
Abstract
Here, as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, for which whole-genome and—for a subset—whole-transcriptome sequencing data from 2,658 cancers across 38 tumor types was aggregated, we systematically investigated potential viral pathogens using a consensus approach that integrated three independent pipelines. Viruses were detected in 382 genome and 68 transcriptome datasets. We found a high prevalence of known tumor-associated viruses such as Epstein–Barr virus (EBV), hepatitis B virus (HBV) and human papilloma virus (HPV; for example, HPV16 or HPV18). The study revealed significant exclusivity of HPV and driver mutations in head-and-neck cancer and the association of HPV with APOBEC mutational signatures, which suggests that impaired antiviral defense is a driving force in cervical, bladder and head-and-neck carcinoma. For HBV, HPV16, HPV18 and adeno-associated virus-2 (AAV2), viral integration was associated with local variations in genomic copy numbers. Integrations at the TERT promoter were associated with high telomerase expression evidently activating this tumor-driving process. High levels of endogenous retrovirus (ERV1) expression were linked to a worse survival outcome in patients with kidney cancer.
Similar content being viewed by others

Main
The World Health Organization estimates that 15.4% of all cancers are attributable to infections and 9.9% are linked to viruses1,2. Cancers that are attributable to infections have a greater incidence than any individual type of cancer worldwide. Eleven pathogens have been classified as carcinogenic agents in humans by the International Agency for Research on Cancer (IARC)3. After Helicobacter pylori (associated with 770,000 cases worldwide), the four most prominent infection-related causes of cancer are estimated to be viral2: HPV4 (associated with 640,000 cases), HBV5 (420,000 cases), hepatitis C virus (HCV)6 (170,000 cases) and EBV7 (120,000 cases). It has been shown that viruses can contribute to the biology of multistep oncogenesis and are implicated in many of the hallmarks of cancer8. Notably, the discovery of links between infection and cancer types has provided actionable opportunities, such as the use of HPV vaccines as a preventive measure, to reduce the global impact of cancer. The following characteristics have been proposed to define human viruses that cause cancer through direct or indirect carcinogenesis9: (1) presence and persistence of viral DNA in tumor biopsies; (2) growth-promoting activity of viral genes in model systems; (3) dependence of a malignant phenotype on continuous viral oncogene expression or modification of host genes; and (4) epidemiological evidence that a virus infection represents a major risk for the development of cancer.
The worldwide efforts of comprehensive genome and whole-transcriptome analyses of tissue samples from patients with cancer have generated appropriate facilities for capturing information not only from human cells but also from other—potentially pathogenic—organisms or viruses that are present in the tissue. A comprehensive collection of whole-genome and whole-transcriptome data from cancer tissues has been generated within the International Cancer Genome Consortium (ICGC) project PCAWG10, providing a unique opportunity for a systematic search for tumor-associated viruses.
The PCAWG Consortium aggregated whole-genome sequencing (WGS) data from 2,658 cancers across 38 tumor types that have been generated by the ICGC and The Cancer Genome Atlas (TCGA) projects. These sequencing data were reanalyzed with standardized, high-accuracy pipelines to align to the human genome (build hs37d5) and identify germline variants and somatically acquired mutations10. The PCAWG working group ‘Pathogens’ analyzed the WGS and whole-transcriptome sequencing (RNA-sequencing (RNA-seq)) data of the PCAWG consensus cohort (2,656 donors). Focusing on viral pathogens, we applied three independently developed pathogen-detection pipelines ‘Computational Pathogen Sequence Identification’ (CaPSID)11, ‘Pathogen Discovery Pipeline’ (P-DiP) and ‘Searching for Pathogens’ (SEPATH) to generate a large compendium of viral associations across 38 cancer types. We extensively characterized the known and novel viral associations by integrating driver mutations, mutational signatures, gene expression profiles and patient survival data of the same set of tumors analyzed by the PCAWG Consortium.
Results
Identification of tumor-associated viruses
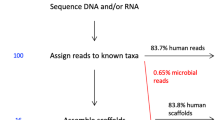
To identify the presence of viral sequences, we explored the WGS data of 5,354 tumor–normal samples across 38 cancer types, and 1,057 tumor RNA-seq data across 25 cancer types (Supplementary Tables 1, 2, 20). In total, 195.8 billion reads were considered for analysis, as they were not sufficiently aligned to the human reference genome in the PCAWG-generated alignment. The remaining reads ranged from 28,036 to 800 million reads per WGS and up to 120 million reads per RNA-seq tumor sample (Fig. 1a, Extended Data Fig. 1a–c). Viral sequences were detected and quantified independently by the three recently developed pathogen-discovery pipelines CaPSID, P-DiP and SEPATH. The estimated relative abundance of a virus was calculated as viral reads per million extracted reads (PMER) at the genus level to improve consistency between pipelines. To minimize the rate of false-positive hits in virus detection, we applied a strict threshold of PMER > 1 supported by at least three viral reads as suggested in previous studies11,12. Virus detection in a sample by at least two pipelines was considered to be a consensus hit. In total, 532 genera were considered for the extensive virus search in at least two of the pipelines (Extended Data Fig. 1d, Supplementary Table 18). Filtering of suspected viral laboratory contaminants was achieved through P-DiP, by examining each assembled contig of viral sequence segments for artificial, non-viral vector sequences and inspecting virus genome coverage across all positive samples (Extended Data Fig. 2a). The most frequent hits prone to suspected contamination were lambdavirus, alphabaculovirus, microvirus, simplexvirus, hepacivirus, cytomegalovirus (CMV), orthopoxvirus and punalikevirus; these were observed across many tumor types (Fig. 1b). For example, mastadenovirus showed an uneven genome coverage that could result from contaminating vector sequences. Therefore, we analyzed the virus detections across sequencing dates (Extended Data Fig. 2b) to assess any batch effect indicative of a contaminant; in mastadenovirus, we identified an association with sequencing date in early-onset prostate cancer regardless of tumor–normal state. We conclude that our mastadenovirus detections are due to a contamination that occurred across projects worldwide for which similar patterns could be identified.

a, Workflow to identify and characterize viral sequences from the WGS and RNA sequencing of tumor and non-malignant samples. Viral hits were characterized in detail by using several clinical annotations and resources generated by PCAWG. The red line represents the median. CNS, central nervous system. b, Identified viral hits in contigs that showed higher viral reads PMER for artificial sequences such as vectors than for the virus. All viruses that occurred in at least 20 primary tumor samples in the same contig together with an artificial sequence are shown. c, Summary of the viral search space used in the analysis grouped by virus genome type. The number of virus-positive tumor samples is indicated in the outer rings (PMER log scale for WGS and RNA-seq data) as detected by any of the pipelines. Taxonomic relations between the viruses are indicated by the phylogenetic tree. dsDNA, double-stranded DNA; dsDNA-RT, double-stranded DNA with reverse transcriptase; dsRNA, double-stranded RNA; ssDNA, single-stranded DNA; ssRNA-RT: single-stranded RNA with reverse transcriptase; ssRNA, single-stranded RNA; dsRNA, double-stranded RNA. The fractions of hits in WGS and RNA-seq data are depicted as stacked bar graphs.
We generally observed a strong overlap of the genera identified across pipelines (Extended Data Fig. 1e, Supplementary Tables 6, 7, 11). From the WGS dataset, we identified 321, 598 and 206 virus–tumor pairs using P-DiP, CaPSID and SEPATH, respectively (Fig. 2a; overlap after random permutation of detections, Extended Data Fig. 3a, Supplementary Tables 3–5). The number of hits derived from the RNA-seq dataset differed between the pipelines (virus–tumor pairs: 101 for P-DiP, 83 for CaPSID, 41 for SEPATH; Fig. 2b, Supplementary Tables 8–10). SEPATH, which used a k-mer approach, detected the lowest number of virus hits and was the least sensitive. Despite this, the identified viruses matched well with the consensus (DNA 90%, RNA 95%). P-DiP, which was based on an assembly and BLAST approach, detected more hits with 59% of the DNA and 54% of the RNA hits in the consensus set, whereas CaPSID, which was the most sensitive, implemented a two-step alignment process complemented with an assembly step and identified 60% (DNA) and 80% (RNA) hits within the consensus set. Although the majority of the virus hits from RNA-seq (n = 61 out of 68 consensus hits based on RNA-seq) overlapped with the WGS data, a lower fraction of detections from the WGS data were present in the RNA-seq data (n = 61 out of 168 of 382 consensus hits based on WGS with available RNA-seq data), emphasizing the importance of DNA sequencing for generating an unbiased catalog of tumor-associated viruses. This difference can also be attributed to the viral life cycle, as viral gene expression can be minimal during incubation or latent phases13. Contrasting virus-positive and virus-negative samples within each organ type shows that the organ system, as expected, has a significant influence, but virus positivity does not (P < 2 × 10−16, analysis of variance modeling of candidate reads that are dependent on organ system and virus positivity; Extended Data Fig. 1c). This indicates that virus-positive tumors were not detected owing to a higher number of candidate reads; this is consistent with the fact that the viral reads in most cases do not substantially contribute to the reads analyzed. In total, 86% of the sequence hits detected in WGS and RNA-seq data were found to be from double-stranded DNA viruses and double-stranded DNA viruses with reverse transcriptase (Fig. 1c, Supplementary Table 19). This could be attributed to (1) a higher frequency of tumor-associated viruses from these genome types3, (2) a larger sequencing dataset for WGS compared with RNA-seq, (3) a potential limitation of our analysis due to DNA and RNA extraction protocols that are less likely to include single-stranded DNA or RNA viruses or (4) the selection bias of tumor entities included in the PCAWG study (Fig. 1c).

