
Abstract
Intraductal papillary mucinous neoplasm (IPMN) is a common pancreatic cystic neoplasm that is often invasive and metastatic, resulting in a poor prognosis. Few molecular alterations unique to IPMN are known. We performed whole-exome sequencing for a primary IPMN tissue, which uncovered somatic mutations in KCNF1, DYNC1H1, PGCP, STAB1, PTPRM, PRPF8, RNASE3, SPHKAP, MLXIPL, VPS13C, PRCC, GNAS, KRAS, RBM10, RNF43, DOCK2 and CENPF. We further analyzed GNAS mutations in archival cases of 118 IPMNs and 32 pancreatic ductal adenocarcinomas (PDAs), which revealed that 48 (40.7%) of the 118 IPMNs but none of the 32 PDAs harbored GNAS mutations. G-protein alpha-subunit encoded by GNAS and its downstream targets, phosphorylated substrates of protein kinase A, were evidently expressed in IPMN; the latter was associated with neoplastic grade. These results indicate that GNAS mutations are common and specific for IPMN and activation of G-protein signaling appears to play a pivotal role in IPMN.
Similar content being viewed by others
Introduction
Intraductal papillary mucinous neoplasm of the pancreas (IPMN) is a cystic neoplasm consisting of dilated ducts lined by neoplastic cells that secrete copious mucin1,2,3. This neoplasm is distinct from pancreatic ductal adenocarcinoma (PDA), a conventional type of pancreatic cancer that usually forms a solid and ill-defined mass. Although IPMN is less frequent than PDA, it is fairly common; in the US, the incidence is reported to be 2.04 per 100000 person-years in the general population and in Japan, IPMN is prevalent in 5% of all registered surgical cases of pancreatic neoplasms with available histological data4,5. Patients with IPMN suffer from acute pancreatitis due to obstruction of the ducts with mucus or pancreatic functional insufficiency resulting from chronic atrophy of the parenchyma6. Moreover, IPMNs are often associated with invasive carcinoma, which leads to a poor outcome. The prognosis of patients with IPMN with an associated invasive carcinoma is a 5-year survival rate of 27%–60%, depending upon the extent and histological type of the invasive component7. Although these distinct and unique features are well-known, molecular alterations specific to IPMN are poorly understood. A better understanding of the molecular alterations specific to IPMN may lead to development of more efficient methods of prevention, early diagnosis and cure of this disease.
In this study, we carried out whole-exome sequencing using DNA obtained from primary IPMN tissue and found a number of previously unidentified mutated genes. Among them, we then focused on mutations in GNAS, a gene encoding the guanine nucleotide-binding protein (G-protein) alpha subunit (Gsα), in archival cases of IPMN and found that the gene was frequently mutated in IPMN. We further studied the clinicopathological relevance of associated molecules of these mutations in G-protein signaling in this neoplasm.
Results
Whole-exome sequencing of IPMN
Whole-exome sequencing was carried out on DNA extracted from primary IPMN tissue, using the solution hybridization-based exon-enrichment method and a massively parallel deep sequencer. The analyzed tumor was an intestinal-type high-grade IPMN with minimal invasion from a 76-year-old Japanese man. Tumor cells and non-tumor cells were separately collected from a frozen sample of primary tissue by laser-capture microdissection. Raw sequence data revealed 68830 single nucleotide polymorphisms (SNPs) and 4139 small insertions and deletions (InDls) in the tumor cells. A series of subsequent qualifications of these data narrowed down these variations into 21 nonsynonymous tumor-specific SNPs and 6 InDls (Fig. 1). We validated these SNPs and InDls by the Sanger method and confirmed that 13 SNPs and 4 InDls were somatic mutations. These confirmed mutated genes were KCNF1, DYNC1H1, PGCP, STAB1, PTPRM, PRPF8, RNASE3, SPHKAP, MLXIPL, VPS13C, PRCC, GNAS, KRAS, RBM10, RNF43, DOCK2 and CENPF (Table 1). Among these mutated genes, we focused on GNAS that is known to be mutated in human tumors, mainly in those of endocrine origin and rarely in other common cancers, including pancreatic cancer.

Processing of data obtained by whole-exome sequencing using a massively parallel deep sequencer.
