
Abstract
Following initial successes in melanoma treatment, immunotherapy has rapidly become established as a major treatment modality for multiple types of solid cancers, including a subset of colorectal cancers (CRCs). Two programmed cell death 1 (PD1)-blocking antibodies, pembrolizumab and nivolumab, have shown efficacy in patients with metastatic CRC that is mismatch-repair-deficient and microsatellite instability-high (dMMR–MSI-H), and have been granted accelerated FDA approval. In contrast to most other treatments for metastatic cancer, immunotherapy achieves long-term durable remission in a subset of patients, highlighting the tremendous promise of immunotherapy in treating dMMR–MSI-H metastatic CRC. Here, we review the clinical development of immune checkpoint inhibition in CRC leading to regulatory approvals for the treatment of dMMR–MSI-H CRC. We focus on new advances in expanding the efficacy of immunotherapy to early-stage CRC and CRC that is mismatch-repair-proficient and has low microsatellite instability (pMMR–MSI-L) and discuss emerging approaches for targeting the immune microenvironment, which might complement immune checkpoint inhibition.
Key points
-
Colorectal cancer (CRC) can be categorized into tumours that are mismatch-repair-deficient or have high levels of microsatellite instability (dMMR–MSI-H; ~15%) and mismatch-repair-proficient or microsatellite instability-low tumours (pMMR–MSI-L; ~85%).
-
dMMR–MSI-H CRC is associated with a high tumour mutation burden and immune cell infiltration.
-
Immune checkpoint inhibitor (ICI) treatment, specifically with monoclonal antibodies targeting programmed cell death 1 (PD1) and cytotoxic T lymphocyte antigen 4 (CTLA4), results in improved survival in metastatic dMMR–MSI-H CRC, but pMMR–MSI-L CRC is largely unresponsive to current ICIs.
-
The FDA has granted accelerated approval to the anti-PD1 antibodies pembrolizumab and nivolumab and to the combination of nivolumab with the anti-CTLA4 antibody ipilimumab for treatment of refractory dMMR–MSI-H CRC.
-
Clinical evaluation of ICIs in first-line metastatic, adjuvant and neoadjuvant settings and in combination with other therapies and research into improved prognostic and predictive biomarkers of ICI response and improved activity in pMMR–MSI-L CRC are ongoing.
-
Beyond PD1 blockade, monospecific and bispecific antibodies, cellular therapies, vaccines and cytokines targeting other immune checkpoint molecules, macrophages and other components of innate immunity are under active investigation.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is a major cause of cancer death worldwide. In developed countries, early detection through screening has improved the 5-year survival of patients with CRC, but ~25% of patients still present with stage 4 disease, and a further 25–50% present with early-stage disease but go on to develop metastatic disease1,2,3,4. The prognosis for patients with metastatic CRC (mCRC) remains poor, with a median 5-year survival of only 12.5% in the USA2. Thus, the development of more effective treatments for patients with this disease is an urgent unmet need. In the past decade, immunotherapy has elicited tremendous excitement owing to its success in achieving long-term durable responses in previously difficult-to-treat solid tumours, such as melanoma and lung cancer. High tumour mutation burden has emerged as a marker of responsiveness to immunotherapy in several tumour types5,6. In CRC, immune checkpoint therapy received regulatory approval in 2017 for the treatment of heavily mutated tumours that are mismatch-repair-deficient (dMMR) or have high levels of microsatellite instability (MSI-H) (termed dMMR–MSI-H tumours). By contrast, current immune checkpoint inhibitors (ICIs) are ineffective in tumours that are mismatch-repair-proficient (pMMR) and are microsatellite-stable (MSS) or have low levels of microsatellite instability (MSI-L) (termed pMMR–MSI-L tumours). In these tumours, low tumour mutation burden and the lack of immune cell infiltration have been posited as mechanisms of immune resistance7,8. In this Review, we describe the rationale for using immunotherapy in select patients with mCRC, discuss available clinical data supporting its use and highlight current clinical approaches and future directions for expanding the scope of immunotherapy in CRC.
Rationale for immunotherapy in CRC
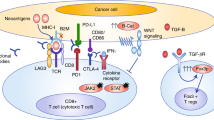
In CRC, T cell infiltration into the tumour bed has long been associated with favourable outcomes, suggesting a possible role for immunoediting in controlling tumour growth7,9,10. The immune system distinguishes self from non-self through the binding of T cell receptors (TCR) on T cells to complexes of peptides with major histocompatibility complex (MHC) class I molecules presented on the surface of all cells, including tumour cells11,12. Recognition of peptide–MHC class I complexes by the TCR alone is insufficient for T cell activation. TCR–MHC signalling pathways are modulated by co-stimulatory or co-inhibitory signals, which tumour cells exploit to escape destruction13,14,15. ICIs target co-inhibitory receptors, such as cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed cell death 1 (PD1) on T cells and other immune cell subpopulations, or their ligands, such as programmed cell death 1 ligand 1 (PDL1) on tumour cells and various immune cells. Thus, ICIs prevent T cell dysfunction and apoptosis and instead enhance T cell activation, potentiating cytotoxic killing of tumour cells (Fig. 1).

Endogenous peptides are processed and presented on major histocompatibility complex (MHC) class I molecules on the surface of all human cells, including cancer cells. The peptide–MHC complex is recognized by T cell receptors (TCRs). The response of the T cell is fine-tuned by a range of co-inhibitory or co-stimulatory signals. The ligands CD80 and CD86 of the B7 family of membrane-bound ligands can bind to the co-stimulatory CD28 and, especially in activated T cells, to cytotoxic T lymphocyte antigen 4 (CTLA4). Similarly, membrane-bound programmed cell death 1 ligand 1 (PDL1) and programmed cell death 1 ligand 2 (PDL2) can engage programmed cell death 1 (PD1), leading to T cell anergy and/or apoptosis. Monoclonal antibodies that bind to either the inhibitory receptors on T cells or their cognate ligands on cancer cells antagonize inhibitory signalling and enable T cell activation and cytotoxic tumour cell killing. Currently, FDA-approved immune checkpoint inhibitors target CTLA4 (ipilimumab), PD1 (pembrolizumab and nivolumab) and PDL1 (atezolizumab and durvalumab). Pembrolizumab and nivolumab, as well as the combination of nivolumab and ipilimumab, are currently approved for colorectal cancer in the USA.