Number of genus hits among tumor samples for the three independent pipelines and the consensus set defined by evidence from multiple pipelines. a, Analysis based on WGS. b, Analysis based on whole-transcriptome sequencing. c, Heat map showing the total number of viruses detected across various cancer entities. The sequencing data used for detection are indicated among the total number of hits (WGS, blue; RNA sequencing, green). The fraction of virus-positive samples is shown at the top and the type of non-malignant tissue used in the analysis is indicated if more than 15% of the analyzed samples are from a respective tissue type (solid tissue, lymph node, blood or adjacent to primary tumor). d, t-SNE clustering of the tumor samples based on PMER of their consensus virome profiles, using Pearson correlation as the distance metric. Major clusters are highlighted by indicating the strongest viral genus and the dominant tissue types that are positive in that cluster. Dot size represents the viral reads PMER.
The virome landscape across 38 distinct tumor types
We used a consensus approach that resulted in a reliable set of 389 distinct virus–tumor pairs from WGS and RNA-seq data (Fig. 2a–d). Overall, 23 virus genera were detected across 356 patients with cancer (13%). The top-five most-prevalent viruses (lymphocryptovirus, orthohepadnavirus, roseolovirus, alphapapillomavirus and CMV) account for 85% of the consensus virus hits in tumors (n = 329 out of 389). Among these five prevalent virus genera, three have been well described in the literature as drivers of tumor initiation and progression9: (1) lymphocryptovirus (n = 145 samples (5.5%); for example, EBV) is the most common viral infection across a variety of tumor entities that mainly occur in the gastrointestinal tract and shows a much lower prevalence in the matched non-malignant control samples (n = 82 (3%); Fig. 2c); (2) orthohepadnavirus (n = 67 (2.5%); for example, HBV) is—as expected—the most frequent among liver cancer with HBV present in 62 of 330 donors (18.9%); and (3) alphapapillomavirus (discussed below). Lymphocryptovirus (n = 11), orthohepadnavirus (n = 18) and alphapapillomavirus (n = 32) were detected in both RNA-seq and DNA-sequencing data (Fig. 2c, left), of which alphapapillomavirus was the most frequent (32 out of 39 consensus hits). This is consistent with the constitutive expression of viral oncogenes in cancers associated with these viruses, a parameter that supports a direct role in carcinogenesis9. An in-depth analysis of the virus genome equivalents per human tumor genome equivalent, which considers genome sizes, coverage and tumor purity, showed overall low viral genome equivalents even for established tumor viruses (Extended Data Fig. 3c, Supplementary Table 12). Evidence of a mouse mammary tumor virus (MMTV, PMER = 3.4) was detected in one renal carcinoma sample and in none of the 214 analyzed breast cancer samples. Previous work has suggested that MMTV may have a role in breast cancer but our comprehensive search of viral sequences could not identify any MMTV-positive case in breast cancer that would support this claim.
Roseolovirus and alphatorquevirus show a higher number of hits in non-malignant control samples, which were mainly derived from blood cells (Fig. 2c). For example, we identified 59 patients as roseolovirus-positive (human herpesvirus (HHV)-6A, HHV-6B and HHV-7) in their tumors (pancreas, 6%; stomach, 8%; colon/rectum, 8.3%) and 90 patients positive in the non-malignant control samples. Considering the known cell tropism of roseolovirus for B and T cells15, we asked whether immune infiltration would be higher in roseolovirus-positive tumors. However, we could not identify a stronger contribution of immune cells in virus-positive tumor cases as estimated using CIBERSORT14 (false-discovery rate (FDR)-corrected P > 0.05 for pancreas; Extended Data Fig. 4a). Therefore, consistently with current knowledge (reviewed in ref. 16), we cannot confirm a link between roseolovirus and immune-cell content or tumor development. Furthermore, we could not identify actively transcribed viral genes for roseolovirus and alphatorquevirus at the transcriptome level. This is in agreement with the latent state of these viruses in blood mononuclear cells15, and their transmission through blood transfusions17. CMV was found, as expected18, after identification and removal of contaminations in both stomach tumors (n = 13) and the adjacent non-malignant tissue (n = 11). In line with a recent publication19, we could not detect CMV in the 294 tumors of the central nervous system (146 medulloblastomas, 89 pilocytic astrocytoma, 41 glioblastomas and 18 oligodendrogliomas) that were analyzed. Therefore, a previously debated role of this virus is not supported. Notably, we did not identify a significant enrichment of co-infection of multiple viruses in any tumor type (Extended Data Fig. 3d).
Incidence of HBV
HBV was most frequently detected in liver cancers (n = 62). Compared with the histopathological gold-standard HBV PCR test20,21 (n = 228), the WGS-based consensus detections had the same high specificity (96.1%) and a high sensitivity (84.0%), indicating that HBV detection using WGS is reliable (Fig. 3a, Extended Data Fig. 4b, Supplementary Table 13). Furthermore, five out of the seven cases that were positive using WGS but negative for HBV PCR showed positivity for HBAg, indicating that the WGS analysis has a high sensitivity. In summary, the precision (85.7%) and recall (84%) for the detection of HBV based on around 30-fold-coverage WGS data were comparable to those of targeted PCR. We confirmed a significant exclusivity between HBV infection and mutations in CTNNB1, TP53 and ARID1A that was found in a larger liver cancer cohort analyzed by high-throughput sequencing (FDR-corrected P = 5.35 × 10−6, 0.0023 and 0.0023, respectively; DISCOVER22)23.

a, HBV detections, validations and driver mutations in liver cancer. The asterisk indicates mutual exclusivity between HBV detection and somatic driver gene mutations. Red boxes represent virus-positive tumor samples, purple boxes show viral genomic integrations, green boxes indicate driver mutations and gray boxes represent missing data. b, Virus detections in gastric cancer samples, indication of virus phase (lytic/latent, dark red) and driver mutations (green). A yellow color indicates donors with virus-positive non-malignant samples. The gray box refers to samples with available RNA-seq data. c, Virus detections (red) and driver mutations (green) in cervix (blue) and head-and-neck cancer (brown). The asterisk indicates mutual exclusivity between alphapapillomavirus detections and somatic driver gene mutations. d, Alphapapillomavirus detection and exposures of mutational APOBEC signatures SBS2 and SBS13. Sample sizes are shown at the bottom. A two-sided Wilcoxon rank-sum test showed a significant difference (P = 0.02) of mutational signature exposure between virus-positive and virus-negative head-and-neck tumor samples. The black line indicates the median for each group. e, Gene expression analysis based a t-SNE map of head-and-neck cancer samples shows a distinct gene expression profile for virus-positive samples. Virus-positive and virus-negative samples are shown as red and gray dots, respectively. f, The violin plot of APOBEC3B gene expression for alphapapillomavirus-positive and alphapapillomavirus-negative samples in cervix and head-and-neck cancer (FDR-corrected two-sided Wilcoxon rank-sum test, P = 1.6 × 10−4). FPKM, fragments per kilobase of transcript per million mapped reads. The center line represents the median, and the upper and lower boundaries of the violin plot refer to the maximum and minimum values, respectively. g, Tumor-infiltrating immune cells as quantified by CIBERSORT using RNA-seq samples from patient with head-and-neck cancer. All four cell types showed significant enrichment of immune cells in virus-positive samples (FDR-corrected two-sided Wilcoxon rank-sum test, n = 24 virus negative versus 18 virus positive). Tukey box plots show the median (the middle line) and the 25–75th percentiles (the box); the whiskers show 1.5× the interquartile range from the lower and upper quartile.