GNAS mutations in archival cases of IPMN and PDA
We further examined mutations in GNAS in 118 archival cases of IPMN and, for comparison, 32 cases of PDA. Somatic mutations in GNAS were frequently found in IPMNs and were identified in 48 (41%) of the 118 cases. All of these mutations involved codon 201 of GNAS, where arginine was substituted by cysteine or histidine (R201C or R201H) (Supplementary Table S1 online). However, the GNAS mutation was not observed in any of the examined cases of PDAs. This association between GNAS mutations and pathological types of tumors was statistically significant (p = 6.58 × 10−7; Fisher's exact test) (Table 2). We also examined somatic mutations in KRAS, a gene known to be commonly mutated in PDAs and IPMNs. Mutations in KRAS were observed in 56 (48%) of the 118 IPMNs and in 25 (78%) of the 32 PDAs. The KRAS mutations were significantly more frequent in PDAs than in IPMNs (p = 0.0016 by Fisher's exact test) (Table 2 and Supplementary Table S1 online). In addition, we examined 25 cultured pancreatic cancer cell lines and found that none of them harbored the GNAS mutation, whereas most of them, i.e., 24 of the 25, harbored KRAS mutations (Supplementary Table S1 online). These results indicate that GNAS mutations are frequent and highly specific for IPMN among pancreatic neoplasms. Moreover, 30 (25%) of the 118 IPMNs harbored concurrent mutations in GNAS and KRAS, indicating a significant co-occurrence of these mutations in IPMNs (p = 0.0057; Fisher's exact test).
G-protein signaling in IPMN and PDA
To identify the biological significance of GNAS in pancreatic neoplasms, we performed immunohistochemical analysis and examined the expression of Gsα in IPMNs and PDAs. We found that Gsα was more frequently and abundantly expressed in IPMNs than in PDAs, i.e., 117 (99%) of the 118 IPMNs and 19 (59%) of the 32 PDAs showed moderate or high expression (p < 0.001; ANOVA; Fig. 2, Table 2 and Supplementary Table S1 online). GNAS mutations were not significantly associated with Gsα expression in IPMNs (p = 0.061 by ANOVA). Phosphorylated substrates of cyclic-AMP (cAMP)-dependent protein kinase/protein kinase A (PKA), a downstream target of Gsα in the G-protein-coupled receptor (GPCR)-cAMP signaling pathway8, were observed in both IPMNs and PDAs, i.e., 92 (78%) of the 118 IPMNs and 23 (72%) of the 32 PDAs showed moderate or high expression (p = 0.425; ANOVA; Fig. 2, Table 2 and Supplementary Table S1 online). In IPMNs, neither GNAS nor KRAS mutations were associated with expressions of phosphorylated substrates of PKA (p = 0.113 for GNAS and p = 0.339 for KRAS by ANOVA).

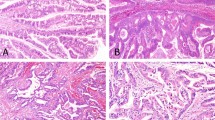
Expression of the G-protein α subunit (B, E) and phosphorylated substrates of protein kinase A (C, F) in intraductal papillary mucinous neoplasm (A–C) and ductal adenocarcinoma of the pancreas (D–F). Diaminobenzidine was used as the chromogen to visualize the immunoreaction in immunohistochemistry (B, C, E, F). Panels (A) and (D) show images of hematoxylin-eosin staining.
Original magnification, ×200.
Clinicopathological relevance of molecular alterations
To understand the clinical significance of mutations in GNAS and KRAS and expression of Gsα and the phosphorylated substrates of PKA in patients with IPMN, we compared these molecular phenotypes and clinicopathological features in our cohort. Mutations in GNAS, KRAS, or both genes did not appear to be associated with any of the clinicopathological features of IPMN, including sex, age, morphologic variation, macroscopic type, mural nodule, grade, or stage (Table 3). Most IPMNs showed strong expression of Gsα and no obvious association with the clinicopathological features (Table 3). In contrast, the expression of phosphorylated substrates of PKA was associated with tumor grade, being more obvious in high-grade and invasive IPMN cases than in low-grade IPMN cases (p = 0.010; ANOVA) (Table 3). In survival analysis, neither the gene mutations, each alone or in combination of GNAS and KRAS, nor the molecular expression showed any statistically significant associations with prognosis of patients in our cohort or those in any subcohorts including those by morphologic type, tumor grades, macroscopic type, or stages, although survival curves between patients with low expression of phosphorylated PKA substrates and those with moderate or high expression of the substrates appeared different (Fig. 3).

Kaplan–Meier survival analyses of patients with intraductal papillary mucinous neoplasms according to molecular alterations and expression profiles.