ICIs were first shown to improve survival in metastatic melanoma and subsequently in non-small-cell lung cancer (NSCLC), leading to FDA approvals for ipilimumab (targeting CTLA4) and pembrolizumab and nivolumab (targeting PD1) for the treatment of these solid tumours16,17,18,19,20,21,22,23,24. Importantly, long-term follow-up data showed that a subset of patients survived for ≥10 years after ipilimumab treatment20. As the potential of immunotherapy to achieve long-term durable responses in some advanced solid tumours was recognized, the need for biomarkers that could distinguish tumours that did or did not respond to immunotherapy became apparent. In a study of the mutational landscapes of human cancer, melanoma and NSCLC were the cancer types with the highest prevalence of mutations25. The correlation between mutation prevalence and immunotherapy response suggested that tumour cells with high mutation burdens generate and present more peptide neoantigens on their MHC class I molecules; thus, these tumours are more likely to be recognized as non-self, in turn priming T cells for activation and cytotoxic killing26,27.
dMMR–MSI-H and pMMR–MSI-L CRC
CRC can be categorized into two discrete groups on the basis of mutation patterns: tumours that have a dMMR–MSI-H signature and high overall mutation burden (>12 mutations per 106 DNA bases) and tumours that have a pMMR–MSI-L signature with a much lower mutation burden (<8.24 mutations per 106 DNA bases)28. Defective DNA mismatch repair (MMR) can be detected either by the lack of immunohistochemical staining of the MMR proteins MLH1, MSH2, MSH6 or PMS2 or by PCR-identified alterations in the lengths of microsatellites between a patient’s tumour and a sample of normal tissue or blood. In the past 5 years, computational analyses of tumour next-generation sequencing were also shown to accurately detect microsatellite instability (MSI) status (mSINGS29, MSIsensor30,31 and MOSAIC32). Assessment for dMMR–MSI-H was initially used to identify patients in whom further germline testing for Lynch syndrome was warranted33. MSI-H is the hallmark of tumours in patients with Lynch syndrome, but the development of dMMR–MSI-H is a sporadic event in ~70–85% of all patients with dMMR–MSI-H tumours owing to somatic defects in MMR gene function, most commonly hypermethylation of the MLH1 promoter. Importantly, dMMR–MSI-H tumours are heavily infiltrated by immune cells, notably CD8+ tumour-infiltrating lymphocytes (TILs), T helper 1 (TH1) CD4+ TILs and macrophages, and have a microenvironment that is rich in type I interferons in comparison with other CRCs7,34,35,36,37,38,39,40,41,42 (Box 1; Fig. 2). However, the extent to which these two features overlap has not been rigorously investigated, and clinical trials investigating ICIs in CRC have not specifically utilized TILs as a predictive biomarker.

Colorectal cancers (CRCs) can be grouped into subtypes by distinguishing those that are mismatch-repair-deficient (dMMR) and have high levels of microsatellite instability (MSI-H) (termed dMMR–MSI-H) and those that are mismatch-repair-proficient (pMMR) and are microsatellite-stable or have low levels of microsatellite instability (MSI-L) (pMMR–MSI-L tumours). a | dMMR–MSI-H CRC shows prominent tumour-infiltrating lymphocytes that are programmed cell death 1 (PD1)+, accompanied by increased levels of programmed cell death 1 ligand 1 (PDL1)+ immune cells that are primarily present at the tumour–stroma interface. This tumour also shows loss of β2-microglobulin expression. pMMR–MSI-L CRC shows conventional morphology with no appreciable tumour-infiltrating lymphocytes. PD1+ lymphocytes in the stroma are rare, and no PDL1 labelling can be seen. This tumour has retained β2-microglobulin expression. Tissue sections are stained with haematoxylin and eosin. Positive immunohistochemistry staining is shown in brown. b | dMMR–MSI-H and pMMR–MSI-L CRCs have distinct tumour microenvironments. dMMR–MSI-H tumour cells are characterized by a high number of genomic mutations and, consequently, present mutated peptides on their major histocompatibility complex (MHC) class I molecules. Complexes of mutant peptides with MHC class I are recognized as foreign neoantigens, triggering immune cell priming and infiltration. Tumour-associated macrophages are an important component of the tumour microenvironment, influencing tumour growth and progression. dMMR–MSI-H CRCs are characterized by high levels of CD8+ T cell infiltration, T helper 1 (TH1) CD4+ T cell infiltration and IFNγ secretion. To evade immune-mediated killing in this T cell-inflamed microenvironment, tumour cells strongly upregulate T cell inhibitory ligands, such as CD80 and CD86 of the B7 family and PDL1, which bind co-inhibitory receptors, such as cytotoxic T lymphocyte antigen 4 (CTLA4) and PD1. Immune checkpoint inhibitors (ICIs) exploit this pre-existing inflamed microenvironment by antagonizing T cell inhibitor signalling, exposing the tumour cells to cytotoxic destruction. By contrast, pMMR–MSI-L tumours do not generate immunostimulatory neoantigens and are characterized by T cell exclusion from the tumour. They express relatively low levels of immune-inhibitory ligands. These features suggest reasons for the differential response of dMMR–MSI-H and pMMR–MSI-L CRCs to ICIs and might be suitable as predictive biomarkers for patient selection. TCR, T cell receptor.
Approximately 15% of all CRCs are dMMR–MSI-H43. Presence of dMMR–MSI-H disease is prognostic, as stage 2 dMMR–MSI-H tumours have a lower risk of recurrence than stage 2 pMMR–MSI-L tumours, with a hazard ratio for overall survival associated with MSI of 0.65 (95% CI 0.59–0.71) in pooled analysis44. Accordingly, stage 4 dMMR–MSI-H tumours constitute only ~2–4% of all mCRCs. Patients with dMMR–MSI-H tumours that metastasize have a dismal prognosis45, but expression of PD1, PDL1 and CTLA4 is substantially upregulated in their cancers39. These observations suggested that dMMR–MSI-H CRCs might respond well to immune checkpoint blockade.
Immunotherapy for dMMR–MSI-H CRC
Studies resulting in immune checkpoint inhibitor approval
In initial studies published between 2010 and 2013, ICIs demonstrated very limited clinical activity in nonselected CRC. An anti-CTLA4 immunoglobulin G2 (IgG2) antibody, tremelimumab, was evaluated in 45 patients with treatment-refractory CRC and resulted in a partial response in 1 individual, but the MMR status of this patient was not known46. In a phase I study of BMS-936559, an anti-PDL1 antibody, in refractory solid tumours, no responses were observed47. Nivolumab, an anti-PD1 antibody, was evaluated in 19 patients, and initially, no responses were reported48; however, 1 of the patients had a response at 21 months, and after re-treatment, this patient achieved a complete response that lasted ≥3 years49,50. This patient had dMMR–MSI-H CRC.
