosed, which cleared with hydration measures. At 4 years of age, he is healthy and developing well. The family is not aware of other family members with similar presentations.
Methods
The family was studied as a part of the Finding of Rare Disease Genes Canada project. Informed consent was obtained from all participating individuals according to the Declaration of Helsinki and the Research Ethics Boards of the Children’s Hospital of Eastern Ontario approved the study. We used standard methods to isolate genomic DNA from peripheral blood and saliva from the family members.
Shortly after recruitment to the Finding of Rare Disease Genes study for whole exome sequencing, the paper by Romi et al2 was published. As the GUCY2C gene was thought to be a likely candidate for this family, all exons of the gene were analyzed using Sanger sequencing under standard conditions (available upon request). Meanwhile, one affected brother (IV-4) had undergone whole exome sequencing. Exome capture and high-throughput sequencing was performed at McGill University and Genome Quebec Innovation Centre (Montréal, Canada). Target enrichment was achieved using the Agilent SureSelect 50 Mb (V3) All Exon Kit and followed by sequencing (Illumina HiSeq), generating >14 Gbp of 100 base pair, paired-end reads per sample. Read alignment, variant calling, and annotation were done as outlined for previous Finding of Rare Disease Genes projects6 with a pipeline based on BWA, Picard, Annovar, and custom annotation scripts. Variants were compared with those previously seen in dbSNP132, the 1000 genomes data set (2012/04 release), or the 6500 NHLBI exomes (Exome Variant Server, NHLBI GO Exome Sequencing Project, Seattle, WA (data downloaded 2012-10-03), as well as in approximately 1000 exomes previously sequenced at the McGill University and Genome Quebec Innovation Centre. The variants have been submitted in the Leiden Open Variation Database (www.lovd.nl/GUCY2C with the identifier 00019772).
Results
Sanger sequencing in the family demonstrated two different rare variants in the GUCY2C gene (NM_004963.3): p.(Ala670Thr) (c.2008G>A) and p.(Cys928Arg) (c.2782T>C) (Figure 1). Neither variant was present in HapMap, nor the 1000 genomes databases, and whole exome sequencing did not suggest any other candidate genes. The proband was homozygous for the p.(Cys928Arg) variant, whereas his affected younger sibling and unaffected mother were compound heterozygous, p.[(Ala670Thr)];[(Cys928Arg)]. The middle brother (IV-3) and the father were heterozygous for the p.(Cys928Arg) variant. DNA was not available from the other family members.
Both variants occur in highly conserved regions of the protein (Table 1) and are predicted to be damaging by Polyphen2 (p.Ala670Thr: 0.746 and p.Cys928Arg: 1.00). The p.Ala670Thr variant results in the substitution of a hydrophobic amino acid to a hydrophilic residue in a kinase homology domain, likely resulting in a conformational change of the protein. The p.Cys928Arg variant results in the substitution of an amino-acid residue that creates disulfide bonds to one that is charged and hydrophilic within the catalytic domain of the GUCY2C protein. The catalytic domain is only functional when in dimer form, thus the removal of a cysteine amino acid may cause a loss of function mutation resulting in decreased production of cyclic GMP and concomitant reduction in cystic fibrosis transmembrane receptor opening and increased risk for MI.
Discussion
This is the third family and the second report of mutations in the GUCY2C gene being associated with MI. This family segregates two different mutations in this gene. Homozygosity for p.(Cys928Arg) in the proband is associated with a more significant phenotype in this family with the MI requiring surgical intervention. Those with the compound heterozygous genotype, had no, or easier to manage manifestations suggesting decreased penetrance and also variable expressivity. Romi et al2 had also observed decreased penetrance in their family 1, who had the mutation in the extracellular ligand binding domain. Although based on a small sample, it appears that there is a genotype/phenotype correlation with those individuals having two variants in the catalytic domain (Romi et al2 family 2 and our proband) presenting with a more severe MI course. Further families will need to be studied to assess whether this observation holds true.
In contrast to MI, an autosomal dominant familial diarrhea syndrome has been reported in a Norwegian family due to a change in the GUCY2C gene.7 This was found to be because of a missense mutation in the catalytic domain (c.2519G>T: p.Ser840Ile), resulting in a gain of function. The authors presented functional analysis of the mutation in cell culture demonstrating increased ligand-mediated activation of guanylate cyclase C with subsequent intracellular accumulation of cyclic GMP. As demonstrated in Table 2, only five changes in the GUCY2C gene have been reported to date, including this current family. However, we expect that there are likely more.
The presence of echogenic bowel on fetal ultrasound in the second trimester was the first clinical indication in this family that the younger sibling (IV-4) could present with MI as a neonate. Fortunately, MI is a time-limited condition and likely related to the timing of expression of GUCY2C, which is most highly expressed in the newborn period8 and there does not appear to be any pulmonary or pancreatic complications. The fact that this is a self-limited condition may explain why there have not been other reports of GUCY2C-associated MI. For members of this extended family, how we counsel on the differential diagnosis of a prenatal presentation of fetal echogenic bowel needs to be expanded to include GUCY2C-associated MI.
As was suggested by Romi et al,2 loss of function mutations in the GUCY2C gene may provide protection during gastrointestinal infections in young individuals due to the retention of fluids. However, further work would need to be done to know if such variants are indeed enriched in either the Lebanese or Bedouin populations.
References
Fakoury K, Durie PR, Levison H, Canny GJ : Meconium ileus in the absence of cystic fibrosis. Arch Dis Child 1992; 67: 1204–1206.
Romi H, Cohen I, Landau D et al: Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am J Hum Genet 2012; 90: 893–899.
Vaandrager AB : Structure and function of the heat-stable enterotoxin receptor/guanylyl cyclase C. Mol Cell Biochem 2002; 230: 73–83.
Krause WJ, Cullingford GL, Freeman RH et al: Distribution of heat-stable enterotoxin/guanylin receptors in the intestinal tract of man and other mammals. J Anat 1994; 184: 407–417.
Lucas KA, Pitari GM, Kazerounian S et al: Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev 2000; 52: 375–414.
Srour M, Hamdan FF, Schwartzentruber JA et al: Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet 2012; 49: 636–641.
Fickerstrand T, Arshad N, Haukanes BI et al: Familial diarrhea syndrome casued by an activating GUCY2C mutation. N Engl J Med 2012; 366: 1586–1595.
Cohen MB, Guarino A, Shykla R, Giannella RA : Age-related differences in receptors for Escherichia coli heat-stable enterotoxin in the small and large intestine of children. Gastroenterology 1988; 94: 367–273.
Acknowledgements
We would like to thank the family for their participation and assistance. Research was supported through the FORGE Canada Consortium with funding provided by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research (CIHR) and the Ontario Genomics Institute (OGI-049).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Smith, A., Bulman, D., Goldsmith, C. et al. Meconium ileus in a Lebanese family secondary to mutations in the GUCY2C gene. Eur J Hum Genet 23, 990–992 (2015). https://doi.org/10.1038/ejhg.2014.236
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.236