Detection of EBV
EBV was detected in many different tumor entities and normal samples (Fig. 2c). When comparing the PMER of EBV in tumor and matched normal samples, we see a stronger contribution in matched normal samples from matched solid tissue or tissue adjacent to the tumor (Extended Data Fig. 4c). For samples that contained reads for EBV in WGS and with available RNA-seq data, the absolute score for immune cells based on CIBERSORT14 was not significantly different between virus-positive and virus-negative samples (FDR-corrected P > 0.05 for colon/rectum, head-and-neck, lymphoid and stomach; Extended Data Fig. 4a). In summary, there is no evidence that the detection of EBV is due to infiltrating immune cells. This indicates the presence of EBV in the respective organs. On the basis of the expression data available for the tumor samples, we identified viral transcripts of the latent as well as lytic phase of the viral life cycle (Fig. 3b, Extended Data Fig. 4d, Supplementary Table 13). Eight of the nine tumors that expressed lytic EBV transcripts were from stomach cancers, confirming the active contribution of EBV to gastric cancer24.
Identification of alphapapillomaviruses
Alphapapillomaviruses were mainly detected in head-and-neck cancers (n = 18 out of 57), cervical cancers (n = 19 out of 20) and in two bladder cancer cases out of 23, in agreement with previous studies4,25,26. There is also supporting evidence for 32 out of 39 alphapapillomavirus hits in the whole-transcriptome data (Fig. 2c). We observed only one HPV subtype per tumor according to the P-DiP results and HPV16 was the dominant type in cervical (n = 11) and head-and-neck (n = 15) tumors, followed by HPV18, which was present in only cervical cancer (n = 6). As reported previously27, HPV33 was identified in head-and-neck (n = 3) and cervical (n = 1) tumors. Different HPV variants, type 6 and 45, were detected in bladder cancer.
In head-and-neck cancer, HPV-positive tumors exhibited an almost complete mutual exclusivity with mutations in known drivers such as TP53, CDKN2A and TERT (FDR-corrected P = 1.73 × 10−5, 1.73 × 10−5 and 0.012, respectively; multiple testing corrected for presented mutations in EBV and HPV, DISCOVER22) (Fig. 3c, Supplementary Table 13), as reported previously25, which could be explained by the mutation-independent inactivation of TP53 due to the human papillomaviruses28,29,30. Furthermore, we found that mutational signature 2 was enriched in alphapapillomavirus-positive cases of head-and-neck cancer31 (FDR-corrected P = 0.02; Fig. 3d, Supplementary Tables 12, 22). In addition, the expression of APOBEC3B is significantly higher in virus-positive head-and-neck cancers compared with virus-negative cancers32 (P = 1.6 × 10−4; Fig. 3f). However, we did not observe enrichment of APOBEC signatures and changes in expression in EBV-positive samples found in the cervix or in other tissues.
Distinct expression profiles between virus-positive and virus-negative tumors in head-and-neck cancer were observed33 (Fig. 3e, Supplementary Table 23). Analyzing the immune cells estimated by CIBERSORT, we identified a significant increase in macrophages and T-cell signals in alphapapillomavirus-positive head-and-neck cancers (P = 0.004, 0.012 and 0.012 for follicular helper, CD8 and regulatory T cells, respectively, and P = 0.018 for M1 macrophages; FDR corrected for all viruses and cell types tested; Fig. 3g, Supplementary Table 24). Our integrative analysis of HPV reconfirms many of the findings related to HPV infection, illustrating the potential of our systematic approach in identifying and characterizing tumor-associated viruses.
Activation of endogenous retroviruses linked to outcome
Human endogenous retroviruses (HERV) are integrations in the human DNA that originate from infection of germline cells by retroviruses over millions of years34 and contribute over 500,000 individual sites, or 2.7% of the overall sequence the human genome35,36. ERVs were identified by all three pathogen-detection pipelines but were filtered by CaPSID and SEPATH. In addition, an alignment-based approach was used to detect HERV sequences that were embedded in the human reference genome that could be missed by the pipelines by focusing only on non-human reads. In this study, we quantified the expression of HERV-like long terminal repeat retrotransposons that were categorized into several clades by Repbase37 as ERVL, ERVL-MaLR, ERV1, ERVK and ERV (Supplementary Table 14). In comparison to the other HERV families, ERV1 shows the strongest expression on average (Fig. 4a) and ERVK the highest fraction of active loci (Fig. 4b). By analyzing the expression of HERVs, we could identify strong expression of ERV1 in chronic lymphocytic leukemia compared with all other tumor tissues and adjacent normal tissues (Fig. 4c). However, we could not identify a link between transcriptionally active stemness markers (OCT3/4, SOX2 and KLF4) and increased HERV expression, in contrast to a previous report38 (Spearman rank correlation <0.35; Extended Data Fig. 5). New data suggest that expression of HERVs is associated with prognosis in clear cell renal cell carcinoma39. Analyzing HERV expression in relation to patient survival, we found that high ERV1 expression in kidney cancer was linked to worse survival outcome (P = 0.0081; log-rank test; Fig. 4d, Extended Data Fig. 6, Supplementary Table 15).

a, Heat map showing the expression of HERV across all tumor samples. HERV transcripts per million (TPM) were grouped by family and summed up. Hierarchical clustering was performed by family according to Manhattan distance with complete linkage after log2 transformation of HERV TPM expression values.(RCC, renal cell carcinoma). b, Fraction of active loci in the genome with a TPM > 0.2 plotted against the fraction of samples. c, TPM-based expression of the highly expressed HERVs ERV1 and ERVK across tumor types. n, number of analyzed tumor samples. Violin plots are shown; red dots indicate the median. The upper and lower boundaries of the violin plot extend to the maximum and minimum values. d, Survival difference between patients with kidney cancer expressing high (red) and low levels (blue) of ERV1. Kaplan–Meier curve shows the overall survival of patients (n = 113) with high and low levels of ERV1 with a cut-off of 16.3 TPM (log-rank test P = 0.0081). The number of patients at risk is shown at the bottom.
Genomic integration of viral sequences
Viral integration into the host genome has been shown to be a causal mechanism that can lead to the development of cancer40. This process is well-established for HPVs in cervical, head-and-neck and several other carcinomas, and for HBV in liver cancer41,42.
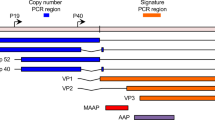
Low-confidence integration events were detected for HHV4 (gastric cancer and malignant lymphoma) and HPV6b (head-and-neck and bladder carcinoma), whereas integration events with high confidence were demonstrated for HBV (liver cancer), AAV2 (liver), HPV16 and HPV18 (in both cervical and head-and-neck carcinoma). Most of these integration events were found to be distributed across chromosomes and a significant number of viral integrations occurred in the intronic (40%) regions whereas only 3.4% of integrations was detected in gene coding regions (n = 84 intronic versus n = 31 other regions excluding intergenic regions, two-sample test for equality of proportions, P = 7.0 × 10–12; Extended Data Fig. 7a–d).
HBV was found to be integrated in 36 liver cancer specimens out of 61 patients who were identified to be HBV positive. Notably, genomic clusters of viral integrations were identified in TERT (number of integration sites within a genomic cluster (NGC) of 6), KMT2B (NGC = 4)—which was recently identified to be a likely cancer driver gene43,44—and RGS12 (NGC = 3) (Extended Data Fig. 7e). Furthermore, two or more integration events in individual samples were observed in the gene (or gene promoter) regions of CCNE1, CDK15, FSIP2, HEATR6, LINC01158 (also known as PANTR1), MARS2 and SLC1A7 (Fig. 5a). Additional events with two integration sites were also detected within a distance of 50 kb from CLMP, CNTNAP2 and LINC00359 genes. Integration events at TERT were found to recur in five different liver cancer samples. One sample had a genomic cluster of three viral integration events within TERT and four samples contained a single integration event in the TERT promoter, or 3′ or 5′ untranslated regions (UTR) (Supplementary Table 17). When comparing gene expression in samples with virus integration to those without, we found that only TERT was overexpressed (fold change ≥2.0) in two liver cancer samples (Fig. 5e). Additional genes with increased expression that were influenced by integration events include TEKT3, CCNA2, CDK15 and THRB (Fig. 5a).