On the basis of the knowledge of the immunogenic microenvironment of MSI-H tumours and the observed impressive tumour response, enthusiasm for immunotherapy in CRC grew, and several studies investigated the therapeutic potential of PD1 inhibitors. A phase II trial (NCT01876511) of the anti-PD1 antibody pembrolizumab was reported in 2015, in which three separate cohorts of patients were treated: dMMR–MSI-H CRCs, pMMR–MSI-L CRCs and dMMR–MSI-H non-CRCs51. Of the 10 patients with dMMR–MSI-H CRC, 4 had a partial response and 5 had stable disease at 20 weeks. At this time point, the median progression-free survival (PFS) and overall survival were not yet reached in the dMMR–MSI-H cohort but were 2.2 months and 5.0 months, respectively, in the pMMR–MSI-L cohort (HR for disease progression or death 0.10 (P < 0.001); HR for death 0.22 (P = 0.05)). In updated results presented at the 2016 American Society of Clinical Oncology (ASCO) Annual Meeting, the response rate was 50% (95% CI 31–69%), and the disease control rate was 89% in the 28 patients with dMMR–MSI-H tumours. At 24 months, PFS was 61%, and overall survival was 66%52. None of the 18 patients with pMMR–MSI-L CRC responded. This study demonstrated the benefit of immune checkpoint blockade in dMMR–MSI-H tumours. Analyses demonstrated that the number of somatic mutations significantly correlated with the chance of achieving a response to therapy (P = 0.02)8. Furthermore, results from an expansion of this study published in 2017 across 12 tumour types demonstrated that pembrolizumab was effective in dMMR tumours regardless of tissue of origin8.
In CheckMate 142 (NCT02060188), another PD1 inhibitor, nivolumab, was tested in 74 patients with dMMR–MSI-H mCRC53. Study results were first published in 2017. At a median follow-up duration of 12 months, 23 (31%) patients achieved an investigator-assessed objective response, and in 51 (69%) patients, disease control for ≥12 weeks was observed. The median PFS was 14.3 months (95% CI 4.3 months to not estimable), and the 12-month PFS was 50% (95% CI 38–61%). The 12 month overall survival was 73% (95% CI 62–82%). Combination of nivolumab with ipilimumab was also evaluated in this trial54,55. Of 30 patients enrolled, 9 patients (33%) achieved an objective response, and 14 patients (52%) achieved stable disease. Updated results of CheckMate 142 in the complete cohort of 119 patients with a median follow-up duration of 13.4 months demonstrated an objective response rate of 55% and tumour burden reduction from baseline in 77% of patients54,55. At this time point, the median PFS was not yet reached, and the 9-month and 12-month PFS values were 76% (95% CI 67.0–82.7%) and 71% (95% CI 61.4–78.7%), respectively. The median overall survival was not reached, and the 9-month and 12-month overall survival values were 87% (95% CI 80.0–92.2%) and 85% (95% CI 77.0–90.2%), respectively. Treatment with combined nivolumab and ipilimumab resulted in an increased rate of drug-related immune-related adverse events: 32% of patients experienced grade 3–4 treatment-related adverse events compared with 20% of patients treated with nivolumab alone54 (Box 2). On the basis of the compelling data in dMMR–MSI-H CRCs, the FDA granted accelerated approval to pembrolizumab in May 2017 and to nivolumab in July 2017 for the second-line treatment of patients with dMMR–MSI-H CRC. To date, no drug or combination has been granted approval by the European Medicines Agency (EMA), pending results of phase III randomized controlled studies.
Current and future studies
Multiple studies are ongoing to evaluate PD1 or PDL1 inhibition in dMMR–MSI-H CRC with the potential of practice-changing results (Table 1). Results of the phase II trial Checkmate 142 evaluating the efficacy and safety of combined nivolumab and low-dose ipilimumab therapy in previously untreated patients with stage 4 dMMR–MSI-H CRC were reported at the 2018 European Society for Medical Oncology (ESMO) Annual Meeting56. In 45 enrolled patients followed-up for a median of 13.8 months, the objective response rate and disease control rate were 60% and 84%, respectively; 7% of patients had a complete response. PFS and overall survival values at 12 months were 77% and 83%, respectively, and the extent of treatment-related adverse events were acceptable56. Keynote-177 is a phase III trial (NCT02563002) evaluating first-line pembrolizumab in stage 4 dMMR–MSI-H CRC57. As of May 2018, 308 patients were enrolled to be randomly allocated to pembrolizumab or investigator’s choice of first-line chemotherapy. The primary end points are PFS and overall survival, and the secondary end point is overall response rate. A further study in the first-line setting is underway (NCT02997228)58. In this trial, 347 patients are planned to be randomly allocated to the anti-PDL1 antibody atezolizumab, first-line combination chemotherapy (comprising 5-fluorouracil, leucovorin and oxaliplatin (FOLFOX) plus the vascular endothelial growth factor antagonist bevacizumab) or the combination of both treatments. The primary trial end point is PFS, and secondary end points include overall survival and objective response rate.
Immunotherapy for pMMR–MSI-L CRC
Unlike in patients with dMMR–MSI-H CRC, immunotherapy alone has not demonstrated a clinical benefit in patients with pMMR–MSI-L CRC, who constitute the vast majority of patients with mCRC. In the pivotal pembrolizumab study, no responses were observed in patients with pMMR–MSI-L tumours51, consistent with the lack of efficacy of immunotherapy in early studies with nonselected patients, most of whom had pMMR–MSI-L tumours. In the Check Mate 142 study, limited responses were seen in pMMR–MSI-L tumours: 1 of 20 patients responded to combination therapy with antibodies blocking PD1 and CTLA4 (ref.54). The lack of recruitment of immune cells to the tumour seems to be the fundamental obstacle to efficacy. Combination treatment of PD1 inhibitors and modulators of other immune checkpoint molecules, such as CTLA4, might be beneficial in a small subset of patients with pMMR–MSI-L tumours, but alternative approaches of immune modulation are required for the majority of patients with this CRC subtype.
MEK and PDL1 inhibition
Preclinical data suggest several opportunities for combination therapy. In addition to direct pro-proliferative effects on tumour cells, activation of the RAS–MAPK pathway has been associated with decreased T cell infiltration into tumours; conversely, in preclinical models, inhibition of MEK, a downstream effector of this pathway, induced IFNγ-dependent HLA and PDL1 upregulation and synergized with PD1 inhibition to augment antitumour activity59,60. On the basis of these data, a phase I study (NCT01988896) of the MEK inhibitor cobimetinib and PDL1 inhibition with atezolizumab was initiated61. The study includes an expansion cohort of patients with KRAS-mutant CRC, and of the 23 patients enrolled when preliminary data were reported in 2016, 4 patients (17%) had a partial response. Three of the four responders had confirmed pMMR–MSI-L CRC, and the status of the other patient is unknown. Updated results were presented in 2018, showing a tolerable safety profile and partial responses in 7 of 84 patients enrolled (8%; 3 pMMR–MSI-L, 1 MSI-L and 3 unknown status) at a median follow-up duration of 14.3 months62. These exciting data led to a phase III randomized trial of cobimetinib plus atezolizumab versus atezolizumab only or regorafenib in patients with refractory pMMR–MSI-L CRC (NCT02788279)63. However, in results reported at the 2018 ESMO World Congress on Gastrointestinal Cancer, the study failed to meet its primary end point: atezolizumab and cobimetinib combination therapy and atezolizumab monotherapy failed to demonstrate statistically significant prolonged overall survival compared with regorafenib64. Ongoing studies of combining immunotherapy and targeted therapy in patients with pMMR–MSI-L CRC include a phase II study of combined cobimetinib, nivolumab and ipilimumab treatment (NCT02060188)65 and another phase Ib study of cobimetinib, atezolizumab and bevacizumab combination (NCT02876224)66. In addition, several trials are investigating the combination of MEK inhibition with PD1 and chemotherapy (Table 2).