Discussion
Using whole-exome sequencing, we identified a number of somatically mutated genes that were previously unidentified in IPMN. We further examined mutations in GNAS in archival cases of IPMN and found that GNAS mutations occurred very frequently in this neoplasm. On the other hand, no GNAS mutations were found in any of the examined cases of PDA. Gsα and phosphorylated substrates of PKA were evidently expressed in IPMNs and the latter was associated with the pathological grade of the neoplasm. These results indicate that mutations in GNAS are common and specific for IPMN and activation of G-protein signaling seems to contribute to development and progression of IPMN.
Whole-exome sequencing has recently emerged as a powerful tool to identify unknown cancer-associated genes. Using next-generation sequencing technology employing a massively parallel deep sequencer, it is truly affordable for individual researchers like us to perform whole-exome sequencing in a single laboratory. A key issue for next-generation sequencing is data processing. Because of the short sequencing reads, data from next-generation sequencing must be mapped on a reference sequence. This mapping task is quite challenging because of the complexity of the human genome, which contains numerous redundant sequences. Therefore, a significant number of errors occur in the mapped sequencing data. In this current study, we developed a stepwise method of qualifying the data, as listed in Fig. 1 and successfully identified somatic mutations. For this, we employed values of variation reads/all reads and total number of reads/coverage. These values were useful in dismissing false data. This qualification method is useful for the currently employed system, using the SOLiD system and Bioscope software, but it may also be usefully applied for the extraction of somatic mutations from next-generation sequencing data.
In this study, most of the somatically mutated genes identified in IPMN by whole-exome sequencing were previously unidentified as susceptible genes in cancer. These genes, listed in Table 1, serve a variety of functions and some of them are demonstrated to be rarely mutated in cancer, as discussed below, according to the catalog of somatic mutations in human cancers (COSMIC) (http://www.sanger.ac.uk/genetics/CGP/cosmic/)9. Roles of these mutated genes in cancer phenotypes are largely unknown. KCNF1 encodes a potassium channel protein and is not known for any association with cancer10. DYNC1H1 is a large gene composed of 78 exons encoding dynein 1 heavy chain comprising 4646 residues. Dynein is a microtubule-associated ATPase and interference of its function results in a block of spindle formation11. DYNC1H1 is somatically mutated in pancreatic neuroendocrine neoplasms, ovarian cancer and glioblastoma multiforme12,13. PGCP encodes blood plasma glutamate carboxypeptidase and has not been reported to be mutated in cancer14. STAB1 encodes a large transmembrane scavenger receptor and is mutated in breast and colon cancers and glioblastoma multiforme13,15,16. PTPRM encodes a type IIB receptor protein tyrosine phosphatase involved in regulating adhesion by dephosphorylating components of cadherin-catenin complexes17. PTPRM is mutated in lung and ovarian cancers and glioblastoma multiforme13,18. PRPF8 encodes pre-mRNA splicing factor and is mutated in lung and ovarian cancers19,20. RNASE3 encodes ribonuclease A3/eosinophil cationic protein that is toxic for pathogen and host tissues21. RNASE3 has not been reported to be mutated in cancer. SPHKAP encodes A-kinase anchoring protein implicated in spatial localization of protein kinase A and signal transduction and is mutated in lung and ovarian cancers22. MLXIPL encodes a glucose-responsive transcription factor ChREBP that is required for efficient cell proliferation of tumor cells23. MLXIPL is mutated in pancreatic cancer. VPS13C encodes a member of the vacuolar protein sorting-associated 13 gene family that is a homologue of a yeast protein involved in trafficking of membrane proteins between the trans-Golgi network and the prevacuolar compartment24. VPS13C is mutated in ovarian cancer. PRCC encodes a protein associated with pre-mRNA splicing factor and is known to comprise a fusion gene with TFE3 observed in papillary renal cell carcinoma25. PRCC is mutated in glioblastoma multiforme13. RBM10 is a member of the RNA binding motif gene family and is identified as a susceptibility gene in the cleft palate26. RBM10 is mutated in breast, colon, ovary, pancreas and prostate cancers, although none of these are frameshift mutations like the one we identified, but one nonsense mutation has been reported20,27. RNF43 encodes an E3 ubiquitin ligase that is overexpressed in colorectal cancer28 and mutated in ovarian cancer. DOCK2 encodes a member of the CDM protein family and regulates cell motility and cytokine production through the activation of Rac in mammalian hematopoietic cells29. DOCK2 is mutated in ovarian and pancreatic cancers. CENPF encodes a component of the centromere-kinetocore complex and is overexpressed in breast cancer30. CENPF is also mutated in colon and ovarian cancers and glioblastoma multiforme13,16.
