
Abstract
Integration of the intestinal epithelium and the mucosal immune system is critical for gut homeostasis. The intestinal epithelium is a functional barrier that secludes luminal content, senses changes in the gut microenvironment, and releases immune regulators that signal underlying immune cells. However, interactions between epithelial and innate immune cells to maintain barrier integrity and prevent infection are complex and poorly understood. We developed and characterized a primary human macrophage-enteroid co-culture model for in-depth studies of epithelial and macrophage interactions. Human intestinal stem cell-derived enteroid monolayers co-cultured with human monocyte-derived macrophages were used to evaluate barrier function, cytokine secretion, and protein expression under basal conditions and following bacterial infection. Macrophages enhanced barrier function and maturity of enteroid monolayers as indicated by increased transepithelial electrical resistance and cell height. Communication between the epithelium and macrophages was demonstrated through morphological changes and cytokine production. Intraepithelial macrophage projections, efficient phagocytosis, and stabilized enteroid barrier function revealed a coordinated response to enterotoxigenic and enteropathogenic E. coli infections. In summary, we have established the first primary human macrophage-enteroid co-culture system, defined conditions that allow for a practical and reproducible culture model, and demonstrated its suitability to study gut physiology and host responses to enteric pathogens.
Similar content being viewed by others

Introduction
Coordinated interaction between the intestinal epithelium and immune cells is required to maintain proper barrier function and mucosal immunity that can prevent infection in the human gut1. Despite the development of several animal and human models proposed to study the cellular and molecular events occurring at the intestinal interface, there remains a need for a practical and reproducible in vitro model that employs human primary tissue to confirm and advance our current understanding of gut physiology and mucosal immunology2. Indeed, while animal models have contributed to our understanding of gut immunology, the biological differences between human and other mammalian species limit the relevance of the data generated through these systems3. Ex vivo experimentation using human intestinal mucosa is a viable approach but limited by tissue complexity (the system cannot be reduced to dissect interactions between discrete cell types), insufficient amount or quality of tissue available, and the need for repeated access to biopsy samples from human donors4. While models composed of human primary isolated intestinal and immune cell types have been used to interrogate interactions between specific cell types, the challenges regarding tissue availability (particularly from healthy individuals), poor tissue characterization, and short viability remain an obstacle that limits their use5,6. Immortalized stable human cell lines such as Caco-2, T-84, and HT-29 cells with one or more immune cell types in assorted co-culture formats7,8,9 have also been applied to address questions regarding epithelial-immune cell interactions and communication. The human neonatal small intestinal line H4 is an alternative non-transformed model of epithelial cells, although it is unclear whether these cells retain the characteristics of the intestinal tissue in vivo. It is also unlikely that one cell type would reflect its diverse composition and complexity10,11. Hence, the development of an epithelial-immune co-culture model derived entirely from primary human tissue that does not require fresh patient samples for each study and exhibits normal intestinal features is a necessary advancement for studying coordinated epithelial immune response under normal or pathogenic conditions12. Human enteroids provide an attractive framework for developing in vitro models of gut architecture and function using primary human tissue13,14,15,16. Enteroids and colonoids derived from LGR5+ stem cells or LGR5+-containing crypts from the small intestine and colon17, respectively, contain the four major human intestinal epithelial cell lineages with distinct roles in gut homeostasis and immune modulation (i.e. absorptive enterocytes, mucus-producing Goblet cells, hormone-producing enteroendocrine cells, and antimicrobial molecule-producing Paneth cells). Further, enteroids and colonoids recapitulate important aspects of human intestinal physiology18,19. Enteroid monolayers that allow experimental access to both apical and basolateral sides of the epithelial cells have been recently developed20,21. These monolayers provide a structure for basolateral addition of relevant cell types, such as immune cells, to interrogate specific physical interactions, paracrine communication, and molecular mechanisms underlying host responses to apical stimuli.
In this work, we describe the development and characterization of a macrophage-enteroid co-culture model consisting of human enteroid monolayers and human monocyte-derived macrophages (one the most prominent cell types participating in innate and adaptive host defenses) and its successful application to investigate intestinal epithelial and macrophage interactions, and their responses to enteric pathogens.
Results
Enteroid monolayers recapitulate features of human small intestinal physiology ex vivo
In order to develop a primary human small intestinal model for the purpose of studying epithelial-macrophage interactions and signaling, we first established human enteroid monolayers according to previously described methods21 and examined their capacity to recreate a polarized epithelial barrier with the unique features of the normal human intestine. Enteroid cysts (three-dimensional [3D] structures) were seeded onto permeable membrane supports and grown to confluency (Fig. 1a,b) to generate monolayers (two-dimensional [2D] structures). The resulting monolayers allow for controllable access to both apical and basolateral surfaces of the intestinal epithelial cells (Fig. 1b). Previous studies established that the removal of Wnt3A from enteroid and colonoid cultures leads to the differentiation of LGR5+ stem cell-containing enteroid cultures (referred to as the non-differentiated [ND] state) into the distinct cell lineages present in the human intestinal epithelium generating the “villus-like” (referred to as differentiated [DF] state) epithelium13,21,22. In this study, enteroid monolayers, derived from duodenum and jejunum of multiple healthy donors, were maintained in either Wnt3A-containing non-differentiation media (NDM) or Wnt3A-free differentiation media (DFM)23 to examine the morphological and functional features of the ND and DF enteroid monolayers. Confocal microscopy of fluorescently labeled actin and wheat germ agglutinin (WGA; indicating glycocalyx-rich cell membrane) showed that both conditions resulted in an organized and polarized epithelium (Fig. 1c) consistent with previously published data13,14. Enteroid differentiation (five days in DFM) was associated with defined brush border formation demonstrated by apical actin labeling (Fig. 1c, right panel) and a significant increase in cell height when compared to the ND monolayers (Fig. 1d). DF, but not ND, monolayers also exhibited a significant increase in barrier function transepithelial electrical resistance (TER) (Fig. 1e). Immunofluorescent labeling of ND enteroid monolayers showed cellular characteristics of intestinal “crypts” evidenced by lysozyme-positive Paneth cells (Fig. 1f, upper panel), whereas the DF enteroid monolayers showed cellular characteristics of intestinal “villi” as evidenced by the presence of phospho-ezrin-positive enterocytes, chromogranin A-positive enteroendocrine cells, and Muc2-positive goblet cells (Fig. 1f). Paneth cells have a particularly long lifetime24 and were still present in DF enteroids, probably remnant from ND structures (data not shown).