Bispecific antibody therapy
Bispecific antibodies are a new class of engineered agents with the ability to bind to two separate targets. CEA-TCB (also known as RG7802 or RO6958688) is a T cell bispecific antibody that simultaneously binds carcinoembryonic antigen (CEA) on tumour cells and CD3 on T cells, thus crosslinking cancer cells and T cells and leading to T cell engagement and activation independent of pre-existing immunity, T cell infiltration and tumour inflammation. CEA-TCB is being explored in two ongoing phase I studies: as a monotherapy (NCT02324257)67 and in combination with atezolizumab (NCT02650713)68. Following the data cut-off point in March 2017, encouraging clinical activity was reported in patients with metastatic MSS CRC treated with CEA-TCB monotherapy, which was enhanced by combination with atezolizumab69. In the combination therapy group, the response rate was 18% (n = 2), and stable disease was observed in 7 patients (64%), for an overall disease control rate of 82%. Overall, toxic effects were manageable. CEA-TCB is the first T cell bispecific antibody to show efficacy in solid tumours and in MSS CRC in particular. These studies are ongoing69.
Chemotherapy and antiangiogenic combinations
Preclinical data in lung cancer models demonstrated sensitization of tumours to checkpoint blockade through chemotherapy70. The immunomodulatory potential of bevacizumab and antiangiogenic agents was observed in a trial in patients with melanoma that combined ipilimumab with bevacizumab treatment71. In this study, the addition of bevacizumab increased CD8+ T cell infiltration into the tumour compared with ipilimumab alone. Combinations of immune checkpoint blockade with bevacizumab treatment are under investigation, and preliminary results indicate activity. In preliminary data presented as an abstract from one trial of the combination of atezolizumab plus bevacizumab with or without chemotherapy (NCT01633970), 14 patients with refractory pMMR–MSI-L CRC were treated with atezolizumab plus bevacizumab; 1 patient (7%) had an objective response, and 9 patients (64%) had stable disease72,73. Subsequent correlative analysis showed that CD8+ T cell infiltration and PDL1 expression were increased in tumours following chemotherapy administration with or without atezolizumab and bevacizumab74. Several ongoing studies are investigating the combination of ICIs with antiangiogenic agents and chemotherapy (Table 2).
Radiotherapy combinations
As radiation causes DNA damage and probably generates an enlarged neoantigen repertoire for T cell priming, the immunogenic potential of radiotherapy is an active area of investigation75. Interim results of a study investigating pembrolizumab combined with either radiofrequency ablation or external beam radiation (NCT02437071) in patients with CRC were reported in 2016. Of 22 patients who received external beam radiotherapy, 1 patient responded, and no responses were observed in the ablation arm. In patients with melanoma, tumour regression was demonstrated for the combination of radiotherapy with dual immune checkpoint blockade (anti-CTLA4 and PDL1)76. Dual immune checkpoint blockade of CTLA4 and PDL1 in combination with radiotherapy or radiofrequency ablation is currently under investigation (NCT03122509)77 (Table 2).
Biomarkers of response to immunotherapy
Mutational load and neoantigens
Presence of dMMR–MSI-H in colorectal tumours, as well as in other solid tumours, is a clear biomarker for potential response to immunotherapy, but identification of more precise and reliable predictive biomarkers continues to be an unmet clinical need. The relationship between mutational load and response to immunotherapy was initially described in melanoma for CTLA4-blocking antibody treatment and subsequently in NSCLC, in which an increased nonsynonymous mutation (that is, mutations that alter the peptide sequence) burden was associated with improved response to the anti-PD1 antibody pembrolizumab26,78,79. Mutational burden is certainly an important marker of potential response, but high mutational burden alone does not seem to be sufficient for driving immunotherapy response. The nearly 20-times-higher mutation burden in dMMR–MSI-H compared with pMMR–MSI-L CRCs results in the generation of neo-epitopes, which are thought to trigger and be a target for host antitumour immune responses via T cell infiltration and cytolytic activity80,81,82. Somatic mutational load and neoantigen density correlate with benefit from immune checkpoint blockade in many malignancies, with data suggesting that the high density of mutation-associated neoantigens (MANAs) generated results in T cell diversity83. Furthermore, immune responses might be driven by specific generated MANAs. For example, high levels of clonal neoantigens seem to be markers of response, whereas subclonal neoantigens, usually resulting from tumour heterogeneity, might predict resistance to immunotherapy84,85.
Interestingly, a high mutational load might not always be necessary to drive immunotherapy response. Evaluation of the presence of tumour-infiltrating CD3+CD8+ lymphocytes, through assignment of an immunoscore based on the density and the location of subsets of T cells, was prognostic of clinical outcome in patients with early-stage CRC, performing better than MSI and MMR status7,86,87. High immunoscores were also reported in pMMR–MSI-L CRCs, raising the question of whether immunophenotyping might enable prediction of immunotherapy benefit. Hence, combining the immunogenic features of the tumour microenvironment with mutational burden might be more precise in predicting immunotherapy response than either feature alone. Further validation of immunophenotyping as a predictive biomarker of immunotherapy response, specifically in metastatic disease, is needed for broad clinical utility.
POLE proofreading domain mutations
In addition to hypermutation of tumours caused by the dMMR–MSI-H pathway, large-scale genomic studies have revealed that tumours with mutations in the POLE exonuclease domain also result in a remarkably hypermutated somatic profile, which is commonly referred to as an ultramutated phenotype28,88. POLE mutations in CRC have been well described. Recurrent mutations such as R286R, R286H, V411L and S459F are present in 1–2% of CRC tumours, with rare occurrence in mCRC89. In contrast to dMMR–MSI-H tumours, the mutation rate in POLE-mutated tumours often exceeds 100 per 106 DNA bases. In the majority of CRCs, these mutations are somatic events; however, germline mutations in the exonuclease domain of POLE, and to a lesser extent in that of POLD1, can also occur and are characterized by the presence of colonic polyposis, early-onset CRCs and potential risk of other cancers90. POLE-mutated tumours are usually MSS, as the MMR system remains intact, although dMMR–MSI-H tumours in the setting of a POLE mutation, resulting from acquired somatic mutations in MMR genes, have been described91,92. Similar to immunogenic dMMR–MSI-H tumours, POLE-mutated CRCs also display increased CD8+ lymphocyte infiltration, expression of cytotoxic T cell markers and effector cytokines and upregulation of genes encoding immune checkpoints, such as PD1, PDL1 and CTLA4 (ref.93). With respect to clinical characteristics, patients with POLE-mutated CRCs generally have an excellent prognosis, and these tumours are associated with early disease stage at presentation, male sex, right-sided tumour location and younger age at diagnosis93. Given the similarly enhanced immunogenicity of POLE-mutated CRCs to dMMR–MSI-H CRCs, the therapeutic potential of immune checkpoint blockade in the subset of POLE-mutated CRCs is of particular interest and deserves further investigation. Clinical trials that include investigation of PD1 and PDL1 inhibitors in other POLE-mutated malignancies are currently ongoing (NCT02912572 (endometrial cancer)94, NCT02899793 (endometrial cancer)95 and NCT02658279 (malignant glioma)96).