Gsα encoded by GNAS forms a heterotrimer with ß and γ G-protein subunits, which then couples with a membrane-bound receptor, GPCR. When GPCR is activated by ligand-binding, the receptor catalyzes exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) bound to Gsα and the GTP-bound Gsα dissociates from the receptor and the ßγ subunits. The dissociated Gsα proceeds to activate specific effector molecules including adenyl cyclase, which produces cAMP that can act as a second messenger8. Increased cAMP activates PKA and the activated PKA phosphorylates a variety of target molecules. The activated Gsα hydrolyzes GTP to GDP and is converted into its inactive form. GNAS is known to be mutated in several neoplasms, mainly of the endocrine system and rarely in non-endocrine common cancers like those of the breast, lung and pancreas according to COSMIC database9. Our study has shown that frequent somatic mutations in GNAS occur in a pancreatic non-endocrine neoplasm, namely, IPMN. Common mutations in GNAS observed in IPMN identified by this study as well as endocrine neoplasms reported thus far in elsewhere are R201C and R201H. These mutations are known to cause disruption of the intrinsic hydrolytic activity of Gsα, which results in constitutive activation of its function31. Our findings of frequent GNAS mutations, together with evident expression of Gsα and phosphorylated substrates of PKA in IPMN, indicate that G-protein signaling is activated in this neoplasm. The mutations in GNAS were not associated with any specific clinicopathological features of the neoplasm. GNAS mutations were observed in low-grade tumors as well as in high-grade tumors and invasive tumors. These results suggest that GNAS mutations may be associated more with initiation but less with progression of the neoplasm with some specific clinicopathological features. Instead, although the expression of phosphorylated substrates of PKA was not associated directly with mutational phenotypes of GNAS, it was associated with the grade of IPMN, which suggests that exacerbation of PKA activity apparently independent from GNAS mutations may be necessary for disease progression. Regarding multifocal nature of IPMN2, it may be interesting to examine molecular features in independent lesions in the neoplasm, which was not determined in the current study and is an important issue for further study.
Although mutations in GNAS were common in IPMN, they were not found in PDAs. On the other hand, mutations in KRAS were fairly common in both types of neoplasms. More interestingly, a significant proportion (25%) of IPMNs showed concurrent mutations in GNAS and KRAS. RAS encoded by KRAS is mainly involved in the mitogen-activated protein kinase (MAPK) pathway32, whereas GNAS is involved in the GPCR pathway. These results highlight a critical difference in molecular oncogenesis between these neoplasms; PDAs depend on activation of the RAS-MAPK pathway, whereas IPMNs depend on activation of both the RAS-MAPK and the GPCR pathways. On the other hand, expression of phosphorylated substrates of PKA was observed in PDAs as well. This result suggests that activation of PKA apparently independent from mutant Gsα may play some roles in PDA. Unraveling the details concerning the synergistic effect of both pathways on pancreatic ductal oncogenesis will need to be clarified by experiments employing a genetically engineered mouse model.
The frequent and specific mutations in GNAS found in IPMN may provide a clue to the molecular diagnosis and targeted therapy of this neoplasm. The highly specific redundancy of GNAS mutations in IPMN may allow a sensitive and specific diagnosis of the neoplasm with exclusion of PDA. It may also help to resolve a recent debate regarding whether the prognosis of pancreatic cancer associated with IPMN is better than that of pancreatic cancer alone33. This debate is hampered by the difficulty of precise evaluation of an association between pancreatic cancer and IPMN. Molecular diagnosis using information on any GNAS mutation in pancreatic cancer may help to evaluate such associations and to enable a better prediction of patients' prognosis. The GPCR pathway involving PKA is already known to be a major target of drug development and indeed one-third of current drugs target GPCR pathways34. Among such drugs, BIM-46174 is a specific drug targeting Gsα, which can suppress malignant phenotypes of various cancer cell lines35. These GPCR-targeting drugs might prove useful in treating IPMN, which will be an important area for future study.