a, Integration sites detected in gene regions (including promoter, exon, intron and 5′ UTR regions) are labeled in red for increased gene expression and blue for expression measured. Rows of each heat map designate the nearest genes to the integration sites, and columns represent individual ICGC donor and project IDs. Intragenic HBV integration sites detected in liver cancers (ICGC project codes: LIRI, LIHC and LINC). For TERT and SEMA6D, intergenic integrations are also shown. b, Integration sites detected for HPV16 and HPV18 in head-and-neck (magenta) and cervical (blue) cancers (ICGC project codes: HNSC and CESC). Gene labels with an asterisk indicate HPV18 as opposed to HPV16 viral integrations. c, A local increase in the number of SCNAs was shown in the vicinity of HBV integrations (n = 21 viral integrations in individual patients, P = 7.4 × 10−3; two-sided paired t-test). d, Genomic visualization of the HBV integration sites relative to the TERT gene in five patients with liver tumors. e, The increased gene expression (in FPKM, upper-quartile normalization, UQ) of TERT in two liver tumors with HBV integrations in comparison to the expression of TERT in tumor and non-malignant adjacent tissues. Tumor samples with a non-coding driver mutation are labeled in orange.
There was a significant association between HBV viral integrations and somatic copy-number alterations (SCNAs, Fig. 5c). For samples with HBV integration events, the number of SCNAs was higher on average in the vicinity of viral integration sites (within 1 Mb) compared with samples without HBV integration (mean 4.2 versus 2.3, P = 7.4 × 10−3; two-sided paired t-test). No evidence of an SCNA association was seen for other integrated viruses like HPV16 and HPV18 (Extended Data Fig. 8a,b).
HPV18 integration events were detected in seven tumors in total (Fig. 5b), with the most notable clusters of integration events that affected TALDO1 (NGC = 4) in cervical cancer samples (Extended Data Fig. 7g).
In 20 samples, HPV16 integration events were detected. Genomic clusters of viral integration sites were identified in cervical and head-and-neck cancer samples (Extended Data Fig. 7f). None of these multiple integration events were observed to recur across patients (Fig. 5b). Integration events were also observed in two different long noncoding RNAs (lncRNAs), LINC00111 and the plasmacytoma variant translocation 1 gene (PVT1), an oncogenic lncRNA45,46. Expression of both genes is strongly increased in the cases with HPV16 integration (Extended Data Fig. 8f, Supplementary Table 17).
Using the PCAWG SNV calls10, we found a significant increase in the number of mutations that occurred within ±10,000 bp of high-confidence viral integration sites (average number of mutations per sample, 0.41 (HPV16+) versus 0.14 (HPV16−), P = 0.02; one-sided paired t-test, alternative greater, Extended Data Fig. 8c,d). Notably, the integration sites are—compared with a random genome background—enriched in proximity (<1,000 bp) to common fragile sites (P = 0.0018, Kolmogorov–Smirnov test). These results suggest that HPV16 integration reflects either characteristics of chromatin features that favor viral integration, such as fragile sites or regions with limited access to DNA repair complexes, or the influence of integrated HPV16 on the host genome. Such a correlation was not seen for the integration sites of other viruses (Extended Data Fig. 8e). Finally, a single AAV2 integration event located in the intronic region of the cancer driver gene KMT2B 47 was detected in one liver cancer sample.
Identification of novel viral species or strains
De novo analysis using the CaPSID pipeline has generated 56 different contigs that have been classified into taxonomic groups at the genus level by CSSSCL48. After filtering de novo contigs for their homology to known reference sequences, we identified 29 contigs in 28 different tumor samples that showed low sequence similarity (on average 63%) to any nucleotide sequence contained in the BLAST database. In this respect, our analysis has shown that WGS and RNA-seq can be used to identify isolates from potentially new viral species. However, the total numbers of novel isolates were low in comparison to viral hits to well-defined genera (Fig. 2c). These de novo contigs were not enriched for a specific tumor entity but rather were distributed across cancer types including bladder, head-and-neck and cervical cancers (Extended Data Fig. 9).
Discussion
Searching large pan-cancer genome and whole-transcriptome datasets enabled the identification of a high percentage of virus-associated cases (16%). In particular, analysis of tumor genomes, which were sequenced on average to a depth of at least 30-fold coverage, identified considerably more virus-positive cases than investigations of whole-transcriptome data alone, which is the search space analyzed in most previous virome studies. This is probably mainly due to viruses with no or only weak transcriptional activity in the given tumor tissue. Co-infections, generally believed to indicate a weak immune system, were very rare (Extended Data Fig. 3d). This could, however, also be the result of selection processes during tumorigenesis.
Although universal criteria for a causality of viral pathogens are prone to errors, it is worthwhile to look at individual features that might support a potentially pathomechanistic contribution of a given pathogen. These include aspects that affect the expression of host factors (for example, after viral integration) or the mutual exclusivity of the presence of viral genomes and other host factors, which are already known to have a role in the etiology of a given tumor type. Such aspects need to be carefully considered when discussing what strengthens the potentially pathogenic role of a virus.
Not surprisingly, known tumor-associated viruses, such as EBV, HBV, HPV16 and HPV18, were among the most frequently detected targets. Notably, viral detection based on WGS showed similar performance with respect to precision and recall as a targeted PCR for HBV, indicating that this approach is sensitive to detect viruses. This is particularly true for the common integration verified for HBV, HPV16 and HPV18 in our study. In addition, the common theme of potential pathomechanistic effects by the genomic integration of viruses, which were also supported by the observations of multiple nearby integration sites in a given tumor genome that we report in the present study, has gained further momentum. By analyzing the effect of viral integrations on gene expression, we identified several links to genes nearby the integration site. In this regard, the frequently observed integration of HBV at the TERT promoter accompanied with the transcriptional upregulation of TERT constitutes an intriguing mechanistic example, as the increased activity of TERT is a well-understood driver of carcinogenesis49. Furthermore, we also linked viral integrations to increased mutations (SNVs and SCNAs) nearby the integration site.
The known causal role of HPV16 and HPV18 in several tumor entities, which triggered one of the largest measures in cancer prevention, has been the motivation for extensive elucidation of the pathogenetic processes involved. Nevertheless, comprehensive analyses of WGS and RNA-seq datasets revealed additional novel findings. While we confirmed the exclusivity of HPV infection and TP53, CDKN2A and TERT mutations in head-and-neck tumors, we could also link virus presence to an increase in mutations attributed to the mutational signature 2 (ref. 50). These are explained by the activity of APOBEC, which—among other effects—changes viral genome sequences as a mechanism of cellular defense against viruses51,52. This activation could have an important function in introducing further host genome alterations and, thus, constitute an important mechanism that drives tumorigenesis32,52. In liver cancer, mutations in CTNNB1, TP53 and ARID1A, major primary oncogenes in this cancer type and HBV infections were confirmed to occur significantly mutually exclusive23. Furthermore, the virus-positive head-and-neck cancer samples had a significantly higher abundance of T-cell and M1 macrophage expression signals, which is in agreement with recently described subtypes of head and neck squamous cell carcinoma that differ—among other features—in virus infection and inflammation features.
Methods
Identifying potential pathogenic reads
To reduce the number of reads to be considered for the pathogen search, we identified potential pathogenic reads by using P-DiP (https://github.com/mzapatka/p-dip). On the basis of reads aligned to hg19 by BWA53 or STAR54 using the standard PCAWG approach, we identified read pairs for which at least one read did not map well to the human genome (longest stretch of mapped bases from 20 to 30 bases) and read pairs that were unmapped or mapped to NC_007605 (human herpesvirus 4, which is contained in the 1000 Genomes version of the hg19 human reference genome), and extracted these for further processing. To speed up the extraction, we used bamcollate2 from Biobambam2 (v.2.08) 55 as an input stream to the Python script.
Identification of ERVs
The expression of ERVs was analyzed using RNA-seq data and aligned STAR sequences based on the settings developed within PCAWG (hg19 and Gencode 19). In contrast to the standard pipeline, the reference transcripts from Gencode 19 were enriched by adding HERV locations extracted from RepeatMasker (http://www.repeatmasker.org, rmsk from UCSC, version 17/08/03) and Featurecounts (subread-1.5.3)56 applied to identify reads mapping to the modified reference transcripts. Resulting reads counts were converted into TPM according to Wagner et al.57.