Other biomarkers for anti-PD1 therapy
Various other biomarkers of response to anti-PD1 therapy are currently being explored. Perhaps the most widely investigated marker is tumour PDL1 expression measured by immunohistochemical staining. Interestingly, in some tumour types, such as NSCLC, gastric cancer and gastroesophageal junction tumours, PDL1 expression might be useful as a predictive marker of response to anti-PD1 therapy22,97, but in CRC, PDL1 expression was not found to be associated with response or survival in the registration studies51,98. Markers of resistance to PD1 blockade, such as acquired mutations in JAK1, JAK2 and B2M, have been discovered in patients with cancers such as melanoma99, but their role in patients with CRC is not well defined. Truncating mutations in B2M, encoding β2-microglobulin, lead to impaired MHC class I antigen presentation and generation of immune escape variants that fail to elicit a T cell response. Although clinical data suggest that patients with B2M-mutant dMMR–MSI-H CRCs have a favourable prognosis100, interestingly, acquired B2M mutations were observed in tumours that developed resistance to pembrolizumab8. Mutations that inactivate JAK1 or JAK2 lead to both acquired as well as primary resistance to anti-PD1 therapy in melanoma99,101, but their role in anti-PD1 response in CRC remains to be fully elucidated. Further, the aetiology of dMMR–MSI-H disease (germline versus somatic event) does not seem to be a predictive marker, as tumours arising in patients with Lynch syndrome have similar responses to anti-PD1 therapy as sporadic dMMR–MSI-H tumours8. Various groups have developed gene expression signatures associated with intratumoural cytotoxic T cell infiltration, but these signatures have not been explored as predictive biomarkers in the context of ICI clinical trials in CRC102,103.
Adjuvant and neoadjuvant therapy
Only ~4% of patients with mCRC have dMMR–MSI-H tumours, but this phenotype is present in 12% of patients with stage 3 CRCs, in whom adjuvant chemotherapy (fluorouracil plus oxaliplatin) is routinely administered. In an effort to determine the potential efficacy of immunotherapy in the adjuvant treatment of early-stage CRC, a randomized phase III trial is evaluating the combination of chemotherapy and atezolizumab compared with chemotherapy only in 700 patients with stage 3 dMMR–MSI-H colon cancer (NCT02912559)104,105. In the experimental arm, patients receive FOLFOX plus atezolizumab for 6 months followed by atezolizumab monotherapy for 6 months. The primary trial end point is disease-free survival, and overall survival and incidence of adverse events are the secondary end points.
Exciting preliminary results from an ongoing single-arm study of short-term combination nivolumab and ipilimumab in patients with resectable, early-stage CRC were presented at the 2018 ESMO meeting106. The primary end points of this study are safety and feasibility, and the secondary end points include pathological response. All patients underwent surgery a maximum of 6 weeks after informed consent. All of the seven patients with dMMR–MSI-H tumours achieved a major pathological response, and four of these (57%) had complete responses. In the eight patients with pMMR–MSI-L tumours, no major pathological responses were noted, but significant increases in T cell infiltration were seen after treatment in both dMMR–MSI-H (P = 0.0009) and pMMR–MSI-L (P = 0.018) tumours. If these results are corroborated in expanded cohorts with long-term follow-up, they raise the intriguing question whether surgical resection, which has long been the mainstay of curative-intent treatment for early-stage CRC, might be safely avoided in a select subgroup of patients with dMMR–MSI-H CRC.
Primary prevention
In addition to opportunities in the treatment of existing tumours, the advent of immunotherapy also provides the possibility of precision prevention oncology for individuals with germline mutations in MMR genes, diagnostic of Lynch syndrome. Patients with Lynch syndrome have an increased lifetime risk of several malignancies, for example, CRC and endometrial, ovarian, gastric, pancreatic and ureteral cancer107. Cancer screening or risk-reducing surgical interventions, such as colonoscopy or prophylactic hysterectomy and oophorectomy, are part of the management considerations for these individuals108, but immune-response-based prevention is also being explored for risk reduction. The applicability of ICIs, including anti-PD1 or anti-PDL1 agents, for cancer prevention is currently limited by drug toxicity; however, other immunological intervention strategies including vaccines are being explored. Defective MMR results in the accumulation of frameshift mutations at microsatellite tracts, with the generation of frameshift-mutation-derived peptides (FSPs) when the mutation is in the coding regions of the genome109. Compared with neoantigens resulting from single-nucleotide alterations, FSP-based neoantigens can encompass long antigenic amino acid stretches that contain multiple immunologically relevant neoepitopes and can be highly immunogenic110,111. Given that microsatellite tracts occur at specific loci, FSPs have predictable sequences, providing an opportunity for targeted vaccine development strategies. For example, vaccination with three commonly mutated FSP antigens was tested in a phase I/IIa trial in patients with stage 3 or 4 dMMR–MSI-H CRC following completion of standard chemotherapy112. In 22 enrolled patients, FSP-specific immune responses were induced in all vaccinated patients, with 1 heavily pretreated patient with bulky metastases demonstrating stable disease for 7 months. Although these are early data, FSP vaccination is a promising approach for improving the prognosis of patients with dMMR–MSI-H cancers in the adjuvant setting or potentially for cancer prevention in individuals with Lynch mutations but without a cancer diagnosis.
Beyond PD1 blockade
T cell checkpoint modulation
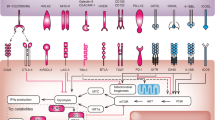
PD1 blockade is currently the only FDA-approved immunotherapeutic strategy for CRC, but PD1–PDL1 interaction is only one of several immune checkpoint interactions that regulate T cell activation in the tumour microenvironment (Fig. 2). On the basis of promising preclinical data113,114,115,116, molecules that block other T cell checkpoint inhibitors (for example, T cell immunoglobulin mucin receptor 3 (TIM3; also known as HAVCR2), lymphocyte activation gene 3 protein (LAG3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT)) are in clinical trials for various advanced malignancies, including CRC117 (Table 3). Complementing immune checkpoint blockade, molecules that promote T cell differentiation, survival and proliferation are being studied either as single agents or in combination with checkpoint blockade (Fig. 3; Table 3). Several of these molecules are antibody agonists of the co-stimulatory group of the TNF receptor superfamily (for example, CD27, OX40 (also known as CD134), 4-1BB (also known as CD137), glucocorticoid-induced TNF receptor-related gene (GITR; also known as CD357) and CD40)118,119,120.