During preparation of this manuscript, we became aware that Wu et al. had published a report regarding GNAS mutations in IPMN36. They found that 66% of 132 IPMNs harbored a GNAS mutation, 81% harbored a KRAS mutation and slightly more than half (51%) harbored both GNAS and KRAS mutations, whereas at least 1 of the 2 genes was mutated in 96.2%. The frequency of these mutations appeared to be higher than that in our cohort. This might be due to methodological differences in the detection of mutations. Wu et al. used cystic fluid samples and the ligation assay for detecting mutations, which might be more sensitive than the traditionally conventional method we employed using DNA from dissected paraffin-embedded tissues and Sanger sequencing for analyzing archival cases, because a low population of mutated genes could be uncovered by their method, with allele frequencies as low as 1% according to their data. Moreover, they observed that the prevalence of KRAS mutations was higher in lower-grade lesions, whereas the prevalence of GNAS mutations was somewhat higher in more advanced lesions. These associations seem to be inconsistent with our data showing no particular association between the mutations and tumor grade. These differences might be due to not only the methodological difference but also differences in features of examined cohorts. In comparisons of some of features available between the two cohorts, the mean age of studied patients and morphologic types of IPMN samples seemed to be different. The mean age was 65.09 years-old in ours while 69.73 years-old in Wu's (P < 0.001 by ANOVA). In Wu's study, morphological types were available for 72 samples and they were consisted of 72% of gastric type-IPMNs, 18% of intestinal type-IPMNs, 10% of pancreatobiliary type-IPMNs and no oncocytic type-IPMN, which was different from the distribution of ours (P = 0.022 by Chi-square test). On the other hand, macroscopic types, tumor grades and sex distribution did not seem to be different. Besides these features, ethnic background of studied populations might be different. Resolving of these inconsistencies will be an important issue of future study.
Methods
Tissues
A frozen tissue sample of intraductal papillary mucinous neoplasm, obtained during surgery at the Tokyo Women's Medical University Hospital, was used for whole-exome sequencing. The patient analyzed provided written consent for use of tissues and clinical information for research involving whole-genome sequencing. Formalin-fixed, paraffin-embedded tissues of archival cases of 118 IPMNs and 32 PDAs of the pancreas operated upon at the Tokyo Women's Medical University Hospital were used for additional analysis of somatic mutations of specific genes and for immunohistochemistry. Clinicopathological features of the patients were listed in Table 3 and Supplementary Table 1. These studied IPMN patients were 73 men and 45 women with the mean age of 65.1 years-old. The IPMN tissues were of 64 gastric types, 40 intestinal types, 9 pancreatobiliary types and 5 oncocytic types in morphological types; 48 branch-duct types, 37 main-duct types and 33 mixed types in macroscopic types; 58 low-grade tumors, 26 high-grade tumors and 47 invasive tumors in grade of tumors; and 56 of stage 0A indicating low-grade tumors, 26 of stage 0, 6 of stage IA, 8 of stage IB, 7 of stage IIA and 15 of stage IIB according to the Union for International Cancer Control staging system, which were determined as described previously7. Invasive IPMNs were determined as invasive carcinomas derived from IPMN according to criteria published in elsewhere33. This study was approved by the Ethical Committee of the Tokyo Women's Medical University.
Whole-exome sequencing
Methanol-fixed, hematoxylin-stained frozen sections were prepared from the frozen tissue. Tumor cells were microdissected from each section using a PALM Microbeam system (Carl Zeiss MicroImaging GmbH, Jena, Germany). Genomic DNA extraction from collected tissues was carried out using the ChargeSwitch® gDNA Mini Tissue kit (Life Technologies, Carlsbad, CA). The extracted DNA was used for construction of a library using a SOLiD Fragment Library Construction Kit (Life Technologies). The library DNA was subjected to whole-exome enrichment using a Sureselect Human All Exon Kit (Agilent Technologies Inc., Santa Clara, CA). The enriched library DNA was then sequenced using the SOLiD System, a deep sequencer employing the massively parallel sequencing method and analyzed using Bioscope software (Life Technologies). Sequence data were mapped on Human Genome Reference, GRCh37/hg19 (The Genome Reference Consortium; http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/index.shtml). All procedures were performed according to the manufacturers' instructions.