The SEPATH pipeline
Our starting point is to take reads that are not mapped to the human genome, using the extracted potentially pathogenic reads. Low quality bases (q < 30) were trimmed from the read ends and the TruSeq indexed adapter and TruSeq universal adapter were removed using Cutadapt (v.1.8.1)58. Reads less than 32 bp were discarded. Additional filtering was performed to remove reads that contained more than 5% of Ns or those with low complexity (dust method with maximum score of 10) by using Prinseq (v.0.20.3)59. Metagenomic Phylogenic Analysis (MetaPhlAn)60,61 was then applied to identify and quantify the presence of bacterial and viral populations. MetaPhlAn comes with a curated marker database of around 1 million unique clade-specific marker genes identified from reference genomes (version 2.0 of the database was used). Reads were aligned against the unique marker gene database by using Bowtie2 (v.2.2.1)62 with presets set to sensitive. Reads were then counted and normalized giving an estimation of the relative abundance for each level of the phylogenetic tree.
Detection and analysis of microbial infectious agents by NGS P-DiP
The assembly-based pipeline (P-DiP) was further developed based on a version implemented by M.A. and A.G.63. In summary, the pipeline runs preprocessing, assembly and BLAST searches and stores processing details and final results in a postgreSQL database. For the WGS and RNA-seq analyses, we started with the potentially pathogenic reads extracted from the BWA-aligned WGS BAM files. As a first step, reads were trimmed based on quality using trimmomatic. Thereafter, host reads were subtracted by aligning to the human reference genome (WGS: hg19 excluding NC_007605 and hs37d5 and adding phiX; RNA sequencing: Homo_sapiens.GRCh37.dna.primary_assembly) using Bowtie2 (v.2.2.8)62. Trinity (v.2.0.6)64 was used for the read assembly of WGS reads that were not aligned by Bowtie with sufficient quality (not aligned with --very-fast (-D 5 -R 1 -N 0 -L 22 -i S,0,2.50) to Homo_sapiens.GRCh37.ncrna, Homo_sapiens.GRCh37.cdna.all or PhiX); for the RNA-seq data we applied idba assembler (v.1.1.3)65. Assembled contigs were filtered by size (minimal length of 300 bp). Abundance was estimated by remapping all of the reads that did not align to the human reference to the assembled contigs by using Bowtie2. Putative PCR duplicates identified by mapping location were removed from the abundance count. The taxonomic classification of the size-filtered contigs was performed using the BLAST+ package (v.2.2.30)66 and nucleotide databases nt (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nt.gz, accessed 15 May 2015) and nr (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz, accessed 20 April 2015). For the extraction of pathogen hits R-scripts were used to filter the BLAST results (https://github.com/mzapatka/p-dip). In summary, for each of the contigs, the best BLAST hits for each segment of the contig were considered and the reads aligning to these segments identified. Potential contaminants were defined based on the taxonomy annotation in NCBI taxonomy. Any taxonomy ID below plasmids (36,549), transposons (2,387), midivariant sequences (31,896), insertion sequences (2,673), artificial sequences (81,077) and synthetic viruses (512,285) was annotated as potential contamination. Segments with higher read counts of these sequences compared to pathogen hits were flagged as contaminants and not further considered.
CaPSID description of the analysis workflow
The metagenomic analysis pipeline of CaPSID11 starts by first processing a BAM file that contains the reads sequenced from a tumor (or normal) sample aligned to the human reference sequence (GRCh37/hg19). Reads that did not map to the human reference were extracted and filtered for low complexity and quality using the SGA67 preprocessing module and then aligned in single-end mode using the Bowtie2 aligner62 to 5,652 NCBI68 viral reference sequences (RefSeq) and a filter sequence reference database composed of 5,242 bacterial and 1,138 fungal reference sequences that were also downloaded from the NCBI. To improve the sensitivity and specificity with which viral sequences were detected, reads that did not map to any reference with Bowtie2 were realigned to the same viral RefSeq database, using the more-sensitive aligner SHRiMP2 in local alignment mode69. At the completion of this two-step alignment process, reads that aligned to viral reference sequences were annotated using the information stored in the genome database of CaPSID, which contains full NCBI GenBank and taxa information. Using information from each aligned read, CaPSID then calculates the following four metrics: (1) the total number of reads (or hits) that aligned across any given viral genome, (2) the total number of reads that aligned only across gene regions within any given viral genome, (3) the total coverage across each viral genome and (4) the maximum coverage across any of the genes in a given viral genome.
Filtering of viral candidates with low significance
For the analysis of the tumor WGS or RNA-seq samples, CaPSID reports candidate sequences from dozens of different viral genomes, some of which are not related to the cancer phenotype. Some of these reported viral hits are also due to a series of experimental and computational artifacts. To reduce the number of potential false-positive hits, the CaPSID pipeline flags viral genomes that could be the result of artifacts present in the sequencing data or those with no obvious relation to cancer phenotype and that could be filtered in subsequent steps. The following criteria were used to flag and filter for potential viral candidates: (1) flag viral candidates with low coverage, (2) flag bacteriophage viral genome sequences, (3) report only viral candidates with a read composition different from the one expected when generated from the host’s reference GRCh37/hg19 sequence, (4) flag viral candidates that are typically not known to infect humans and those with low read abundance and/or low overall alignment read accuracy.
In the first step, CaPSID flagged viral genomes with low read count and/or coverage using three metrics, including total number of uniquely aligned reads <3, total genome coverage <10% and maximum gene coverage <50%. Viral genomes with low read count can arise as a result of (1) low read/transcript abundance in the human sequenced sample, (2) unspecific alignment between sequenced short reads (for example, low complexity reads) and viral reference sequences and (3) for RNA-seq library preparation in which highly expressed transcripts generally dominate over low abundance targets. To limit the reporting of viral genomes with very low coverage, we chose to flag all genomes for which the maximum gene coverage was <50%. As this lower bound on the maximum gene coverage applied to individual genes and not to the complete viral genome, it appears to be unlikely that viruses with such low coverage are biologically important. The second step in our filtering approach was to flag bacteriophage viral genomes that are most likely not related to any cancer phenotype. Bacteriophages are detected as a result of the presence of bacteria (or bacterial contamination) in human sequenced samples. The third step was used to determine whether the genome coverage observed for each viral candidate was different from the one expected to arise from reads that originated exclusively from the human reference DNA GRCh37/hg19 sequence. To build the CaPSID background model, we used the ART NGS read simulator. The entire GRCh37/hg19 sequence reference file is first fed to the ART70 simulator (parameters: art_illumina [Illumina platform] -l [read length=100 bp] -f [the fold of read coverage to be simulated=100] with default values for indels and substitution rates), which then generates single-end (or paired-end) reads and base quality values.
Reads simulated by ART were then aligned to the viral reference sequence database using the same alignment approach for reads that originated from tumor samples (see above). CaPSID then calculated the four metrics for the GRCh37/hg19 background model using the alignment information from simulated reads that aligned to viral reference sequences. The fourth step consisted of flagging viral candidates that were typically not known to infect humans using a dictionary of around 130 terms that we compiled from a database of all viruses known to infect humans. In addition to the above filtering criteria, CaPSID also considered the read abundance associated with each viral candidate sequence (abundance is expressed in terms of aligned reads in parts-per million of total number of unmapped reads) and the average read percentage identity with which reads aligned to a given viral candidate reference sequence.
De novo assembly and taxonomic classification of contigs
The purpose of this analysis step is to attempt to characterize potential novel viral sequences at the species or subspecies level. Unaligned reads that could not be aligned to any of the filter/host or viral reference sequences were assembled into contigs using the IDBA algorithm65. Assembled contigs were then masked for repeat regions by using RepeatMasker and then filtered for their size and read coverage (contig length ≥500 bp and coverage >5×). Resulting contigs were then assigned to taxonomic groups at the genus level by using the CSSSCL algorithm48. Contigs lacking sequence homology to reference sequences contained in the CaPSID or BLAST nucleotide databases with percentage identity <90% were then selected as suggestive of the presence of new viral strains/isolates or species.