Many drugs that modulate immune checkpoints are currently in clinic trials in patients with colorectal cancer (CRC) (Table 3). These agents act as antagonists of T cell inhibitory signals or agonists of T cell activating signals by targeting ligands expressed on the surface of tumour cells or antigen-presenting cells, receptors on the surface of T cells or metabolites and cytokines that serve as paracrine signalling molecules. 4-1BBL, 4-1BB ligand; CD40L, CD40 ligand; CTLA4, cytotoxic T lymphocyte antigen 4; ICOS, inducible T cell co-stimulator; ICOSL, inducible T cell co-stimulator ligand; IDO, indoleamine 2,3-dioxygenase; GITR, glucocorticoid-induced TNF receptor-related gene; GITRL, glucocorticoid-induced TNF receptor-related gene ligand; LAG3, lymphocyte activation gene 3 protein; MHC, major histocompatibility complex; OX40L, OX40 ligand; PD1, programmed cell death 1; PDL1, programmed cell death 1 ligand 1; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM3, T cell immunoglobulin mucin receptor 3.
Cellular immunotherapy
Adoptive cell therapy (ACT) is an exciting emerging modality for treating CRC. ACT involves the collection of T cells from tumours, lymph nodes or peripheral blood of patients, in vitro expansion and transfer of tumour-destroying T cells into patients121. In vitro, tumour-targeting T cells can be selected for binding to tumour antigens or engineered to express chimeric antigen receptors (CARs) that improve T cell recognition of tumour proteins other than MHC class I122. T cells can also be engineered to secrete cytokines or express immunostimulatory ligands that further potentiate their efficacy (termed armoured CAR T cells)123, for example, IL-12, IL-7 receptor and lipid nanoparticles containing IL-15 (ref.124).
Several groups have investigated CEA, which is overexpressed in many CRCs, as a target for ACT125,126,127. In a small study, CAR T cells targeting CEA were administered to three patients with mCRC125. One of the patients had an objective response in lung and liver metastases, and serum CEA levels declined in all three patients. However, all three patients developed severe colitis as a dose-limiting toxic effect. In another study, 7 of 10 patients with heavily treated mCRC had stable disease 4 weeks after CAR T cell infusion, and 2 patients experienced tumour shrinkage127. At 30 weeks, 2 patients continued to have stable disease. In this study, treatment was well tolerated, with no reports of colitis.
In an intriguing advance, researchers in one study collected TILs from a patient with lung metastases from KRASG12D-mutant CRC and selected clonal T cell cultures with the highest specific CD8+ T cell reactivity to KRASG12D for adoptive transfer into the patient128. All seven lung metastases regressed, and the patient had a partial response according to Response Evaluation Criteria in Solid Tumours (RECIST) at 9 months, when 1 lesion progressed. This lesion was resected, and the patient was clinically disease free for 3 months following resection. This early success suggests an exciting new strategy for therapeutically targeting mutant transcription factors that drive CRC growth, which has been difficult to achieve to date.
To date, CAR T cell therapy has been successfully used in the treatment of B cell malignancies, in which treatment with CAR T cells targeting the B cell antigen CD19 resulted in complete responses and has gained approval from the FDA and the EMA Committee for Medicinal Products for Human Use129,130. However, the applicability of CAR T cell therapy to solid tumours, such as CRC, remains to be proved. Concerns regarding the limited extent of infiltration of adoptively transferred T cells into the dense microenvironment of solid tumours and potential safety concerns related to systemic cytokine release syndrome and on-target–off-tumour effects against normal epithelial cells remain to be resolved131,132. In the real-world clinical setting, the feasibility and affordability of highly individualized, technically sophisticated cell manipulation approaches also present a challenge.
Other approaches
Vaccines and immunostimulatory cytokines were first investigated as potential CRC immunotherapeutics >30 years ago but have not changed clinical practice to date133,134. The efficacy of PD1 blockade in dMMR–MSI-H CRC has provided definitive evidence of the clinical utility of immunotherapy and, hence, has renewed interest in these approaches, both as single agents and in combination with PD1 inhibition. Several trials are ongoing that investigate oncolytic bacteria or viruses or peptide, tumour, virus or dendritic cell antigens in combination with various adjuvants with the goal of improving tumour immunogenicity in the adjuvant and metastatic settings in CRC and other solid tumours. As crucial drivers of inflammation conducive to tumour progression, tumour-associated macrophages are attractive targets to complement current PD1-targeted and PDL1-targeted ICIs135. A key target is colony-stimulating factor 1 receptor (CSF1R), which is expressed on mononuclear phagocytes and dimerizes upon binding to colony-stimulating factor 1 (CSF1) or IL-34, activating macrophage proliferation and function136,137. CSF1R-specific inhibitors and other macrophage modulators are currently being investigated in clinical trials for solid tumours including mCRC138, either as single agents or in combination with chemotherapy or ICIs (for example, NCT02777710)139.
Conclusions
Owing to their high mutation burden, dMMR–MSI-H CRCs present peptide neoantigens on MHC class I molecules and, therefore, prime T cells to recognize them as foreign. Building on initial successes in other heavily mutated tumour types such as melanoma, monoclonal antibodies that block immune checkpoints prevent T cell anergy, promote cytotoxic CD8+ T cell destruction of tumours and can induce long-term durable responses in some patients with an increasing range of malignancies. This strategy has proved remarkably successful in the small subset of patients with mCRC whose tumours are characterized by a dMMR–MSI-H phenotype, resulting in FDA approval in 2017 of two checkpoint inhibitors: pembrolizumab and nivolumab for the treatment of dMMR–MSI-H mCRC. Indeed, pembrolizumab is now FDA-approved for the treatment of all dMMR–MSI-H metastatic solid tumours, becoming the first biomarker-based, tumour type-agnostic treatment for cancer8.
The success of PD1 inhibitors in achieving durable responses in some patients with dMMR–MSI-H CRC heralds the dawn of a new era in the treatment of patients with a subtype of mCRC. However, not all patients with dMMR–MSI-H respond to current ICIs. Further insight into the mechanisms of immunotherapy resistance is needed, and biomarkers that predict therapy response in patients with dMMR–MSI-H CRC are required. Beyond dMMR–MSI-H CRC, the critical challenge is to develop strategies for targeting pMMR–MSI-L CRCs, which constitute the vast majority of mCRC cases and for which current immunotherapy approaches have been largely unsuccessful. The convergence of progress in several scientific and medical fields is providing unprecedented insight into the relationship between cancer cells and their immune microenvironment, including histopathology, genomics, immune profiling, single-cell transcriptomics, TCR characterization, neoantigen prediction, ex vivo T cell manipulation, the growing armamentarium of immune checkpoint modulators, cellular therapies and vaccines, and cutting-edge clinical trials providing real-world human data on mechanisms of immune resistance. Hopefully, this surge of knowledge will lead to improved pharmacological strategies to overcome primary resistance to immunotherapy of pMMR–MSI-L CRC.
References
Garborg, K. et al. Current status of screening for colorectal cancer. Ann. Oncol. 24, 1963–1972 (2013).
Siegel, R., Desantis, C. & Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 64, 104–117 (2014).
Edwards, B. K. et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 116, 544–573 (2010).