Sanger sequencing
For validation of the whole-exome sequencing data, DNA was amplified by polymerase chain reaction (PCR) using the primers listed in Supplementary Table S2 online and AccuPrime PCR system (Life Technologies). The amplified products were treated with ExoSAP-IT (GE Healthcare, Chalfont St Giles, Buckinghamshire, UK) and sequenced using Bigdye Terminator and a 3130xl Genetic Analyzer (Life Technologies). For examination of mutations in GNAS and KRAS in archival cases of IPMNs and PDAs, tumor tissues were manually dissected from sections of formalin-fixed and paraffin-embedded tissues. Genomic DNA was extracted using the ChargeSwitch® gDNA Mini Tissue kit (Life Technologies). Portions of exons 8 and 9 of GNAS and exons 2, 3 and 4 of KRAS were amplified and sequenced as described above using the primers listed in the Supplementary Table 2.
Immunohistochemistry
Indirect immunohistochemical staining of paraffin-embedded tissues using the streptavidin and biotin system was performed by using Histofine SAB-PO kit (Nichirei Biosciences Inc., Tokyo, Japan) as described previously37. The antibodies employed included a rabbit polyclonal anti-GNAS (LifeSpan Biosciences, Inc., Seattle, WA) and a rabbit polyclonal anti-Phospho-(Ser/Thr) PKA Substrate (Cell Signaling Technology, Beverly, MA). To check specificity of staining, a negative control staining without specific primary antibody was carried out. Immunohistochemical results were evaluated as follows: The intensity score (IS) was evaluated by comparing staining in tumor and islets of Langerhans and graded as 0 for no staining (completely negative or extremely faint similar to non-specific staining of surrounding stromal tissue), 1 for evident staining (definitely positive but weaker than that in islets of Langerhans) and 2 for strong staining (densely positive as that in islets of Langerhans). The proportional score (PS) was graded as 0 for staining in <5% of the area, 1 for staining in ≥5% but < 30% of the area, 2 for staining in ≥ 30% but < 70% of the area and 3 for staining in ≥70% of the area. The final score (FS) was calculated and graded as follows: low (FS 1), IS × PS = 0 and 1; moderate (FS 2), IS × PS = 2 and 3; and high (FS 3), IS × PS = 4 and 6. The scoring was made by Y. K. and T.F. independently. The concordance ratio was >95% in each case. Differences in opinion were resolved by re-evaluating the sections and by reaching a consensus.
Statistics
Statistical analysis was performed using PASW Statistics (version 18.0; SPSS Inc. Chicago, IL). P values < 0.05 were considered statistically significant.
References
Ohhashi, K. et al. Four cases of mucous secreting pancreatic cancer. Prog Digest Endosc 20, 348-51 (1982).
Furukawa, T., Takahashi, T., Kobari, M. & Matsuno, S. The mucus-hypersecreting tumor of the pancreas. Development and extension visualized by three-dimensional computerized mapping. Cancer 70, 1505–13 (1992).
Hruban, R. H. et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol 28, 977–987 (2004).
Khan, S., Sclabas, G. & Reid-Lombardo, K. M. Population-based epidemiology, risk factors and screening of intraductal papillary mucinous neoplasm patients. World J Gastrointest Surg 2, 314–8 (2010).
Tanaka, M. Pancreatic Cancer Registry Report 2007. Suizo 22, e1–e427 (2007).
Tanaka, M. et al. International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology 6, 17–32 (2006).
Furukawa, T. et al. Prognostic relevance of morphological types of intraductal papillary mucinous neoplasms of the pancreas. Gut 60, 509–16 (2011).
Dhanasekaran, D. N. Transducing the signals: a G protein takes a new identity. Sci STKE 2006, pe31 (2006).
Forbes, S. A. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39, D945–50 (2011).
Su, K. et al. Isolation, characterization and mapping of two human potassium channels. Biochem Biophys Res Commun 241, 675–81 (1997).
Vaisberg, E. A., Koonce, M. P. & McIntosh, J. R. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J Cell Biol 123, 849–58 (1993).
Jiao, Y. et al. DAXX/ATRX, MEN1 and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–203 (2011).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–12 (2008).
Gingras, R. et al. Purification, cDNA cloning and expression of a new human blood plasma glutamate carboxypeptidase homologous to N-acetyl-aspartyl-alpha-glutamate carboxypeptidase/prostate-specific membrane antigen. J Biol Chem 274, 11742–50 (1999).
Zhang, J. et al. A novel GGA-binding site is required for intracellular sorting mediated by stabilin-1. Mol Cell Biol 29, 6097–105 (2009).
Wood, L. D. et al. The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–13 (2007).
Aricescu, A. R. et al. Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science 317, 1217–20 (2007).
Jones, S. et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–31 (2010).