Defining consensus hits
Identification of the consensus hits was achieved by optimizing two features of the individual genus hits: PMER 1 as cut-off (Supplementary Note) and percentage identity >90%. The 90% percentage identity threshold was determined based on our benchmarking study12 that indicated that an alignment-based approach can still accurately characterize viral sequences with up to 10% mutation rate (compared with sequences stored in a reference database). Lowering the threshold, with which short reads align to any given reference sequence below 90% identity on average, results in a drop of sequence coverage due to a high attrition rate of aligned reads, lowering the detection rate and thus providing more uncertain characterizations of viral candidates. Notably, there was no difference in the PMER distribution of common hits across the three pipelines, indicating that a common detection cut-off is reasonable (Extended Data Fig. 3b).
The consensus set was restricted to genera that were covered in at least two detection pipelines (Extended Data Fig. 1b). Notably, we could not detect any more hits with high PMER using the unique search space of P-DiP, indicating that almost all of the viral hits from individual pipelines were also screened by another pipeline.
Virus integration detection analysis
A subset of viral candidates identified to be present in tumor samples by the CaPSID analysis pipeline (parameters used: PMER ≥ 1.1 and genome coverage > simulated background model) was selected for the detection of viral integration events using the VERSE71 algorithm. This subset of viruses included: herpesviruses (HHV1, HHV2, HHV4, HHV5, HHV6A/B), simian virus 40 (SV40) and 12 (SV12), human immunodeficiency virus (HIV1), human and simian T-cell lymphotropic virus type 1 (HTLV1 and STLV1), BK polyomavirus (BKP), human parvovirus B19, mouse mammary tumor virus, murine type C retrovirus, Mason–Pfizer monkey virus, HBV, HPV (HPV16, HPV18 and HPV6a) and AAV2. Below we describe the steps used for the viral integration detection analysis.
Viral integration events in the host can be detected by using paired-end NGS technologies that facilitate the detection of genomic rearrangements, as well as gene fusions and novel transcripts. VERSE is capable of determining virus integration sites within a single base resolution by requiring the presence of both chimeric and soft clipped reads. In addition, VERSE improves the detection through customizing reference genomes and was shown to substantially enhance the sensitivity of the detection of virus integration sites71. VERSE categorizes its predictions into one of two classes: (1) a high confidence hit with a single base resolution—if there was a sufficient number of soft-clipped reads to support an integration locus so that CREST was able to detect it; or (2) a low confidence hit with a 10-bp resolution for which CREST failed to detect an integration event because of the lack of high-quality soft-clipped reads.
To further limit the false-positive rate associated with viral integration sites, we compared results obtained with VERSE to those from a previous study72. Out of 64 WGS liver cancer samples with HBV integration events that were reported previously72, 50 were part of the PCAWG dataset analyzed in this study. Of those, 45 out of 50 tested positive for HBV when analyzed by CaPSID (filtering criteria used: PMER ≥ 1.0, genome coverage > host background model and read percentage identity ≥89%). In addition, 50 of these WGS samples had 23 matching whole-transcriptome samples and 22 of these were identified to be positive for HBV by CaPSID (filtering criteria used: maximum gene coverage ≥50%, read percentage identity ≥89% and PMER ≥ 1.0). By combining WGS and RNA-seq tumor samples, 47 out of 50 samples tested positive for HBV when analyzed by CaPSID.
Using VERSE, virus integration sites were detected in 28 out of 47 (60%) of these. This result indicates that for a subset of viral integration events, VERSE might be a more stringent approach compared to previously used methods72. This can be explained by the fact that VERSE requires both the presence of paired-end chimeric and soft clipped reads whereas the previously described method72 relied only on paired-end reads. To explore these results further, we compared integration sites obtained with VERSE and those described previously72 with an overlapping window of 10 bp. Our analysis indicates that among 23 integration sites identified by VERSE in RNA-seq data and that overlap with the previously published results72, 91% were classified with high confidence hits and only 9% with low (N total overlap = 23, high = 21 (91%) and low = 2 (9%)). However, a similar result was not observed for integration events found using WGS data (N total overlap = 14, high = 6 (43%), low = 8 (57%)), for which the proportion of integration events classified as high and low was similar.
Thus, our analysis indicates that one important factor for improving the agreement between these two datasets is the confidence level assigned by VERSE to each candidate integration site—but only in the case when integration sites are detected using RNA-seq data. To reduce the potential number of false-positive hits, we decided to use all integration sites predicted by VERSE when these were obtained using WGS data and only high-confidence calls when using RNA-seq data.
Contaminations
On the basis of the presence of vector sequences in the contig assembled by P-DiP and the background model from CaPSID, we could identify which virus hits originated from common laboratory contaminants or were due to sequence similarities to the human genome. In addition, we filtered known contaminants (see below). For P-DiP, we filtered all hits that did not have more target reads than any artificial sequence (excluding artificial viruses) on an individual contig region. Hits caused by vector and other artificial sequences were identified by analyzing the assembled contigs for combined hits to viral pathogens and artificial sequences. Checking viral hits that occurred at least 40 times in such a contig, we could clearly separate contaminants from viral pathogens.
The gammaretrovirus hits (NCBI taxonomy ID 153135; species, murine leukemia virus) were also marked as artifacts, on the basis of the additional BLAST hits of the corresponding contigs to the Mus musculus genome by P-DiP as well as the background model of the CaPSID pipeline, which was designed to limit the number of spurious hits. Most of the frequent virus hits prone to contamination by artificial sequences were lambdalikevirus, alphabaculovirus, microvirus, simplexvirus, hepacivirus, CMV, orthopoxvirus and punalikevirus. However, restricting to at least 1 PMER for the potential virus hit contaminants reduced these to one CMV case.
Filtering contaminants
We filtered all Microviridae (taxonomy ID 10841) because of the phix174 spike-in used during sequencing. Caudovirales (taxonomy ID 28883), tailed bacteriophages, were removed as they typically infect bacterial hosts. Baculoviridae were filtered because these infect insect cells and are commonly used in the laboratory. The virus coverage was analyzed by aligning the potentially pathogenic reads with BWA-mem to the human hg19 reference genome after adding the respective virus reference sequence that was most frequently detected within the genus. Coverage was thereafter calculated base specific using BEDTools coverage. As we identified EBV in all 14 normal blood controls from ovarian cancer that were EBV immortalized, these were removed from the virus hits.
Integration of external PCAWG datasets
We tested for mutual exclusivity, for example, between virus detections and driver gene mutations by applying DISCOVER22. On the basis of the gene expression data, immune-cell proportions were analyzed by CIBERSORT14. For survival analysis, Cox proportional hazards analysis was performed using R libraries ‘survival’ and ‘survminer’ for the figures. The optimal cut points were identified by maxstat using a previously described method73 (library maxstat).
Virus load
The viral load in relation to the human genome equivalents was calculated based on the human bases sequenced (read length × number of reads mapped to the human genomes), tumor sample purity (if available or 100% otherwise) assuming a ploidy of two and using a human genome size of 2,897,310,462 bases (the mappable part of the human genome). This number of human genome equivalents was then related to the viral genome equivalents that were calculated based on the number of identified viral reads, read length and virus genome size.
Human research participants
The ethics oversight for the PCAWG protocol was undertaken by the TCGA Program Office and the Ethics and Governance Committee of the ICGC. Each individual ICGC and TCGA project that contributed data to PCAWG had its own local arrangements for ethics oversight and regulatory alignment.
Statistics
If not specified otherwise, we used two-sided Wilcoxon rank-sum tests for groups with n > 3. Further details can be found in the Nature Research Reporting Summary.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Somatic and germline variant calls, mutational signatures, subclonal reconstructions, transcript abundance, splice calls and other core data generated by the ICGC/TCGA PCAWG Consortium are described in an associated paper10 and are available for download at https://dcc.icgc.org/releases/PCAWG. Additional information on accessing the data, including raw read files, can be found at https://docs.icgc.org/pcawg/data/. In accordance with the data-access policies of the ICGC and TCGA projects, most molecular, clinical and specimen data are in an open tier that does not require access approval. To access potentially identifying information, such as germline alleles and underlying sequencing data, researchers will need to apply to the TCGA Data Access Committee (DAC) via dbGaP (https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login) for access to the TCGA portion of the dataset, and to the ICGC Data Access Compliance Office (DACO; http://icgc.org/daco) for the ICGC portion. In addition, to access somatic SNVs derived from TCGA donors, researchers will need to obtain dbGaP authorization. Datasets described specifically in this manuscript can be found in the Supplementary Tables.