Sargent, D. et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J. Clin. Oncol. 27, 872–877 (2009).
Samstein, R. M. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30, 44–56 (2019).
Galon, J. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Pages, F. et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 353, 2654–2666 (2005).
Galon, J., Fridman, W. H. & Pages, F. The adaptive immunologic microenvironment in colorectal cancer: a novel perspective. Cancer Res. 67, 1883–1886 (2007).
Khalil, D. N., Smith, E. L., Brentjens, R. J. & Wolchok, J. D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 13, 273–290 (2016).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Townsend, S. E. & Allison, J. P. Tumor rejection after direct costimulation of CD8+T cells by B7-transfected melanoma cells. Science 259, 368–370 (1993).
Wei, S. C., Duffy, C. R. & Allison, J. P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086 (2018).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Robert, C. et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 (2011).
Robert, C. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330 (2015).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Schadendorf, D. et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 33, 1889–1894 (2015).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Garon, E. B. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015).
Brahmer, J. et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Salipante, S. J., Scroggins, S. M., Hampel, H. L., Turner, E. H. & Pritchard, C. C. Microsatellite instability detection by next generation sequencing. Clin. Chem. 60, 1192–1199 (2014).
Niu, B. et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30, 1015–1016 (2014).
Middha, S. et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis. Oncol. 1, 1–17 (2017).
Hause, R. J., Pritchard, C. C., Shendure, J. & Salipante, S. J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 22, 1342–1350 (2016).
Giardiello, F. M. et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 147, 502–526 (2014).
Alexander, J. et al. Histopathological identification of colon cancer with microsatellite instability. Am. J. Pathol. 158, 527–535 (2001).
Dolcetti, R. et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am. J. Pathol. 154, 1805–1813 (1999).
Smyrk, T. C., Watson, P., Kaul, K. & Lynch, H. T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 91, 2417–2422 (2001).
Young, J. et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am. J. Pathol. 159, 2107–2116 (2001).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Llosa, N. J. et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 5, 43–51 (2015).
Graham, D. M. & Appelman, H. D. Crohn’s-like lymphoid reaction and colorectal carcinoma: a potential histologic prognosticator. Mod. Pathol. 3, 332–335 (1990).
Jass, J. R. et al. Morphology of sporadic colorectal cancer with DNA replication errors. Gut 42, 673–679 (1998).
Nagorsen, D. et al. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: relation to local regulatory T cells, systemic T cell response against tumor-associated antigens and survival. J. Transl Med. 5, 62 (2007).
Boland, C. R. et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 58, 5248–5257 (1998).
Popat, S., Hubner, R. & Houlston, R. S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 23, 609–618 (2005).
Venderbosch, S. et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin. Cancer Res. 20, 5322–5330 (2014).
Chung, K. Y. et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 28, 3485–3490 (2010).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Brahmer, J. R. et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175 (2010).
Lipson, E. J. et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 19, 462–468 (2013).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Le, D. T. et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. J. Clin. Oncol. 34, 103 (2016).
Overman, M. J. et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 18, 1182–1191 (2017).
Overman, M. J. et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 36, 773–779 (2018).
Andre, T. L. et al. Nivolumab+ipilimumab combination in patients with DNA mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC): first report of the full cohort from CheckMate-142. J. Clin. Oncol. 36 (Suppl.), 553 (2018).
Lenz, H.-J. J. et al. Durable clinical benefit with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line therapy in microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). Ann. Oncol. 29 (Suppl.), LBA18_PR (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02997228 (2019).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02563002 (2018).
Liu, L. et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 21, 1639–1651 (2015).
Ebert, P. J. R. et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621 (2016).
Bendell, J. C. et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 34 (Suppl.), 3502 (2016).
Bendell, J. C. B. et al. A phase 1b study of safety and clinical activity of atezolizumab (A) and cobimetinib (C) in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 36, 560 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02788279 (2019).
Bendell, J. et al. Efficacy and safety results from IMblaze370, a randomised Phase III study comparing atezolizumab+cobimetinib and atezolizumab monotherapy versus regorafenib in chemotherapy-refractory metastatic colorectal cancer. Ann. Oncol. 29, LBA–004 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02060188 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02876224 (2019).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02324257 (2019).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02650713 (2019).
Segal, N. H. S. et al. Phase-I studies of the novel carcinoembryonic antigen T cell bispecific (CEA-CD3 TCB) antibody as a single agent and in combination with atezolizumab. Ann. Oncol. 28 (Suppl. 5), 122–141 (2017).
Pfirschke, C. et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 44, 343–354 (2016).
Hodi, F. S. et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2, 632–642 (2014).
Hochster, H. S. B. et al. Efficacy and safety of atezolizumab (atezo) and bevacizumab (bev) in a phase Ib study of microsatellite instability (MSI)-high metastatic colorectal cancer (mCRC). J. Clin. Oncol. 35 (Suppl.), 673 (2017).
Bendell, J. C. et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or FOLFOX in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 33, 704 (2015).
Wallin, J. et al. Clinical activity and immune correlates from a phase Ib study evaluating atezolizumab (anti-PDL1) in combination with FOLFOX and bevacizumab (anti-VEGF) in metastatic colorectal carcinoma. Cancer Res. 76, 2651 (2016).
Park, S. S. et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol. Res. 3, 610–619 (2015).
Twyman-Saint Victor, C. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03122509 (2018).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Tumeh, P. C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015).
Li, B. et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 48, 725–732 (2016).
McGranahan, N. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016).
Anagnostou, V. et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 7, 264–276 (2017).
Mlecnik, B. et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 44, 698–711 (2016).
Pages, F. et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139 (2018).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Yaeger, R. et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33, 125–136 (2018).
Palles, C. et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 45, 136–144 (2013).
Elsayed, F. A. et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 23, 1080–1084 (2015).
Jansen, A. M. et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 24, 1089–1092 (2016).
Domingo, E. et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 1, 207–216 (2016).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02912572 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02899793 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02658279 (2019).
Hersom, M. & Jorgensen, J. T. Companion and complementary diagnostics-focus on PD-L1 expression assays for PD-1/PD-L1 checkpoint inhibitors in NSCLC. Ther. Drug Monit. 40, 9–16 (2017).
André, T. et al. Analysis of tumor PD-L1 expression and biomarkers in relation to clinical activity in patients (pts) with deficient DNA mismatch repair (dMMR)/high microsatellite instability (MSI-H) metastatic colorectal cancer (mCRC) treated with nivolumab (NIVO) + ipilimumab (IPI): CheckMate 142. Ann. Oncol. 28, 484PD (2017).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Koelzer, V. H., Baker, K., Kassahn, D., Baumhoer, D. & Zlobec, I. Prognostic impact of beta-2-microglobulin expression in colorectal cancers stratified by mismatch repair status. J. Clin. Pathol. 65, 996–1002 (2012).
Shin, D. S. et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7, 188–201 (2017).
Bindea, G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795 (2013).