Grainger, R. J. & Beggs, J. D. Prp8 protein: at the heart of the spliceosome. Rna 11, 533–57 (2005).
Kan, Z. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–73 (2010).
Rubin, J. et al. The coding ECP 434(G>C) gene polymorphism determines the cytotoxicity of ECP but has minor effects on fibroblast-mediated gel contraction and no effect on RNase activity. J Immunol 183, 445–51 (2009).
Kovanich, D. et al. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. Chembiochem 11, 963–71 (2010).
Tong, X., Zhao, F., Mancuso, A., Gruber, J. J. & Thompson, C. B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Natl Acad Sci U S A 106, 21660–5 (2009).
Velayos-Baeza, A., Vettori, A., Copley, R. R., Dobson-Stone, C. & Monaco, A. P. Analysis of the human VPS13 gene family. Genomics 84, 536–49 (2004).
Medendorp, K. et al. The renal cell carcinoma-associated oncogenic fusion protein PRCCTFE3 provokes p21 WAF1/CIP1-mediated cell cycle delay. Exp Cell Res 315, 2399–409 (2009).
Johnston, J. J. et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet 86, 743–8 (2010).
Sjoblom, T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–74 (2006).
Shinada, K. et al. RNF43 interacts with NEDL1 and regulates p53-mediated transcription. Biochem Biophys Res Commun 404, 143–7 (2011).
Wang, L. et al. DOCK2 regulates cell proliferation through Rac and ERK activation in B cell lymphoma. Biochem Biophys Res Commun 395, 111–5 (2010).
O'Brien, S. L. et al. CENP-F expression is associated with poor prognosis and chromosomal instability in patients with primary breast cancer. Int J Cancer 120, 1434–43 (2007).
Landis, C. A. et al. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340, 692–6 (1989).
Thatcher, J. D. The Ras-MAPK signal transduction pathway. Sci Signal 3, tr1 (2010).
Yamaguchi, K. et al. Pancreatic ductal adenocarcinoma derived from IPMN and pancreatic ductal adenocarcinoma concomitant with IPMN. Pancreas 40, 571–80 (2011).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nat Rev Drug Discov 5, 993–6 (2006).
Prevost, G. P. et al. Anticancer activity of BIM-46174, a new inhibitor of the heterotrimeric Galpha/Gbetagamma protein complex. Cancer Res 66, 9227–34 (2006).
Wu, J. et al. Recurrent GNAS Mutations Define an Unexpected Pathway for Pancreatic Cyst Development. Sci Transl Med 3, 92ra66 (2011).
Kuboki, Y. et al. Association of epidermal growth factor receptor and mitogen-activated protein kinase with cystic neoplasms of the pancreas. Mod Pathol 23, 1127–35 (2010).
Acknowledgements
This work was supported by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology and by the Program for Promoting the Establishment of Strategic Research Centers, Special Coordination Funds for Promoting Science and Technology, Ministry of Education, Culture, Sports, Science and Technology (Japan), Yoshioka Hiroto Research Fund and Sato Memorial Foundation for Cancer Research.
Author information
Authors and Affiliations
Contributions
TF designed the study and wrote the manuscript. TF, YK, ET, SY and NS prepared and performed experiments. TF, YK, TH, MY, KShim and KShir obtained and analyzed clinical data. TF, YK and NK analyzed experimental data. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Article File
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Furukawa, T., Kuboki, Y., Tanji, E. et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep 1, 161 (2011). https://doi.org/10.1038/srep00161
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00161
This article is cited by
-
Adenosquamous carcinoma coexisting with intraductal papillary mucinous neoplasm of the pancreas: a case report
Journal of Medical Case Reports (2023)
-
Vacuolar protein sorting 35 (VPS35) acts as a tumor promoter via facilitating cell cycle progression in pancreatic ductal adenocarcinoma
Functional & Integrative Genomics (2023)
-
NKX3.1 Expression in Salivary Gland “Intraductal” Papillary Mucinous Neoplasm: A Low-Grade Subtype of Salivary Gland Mucinous Adenocarcinoma
Head and Neck Pathology (2022)
-
Mutant GNAS limits tumor aggressiveness in established pancreatic cancer via antagonizing the KRAS-pathway
Journal of Gastroenterology (2022)
-
A case of ectopic pancreas of the stomach accompanied by intraductal papillary mucinous neoplasm with GNAS mutation
World Journal of Surgical Oncology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