Code availability
The core computational pipelines used by the PCAWG Consortium for alignment, quality control and variant calling are available to the public at https://dockstore.org/search?search=pcawg under the GNU General Public License v.3.0, which enables the reuse and distribution of the pipelines. The pathogen-discovery pipeline P-DiP is available on GitHub (https://github.com/mzapatka/p-dip). CaPSID is available from GitHub (pipeline, https://github.com/capsid/capsid-pipeline; webapp, https://github.com/capsid/capsid-webapp). The taxonomic classifier CSSSCL is available from GitHub (https://github.com/oicr-ibc/cssscl).
Change history
21 March 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41588-023-01316-y
References
Parkin, D. M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 118, 3030–3044 (2006).
Plummer, M. et al. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob. Health 4, e609–e616 (2016).
Bouvard, V. et al. A review of human carcinogens—part B: biological agents. Lancet Oncol. 10, 321–322 (2009).
Muñoz, N., Castellsagué, X., de González, A. B. & Gissmann, L. Chapter 1: HPV in the etiology of human cancer. Vaccine 24, S1–S10 (2006).
Bialecki, E. S. & Di Bisceglie, A. M. Clinical presentation and natural course of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 17, 485–489 (2005).
Hermine, O. et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. N. Engl. J. Med. 347, 89–94 (2002).
Thompson, M. P. & Kurzrock, R. Epstein–Barr virus and cancer. Clin. Cancer Res. 10, 803–821 (2004).
Mesri, E. A., Feitelson, M. A. & Munger, K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe 15, 266–282 (2014).
zur Hausen, H. Oncogenic DNA viruses. Oncogene 20, 7820–7823 (2001).
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature https://doi.org/10.1038/s41586-020-1969-6 (2020).
Borozan, I. et al. CaPSID: a bioinformatics platform for computational pathogen sequence identification in human genomes and transcriptomes. BMC Bioinformatics 13, 206 (2012).
Borozan, I., Watt, S. N. & Ferretti, V. Evaluation of alignment algorithms for discovery and identification of pathogens using RNA-seq. PLoS ONE 8, e76935 (2013).
Nicoll, M. P. et al. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog. 12, e1005539 (2016).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Krug, L. T. & Pellett, P. E. Roseolovirus molecular biology: recent advances. Curr. Opin. Virol. 9, 170–177 (2014).
Eliassen, E. et al. Human herpesvirus 6 and malignancy: a review. Front. Oncol. 8, 512 (2018).
Spandole, S., Cimponeriu, D., Berca, L. M. & Mihăescu, G. Human anelloviruses: an update of molecular, epidemiological and clinical aspects. Arch. Virol. 160, 893–908 (2015).
van de Berg, P. J. et al. Human cytomegalovirus induces systemic immune activation characterized by a type 1 cytokine signature. J. Infect. Dis. 202, 690–699 (2010).
Garcia-Martinez, A. et al. Lack of cytomegalovirus detection in human glioma. Virol. J. 14, 216 (2017).
Fujimoto, A. et al. Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nat. Genet. 42, 931–936 (2010).
Furuta, M. et al. Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers. Oncotarget 9, 25075–25088 (2018).
Canisius, S., Martens, J. W. M. & Wessels, L. F. A. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome Biol. 17, 261 (2016).
Kawai-Kitahata, F. et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 51, 473–486 (2016).
Borozan, I., Zapatka, M., Frappier, L. & Ferretti, V. Analysis of epstein-barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 92, e01239-17 (2018).
Mork, J. et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 344, 1125–1131 (2001).
Li, N. et al. Human papillomavirus infection and bladder cancer risk: a meta-analysis. J. Infect. Dis. 204, 217–223 (2011).
Cao, S. et al. Divergent viral presentation among human tumors and adjacent normal tissues. Sci. Rep. 6, 28294 (2016).
Travé, G. & Zanier, K. HPV-mediated inactivation of tumor suppressor p53. Cell Cycle 15, 2231–2232 (2016).
Werness, B. A., Levine, A. J. & Howley, P. M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248, 76–79 (1990).
Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J. & Howley, P. M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136 (1990).
Henderson, S., Chakravarthy, A., Su, X., Boshoff, C. & Fenton, T. R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 7, 1833–1841 (2014).
Burns, M. B., Temiz, N. A. & Harris, R. S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 (2013).
Schlecht, N. et al. Gene expression profiles in HPV-infected head and neck cancer. J. Pathol. 213, 283–293 (2007).
Nelson, P. N. et al. Demystified. Human endogenous retroviruses. Mol. Pathol. 56, 11–18 (2003).
Paces, J. et al. HERVd: the human endogenous retroviruses database: update. Nucleic Acids Res. 32, D50 (2004).
Pavlícek, A., Paces, J., Elleder, D. & Hejnar, J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: their integration, stability, and distribution. Genome Res. 12, 391–399 (2002).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Ohnuki, M. et al. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl Acad. Sci. USA 111, 12426–12431 (2014).
Smith, C. C. et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Invest. 128, 4804–4820 (2018).
Tang, K.-W. & Larsson, E. Tumour virology in the era of high-throughput genomics. Phil. Trans. R. Soc. Lond. B 372, 20160265 (2017).
Jiang, Z. et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 22, 593–601 (2012).
Hu, Z. et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 47, 158–163 (2015).
Zhao, L.-H. et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 7, 12992 (2016).
Li, X. et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J. Hepatol. 60, 975–984 (2014).
Shen, C.-J., Cheng, Y.-M. & Wang, C.-L. lncRNA PVT1 epigenetically silences miR-195 and modulates EMT and chemoresistance in cervical cancer cells. J. Drug Target. 25, 637–644 (2017).
Tang, K.-W., Alaei-Mahabadi, B., Samuelsson, T., Lindh, M. & Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 4, 2513 (2013).
Nault, J.-C. et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 47, 1187–1193 (2015).
Borozan, I. & Ferretti, V. CSSSCL: a Python package that uses combined sequence similarity scores for accurate taxonomic classification of long and short sequence reads. Bioinformatics 32, 453–455 (2015).
Sung, W. K. et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 44, 765–769 (2012).
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 3, 246–259 (2013).
Wallace, N. A. & Münger, K. The curious case of APOBEC3 activation by cancer-associated human papillomaviruses. PLoS Pathog. 14, e1006717 (2018).
Roberts, S. A. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976 (2013).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint athttps://arxiv.org/abs/1303.3997v2 (2013).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Tischler, G. & Leonard, S. Biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 9, 13 (2014).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285 (2012).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903 (2015).
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Fischer, N. et al. Rapid metagenomic diagnostics for suspected outbreak of severe Pneumonia. Emerg. Infect. Dis. 20, 1072–1075 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Simpson, J. T. & Durbin, R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556 (2012).
Pruitt, K. D., Tatusova, T., Klimke, W. & Maglott, D. R. NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Res. 37, D32–D36 (2009).
David, M., Dzamba, M., Lister, D., Ilie, L. & Brudno, M. SHRiMP2: Sensitive yet practical short read mapping. Bioinformatics 27, 1011–1012 (2011).
Huang, W., Li, L., Myers, J. R. & Marth, G. T. ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594 (2012).
Wang, Q., Jia, P. & Zhao, Z. VERSE: a novel approach to detect virus integration in host genomes through reference genome customization. Genome Med. 7, 2 (2015).
Fujimoto, A. et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 48, 500–509 (2016).
Lausen, B. & Schumacher, M. Maximally selected rank statistics. Biometrics 48, 73–85 (1992).
Acknowledgements
We thank the IT Core Facility at the DKFZ for technical assistance, M. Hain and R. Kabbe for computational support, S. Gerhardt for technical assistance with the validation experiments. We acknowledge the contributions of the many clinical networks across the ICGC and TCGA who provided samples and data to the PCAWG Consortium, and the contributions of the Technical Working Group and the Germline Working Group of the PCAWG Consortium for collation, realignment and harmonized variant calling of the cancer genomes used in this study. We thank the patients and their families for their participation in the individual ICGC and TCGA projects. V.F. and I.B. received support for their work from the Ontario Institute for Cancer Research (OICR) through funding provided by the government of Ontario. A.G. received support for his work from the Leibniz Association (grant number SAW‐2015‐IPB‐2) and the German Center for Infection Research (grant number TTU 01.801). P.L. and A.G. received support for this work from the German Federal Ministry of Education and Research (BMBF BioTop grant number 01EK1502C, ICGC-DE-Mining grant number 01KU1505A-G). D.S.B. and C.S.C. received support from Cancer Research UK C5047/A14835/A22530/A17528, the Dallaglio Foundation, Bob Champion Cancer Trust, The Masonic Charitable Foundation successor to The Grand Charity, The King Family and the Stephen Hargrave Trust. H.M. was supported by a Swiss National Science Foundation grant (number S-87701-03-01).