Chifman, J., Pullikuth, A., Chou, J. W., Bedognetti, D. & Miller, L. D. Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer 16, 911 (2016).
Sinicrope, F. A. O. et al. Randomized trial of FOLFOX alone or combined with atezolizumab as adjuvant therapy for patients with stage III colon cancer and deficient DNA mismatch repair or microsatellite instability (ATOMIC, Alliance A021502). J. Clin. Oncol. 35, TPS3630 (2017).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02912559 (2019).
Grootscholten, C. et al. Neoadjuvant ipilimumab plus nivolumab in early stage colon cancer. Ann. Oncol. 29, LBA37_PR (2018).
Watson, P. et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer 123, 444–449 (2008).
Syngal, S. et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 110, 223–262; quiz 263 (2015).
Schwitalle, Y. et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 134, 988–997 (2008).
Kloor, M. & von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancer. Trends Cancer 2, 121–133 (2016).
Reuschenbach, M. et al. Serum antibodies against frameshift peptides in microsatellite unstable colorectal cancer patients with Lynch syndrome. Fam. Cancer 9, 173–179 (2010).
Doeberitz, M. v. K. et al. Frameshift peptide neoantigens as vaccine targets in microsatellite-unstable cancers. Cancer Immunol. Res. 4, A006 (2016).
Woo, S. R. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Grosso, J. F. et al. LAG-3 regulates CD8+T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Invest. 117, 3383–3392 (2007).
Ngiow, S. F. et al. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 71, 3540–3551 (2011).
Sakuishi, K. et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010).
Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016).
Ward-Kavanagh, L. K., Lin, W. W., Sedy, J. R. & Ware, C. F. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 44, 1005–1019 (2016).
Croft, M., Benedict, C. A. & Ware, C. F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 12, 147–168 (2013).
Brenner, D., Blaser, H. & Mak, T. W. Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 15, 362–374 (2015).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
Miliotou, A. N. & Papadopoulou, L. C. CAR T cell therapy: a new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 19, 5–18 (2018).
Yeku, O. O. & Brentjens, R. J. Armored CAR T cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T cell anti-tumour efficacy. Biochem. Soc. Trans. 44, 412–418 (2016).
Shum, T., Kruse, R. L. & Rooney, C. M. Strategies for enhancing adoptive T cell immunotherapy against solid tumors using engineered cytokine signaling and other modalities. Expert Opin. Biol. Ther. 18, 653–664 (2018).
Parkhurst, M. R. et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 19, 620–626 (2011).
Katz, S. C. et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T cell therapy for CEA+liver metastases. Clin. Cancer Res. 21, 3149–3159 (2015).
Zhang, C. et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Mol. Ther. 25, 1248–1258 (2017).
Tran, E. et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375, 2255–2262 (2016).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
Kochenderfer, J. N. et al. Chemotherapy-refractory diffuse large B cell lymphoma and indolent B cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 33, 540–549 (2015).
Johnson, L. A. & June, C. H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 27, 38–58 (2017).
Newick, K., O’Brien, S., Moon, E. & Albelda, S. M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 68, 139–152 (2017).
Hoover, H. C. Jr. et al. Adjuvant active specific immunotherapy for human colorectal cancer: 6.5-year median follow-up of a phase III prospectively randomized trial. J. Clin. Oncol. 11, 390–399 (1993).
Harris, J. E. et al. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group Study E5283. J. Clin. Oncol. 18, 148–157 (2000).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Lin, H. et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320, 807–811 (2008).
Otero, K. et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 10, 734–743 (2009).
Ngambenjawong, C., Gustafson, H. H. & Pun, S. H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 114, 206–221 (2017).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02777710 (2019).
Pollard, J. W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78 (2004).
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Arlauckas, S. P. et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl Med. 9, eaal3604 (2017).
Ablasser, A. et al. cGAS produces a 2′;-5′;-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Woo, S. R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Corrales, L. et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018–1030 (2015).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT02675439 (2018).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03010176 (2019).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT03172936 (2018).
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J. & Schreiber, R. D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998 (2002).
Freeman-Keller, M. et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin. Cancer Res. 22, 886–894 (2016).
Lo, J. A., Fisher, D. E. & Flaherty, K. T. Prognostic significance of cutaneous adverse events associated with pembrolizumab therapy. JAMA Oncol. 1, 1340–1341 (2015).
Boland, P. M. & Ma, W. W. Immunotherapy for colorectal cancer. Cancers 9, 50 (2017).
Reviewer information
Nature Reviews Gastroenterology & Hepatology thanks T. André, J. Lee and the other anonymous reviewer(s), for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
K.G., Z.K.S., A.C., R.B.M. and N.H.S. researched data for the article. K.G., Z.K.S., A.C., J.S., N.H.S. and L.A.D. made substantial contributions to discussion of the article content. K.G., Z.K.S., A.C., R.B.M., J.S. and N.H.S. wrote the manuscript. K.G., Z.K.S., N.H.S. and L.A.D. reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
R.B.M. is a speaker for Vindico and Medscape and a consultant for Roche. N.H.S. receives research funding from Roche/Genentech, Merck, Bristol-Myers Squibb, MedImmune/AstraZeneca and Incyte and is on the advisory board of Roche/Genentech, Merck, Bristol-Myers Squibb, MedImmune/AstraZeneca, Boehringer Ingelheim and Pfizer. L.A.D. is a founder and shareholder of PapGene and Personal Genome Diagnostics (PGDx) and a consultant for Merck, PGDx and Phoremost. PapGene and PGDx, as well as other companies, have licensed technologies from Johns Hopkins University on which L.A.D. is an inventor. These licences and relationships are associated with equity or royalty payments to L.A.D. L.A.D. is also a member of the board of directors of PGDx and Jounce Therapeutics. The terms of these arrangements are being managed by Johns Hopkins and Memorial Sloan Kettering in accordance with their conflict of interest policies. K.G., Z.K.S., A.C. and J.S. declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov database: https://clinicaltrials.gov/ct2/home
Rights and permissions
About this article

Cite this article
Ganesh, K., Stadler, Z.K., Cercek, A. et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 16, 361–375 (2019). https://doi.org/10.1038/s41575-019-0126-x
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-019-0126-x
This article is cited by
-
Characterizing and forecasting neoantigens-resulting from MUC mutations in COAD
Journal of Translational Medicine (2024)
-
A multi-dimensional approach to unravel the intricacies of lactylation related signature for prognostic and therapeutic insight in colorectal cancer
Journal of Translational Medicine (2024)
-
New immunotherapy approaches for colorectal cancer: focusing on CAR-T cell, BiTE, and oncolytic viruses
Cell Communication and Signaling (2024)
-
Crosstalk between colorectal CSCs and immune cells in tumorigenesis, and strategies for targeting colorectal CSCs
Experimental Hematology & Oncology (2024)
-
Inhibition of autophagy-related protein 7 enhances anti-tumor immune response and improves efficacy of immune checkpoint blockade in microsatellite instability colorectal cancer
Journal of Experimental & Clinical Cancer Research (2024)