Author information
Authors and Affiliations
Consortia
Contributions
M.Z. and P.L. jointly supervised research. V.F., R.E., C.S.C., M.I., I.B., M.Z. and P.L. conceived and designed the experiments. H.S. performed experiments. M.I., D.S.B., I.B. and M.Z. performed statistical analysis. N.D., M.I., A.G., D.S.B., I.B. and M.Z. analyzed the data. V.F., R.E., C.S.C., H.M., M.A., A.G., D.S.B., I.B. and M.Z. contributed reagents, materials and/or analysis tools. M.I., D.S.B., I.B., M.Z. and P.L. wrote the paper. V.F., A.G., C.S.C., D.S.B., M.I., I.B., M.Z. and P.L. critiqued manuscript for intellectual content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Statistics of analyzed reads from WGS and RNA-seq samples.
a, Number of identified candidate pathogen reads used for WGS analysis in non-tumor samples and for RNA-seq analysis. Red line represents the median. b, Fraction of analysed reads mapped to phiX174 (green) and the human reference genome hg19 (red) and the rest labeled as potentially pathogenic reads (blue). c, Fraction of analysed reads per genome coverage separated for virus positive and negative tumor samples across organ systems. Thick black line represents the median. d, Search space overlap for genera across the three pipelines. e, Hit space overlap for genera across the three pipelines.
Extended Data Fig. 2 Genome coverage of mastadenovirus contamination detected in batches.
a, Coverage of the virus genomes summarizing all mapped reads across all virus-positive tumor/normal samples. Alignment was done using BWA-mem. b, Mastadenovirus-positive samples ordered based on their sequencing date as years, indicating samples from early-onset prostate cancer (EOPC-DE) project across sequencing batches.
Extended Data Fig. 3 The distribution of PMER values for consensus hits across pathogen detection pipelines.
a, Overlap calculated between three pipelines for the cases of shuffled viral hits randomized for their donor. b, PMER distribution of common viral hits detected by all three pipelines. c, Virus genome equivalents in relation to human tumor genome equivalents calculated for each sample positive for the virus. d, Co-infection of viruses detected in individual tumor samples. The fraction of overlap between two viruses were calculated as the number of shared samples divided by the smaller set.
Extended Data Fig. 4 Specific findings for lymphocrypto- and roseolovirus, HBV and EBV.
Overall contribution of immune cells across organ system in samples positive or negative for lymphocryptovirus and roseolovirus. Tukey boxplot indicates the median by the middle line and the 25–75th percentiles by the box. The whiskers were drawn up to the 1.5 interquartile range from the lower and upper quartile. b, Comparison of histopathologically detected HBV in liver cancer with the PMERs detected in WGS. Precision and recall of the PCR based HBV test versus the consensus calls from WGS data. Red dot indicates the PMER cut-off of 1. c, Relation of PMER for EBV detections in tumor and normal samples across organ system and normal tissue type. d, Epstein-Barr virus expression presenting lytic (red) and latent (green) genes across organ systems. Reads were counted after alignment with kallisto to the EBV reference transcriptome (see Methods).
Extended Data Fig. 5 Expression of stem cell markers in relation to HERV expression.
Expression values of KLF4, POU5F1 and SOX2 in relation to transcriptional activity of HERVs (ERV, ERV1, ERVK, ERVL, ERVL.MaLR) for 908 tumor samples. Correlation coefficient (R) presented is calculated using Spearman Rank Correlation.
Extended Data Fig. 6 Overall Survival analysis of endogenous retrovirus expression in different tissue types.
Cut-offs were defined by maxstat R package using log-rank test and. P values were corrected for multiple testing of variable cut-offs using Lau2 method. Analyzed were all tissue types with more than 40 cases and at least 15 events. Number of patients at risk is provided separated by high or low expression groupings based on the tpm cutoff for the respective ERV family provided in the title of individual panels. P-value of the log-rank test is provided for each analysis.
Extended Data Fig. 7 Number of viral integration events as a function of the chromosome and genomic location.
a, Shows the number of viral integration events detected for HBV, HPV16 and HPV18 as a function of the human chromosome. Numbers within each stacked bar plot represent the number of integration events detected for each virus and within each chromosome. b, Shows the percentage of the total number of integration events detected for each chromosome averaged over three viral types shown in panel A. c, Number of viral integration events detected for HBV, HPV16 and HPV18 as a function of the host’s genomic location. Numbers within each stacked bar plot represent the number of integration events detected for each virus and within each genomic location. d, Shows the percentage of the total number of integration events detected within each genomic location averaged over three viral types shown in panel C. e, Shows the number of HBV integration events detected in liver cancers in the host’s gene coding and/or gene promoter regions. Stacked bar plot represents the number of integration events detected within each sample and each gene, each sample is indicated using the color code shown in the legend to the right. f, Shows the number of HPV18 integration events detected in head/neck and cervical cancers in the host’s gene coding or gene promoter regions. g, Shows the number of HPV16 integration events detected in head/neck and cervical cancers in the host’s gene coding and/or gene promoter regions.
Extended Data Fig. 8 Comparison of the somatic copy number alterations (SCNA) and single nucleotide variants (SNVs) for samples with and without HPV and HBV integrations into human genome.
a, Boxplots showing the number of SCNA detected in head/neck and cervical cancers: HPV16+(red) vs HPV16- (grey) samples. SCNAs are calculated using three different distances from the integration site: i) greater than 1 Megabases (Mbp), ii) exactly +/− 1 Mbp away, and iii) below 1 Mbp (n = 17 virus integrations). b, Boxplots showing the number of SCNAs detected in head/neck and cervical cancers with and without HPV18 integrations (n = 8 virus integrations). c, Number of SNVs detected in head/neck and cervical cancers with and without HPV16 integrations. Number of SNVs are calculated using three different ranges for the human genome: i) SNVs within the nearest gene to the virus integration site (maximum: 50Kb), ii) SNVs at the location of the viral integration site in the chromosomal region +/− the position of the second breakpoint located in the viral sequence, and iii) SNVs around 10 kb of the viral integration site. Blue triangles indicate the mean values. (n = 87 virus integrations) d, Number of SNVs detected in liver cancers with and without HBV integrations (n = 109 virus integrations). e. Number of SNVs detected in head/neck and cervical cancers with and without HPV18 integrations (n = 14 virus integrations). In all Tukey boxplots, black line in the middle represents median and the 25–75th percentiles by the box. The whiskers were drawn up to the 1.5 interquartile range from the lower and upper quartile. f, Expression of tumors and normal samples for long noncoding RNAs with and without HPV16 integrations near to the integration site.
Extended Data Fig. 9 Contigs from de novo assembly identified as possibly originating from novel viral species or strains.
Barplot showing the number of contigs obtained using the CaPSID’s de novo assembly step (see Methods) within each genus. Taxonomic classification for each contig was performed using the CSSSCL algorithm. Each of the 29 contigs considered for this plot had to have a sequence homology <90% when aligned to any known sequence contained by the latest nucleotide BLAST database. The legend to the right indicates the following ICGC project codes: BLCA—bladder cancer, CESC—cervical cancer, CLLE—chronic lymphocytic leukemia, HNSC—head and neck, LIHC and LIRI—liver cancer, PBCA—pediatric brain cancer, and STAD—stomach cancer.
Supplementary information
Supplementary Information
Supplementary Note
Supplementary Tables
Supplementary Tables 1–24
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zapatka, M., Borozan, I., Brewer, D.S. et al. The landscape of viral associations in human cancers. Nat Genet 52, 320–330 (2020). https://doi.org/10.1038/s41588-019-0558-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0558-9
This article is cited by
-
Intratumoural microbiota: a new frontier in cancer development and therapy
Signal Transduction and Targeted Therapy (2024)
-
The ALT pathway generates telomere fusions that can be detected in the blood of cancer patients
Nature Communications (2024)
-
Integrated genomic and transcriptomic analysis reveals the activation of PI3K signaling pathway in HPV-independent cervical cancers
British Journal of Cancer (2024)
-
The Microbiome Matters: Its Impact on Cancer Development and Therapeutic Responses
Journal of Microbiology (2024)
-
Detection of HHV-6 Virus in specimen of a ductal pancreatic adenocarcinoma with comparison in tumor and normal tissue
Diagnostic Pathology (2023)