(a) Human small intestinal enteroids cysts derived from biopsies (left panel) were grown as confluent epithelial monolayers (right panel) on permeable inserts. (b) Schematic representation of polarized enteroid monolayer established on permeable insert. (c) Non-differentiated (ND; left panel) and differentiated (DF; right panel) enteroid monolayers from small intestinal biopsies were visualized by confocal microscopy and showed polarized apical surfaces (wheat germ agglutinin [WGA]), green; indicated with black arrowheads in top panels). Mature microvilli were observed on the apical surface of DF enteroid monolayers (actin, red). XZ projection, top panels; XY projection, bottom panels; nuclei, blue. (d) Cell heights significantly increased in DF enteroid monolayers (*indicates p = 3.21 × 10−21). (e) TER increased upon 5 days of differentiation (*indicates p = 2.13 × 10−6; **indicates p = 4.96 × 10−9). (f) Paneth cells (lysozyme, red; top panel) were found in ND enteroid monolayers. Enteroendocrine cells (chromogranin-A, green), enterocytes (phospho-ezrin, green), and goblet cells (MUC2, green) were observed in DF enteroid monolayers. Nuclei, blue; actin, white. (g) Total amount of secreted cytokines released in the apical and basolateral media were quantified following 24 h of culture for IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13 IFN-γ, and TNF-α, and 72 h of culture for TGF-β1, in ND and DF enteroid monolayer cultures. IL-8: *indicates p = 0.0132; **indicates p = 0.0010; ***indicates p = 0.0133; ****indicates p = 0.0009. TGF-β1: *indicates p = 0.0067; **indicates p = 0.0448; ***indicates p = 0.0018. Values are presented as Mean + SEM of at least three independent experiments involving enteroids generated from at least three individual donors with the exception of IFN-γ and IL-6 production in ND cultures that represent Mean +© SEM of two independent experiments with enteroids generated from two different donors (Fig. 1g).
To assess the ability of the enteroids to produce and secrete soluble immunological markers, we investigated the presence of 11 cytokines and chemokines in culture media exposed to the apical and basolateral compartments of enteroid monolayers. IL-1β, IL-2, IL-4, IL-10, IL-13, IL-12p70 and TNF-α were undetectable or present at very low levels (below 1 pg; see Supplementary Table S1). Interestingly, large amounts of TGF-β1 and IL-8 (Fig. 1g), two critical cytokines produced by human intestinal mucosa in vivo25, were produced by the small intestinal enteroid cultures. IFN-γ and IL-6 were produced at levels reaching 50 pg (Fig. 1g). All of them, with the exception of IL-6, were secreted in a polarized manner; higher levels of TGF-β1 were detected in the basolateral compartment, whereas IFN-γ and IL-8 were secreted mainly from the apical surface (Fig. 1g). In addition, while IL-6 did not seem to be influenced by differentiation state, IFN-γ and IL-8 were produced more abundantly by the DF “villus-like” enteroid as compared to the ND “crypt-like” counterpart, and the opposite was true for TGF-β1 production. Together, these results demonstrate that enteroid monolayers maintain appropriate barrier function and cell diversity in addition to secreting cytokines whose production can be spatially measured.
Generation of mature human macrophages to establish a macrophage-enteroid co-culture system
In order to investigate the interaction between intestinal epithelial and immune cells and their role in anti-microbial defenses, we developed, as an initial step, an enteroid monolayer culture system that included macrophages. Macrophages (derived from human peripheral blood monocytes) were selected as the first cell lineage to assemble with the intestinal epithelium because they provide a critical first line of innate host immune defense. To this end, monocytes (Mo) isolated from blood of three healthy donors were differentiated into primary mature but not activated macrophages. Three different monocyte subsets were identified by flow cytometry (i.e. Q1, Q2, and Q3; n = 8, including three different donors) on the basis of cell surface marker expression that included: a major CD14+ CD16− CD64+ CX3CR1low classical population (Q3; 76.3% ± 5.0), a CD14− CD16+ CD64− CX3CR1high non-classical population (Q1; 9.0% ± 3.2), and a CD14 and CD16 double positive intermediate population (Q2; 9.1% ± 1.8) (Fig. 2a, upper panels). Following six days of differentiation in the presence of Macrophage Colony-Stimulating Factor (M-CSF), monocyte-derived macrophages (MΦ) were obtained that exhibited a homogeneous CD14+ CD16low CD64low CX3CR1− phenotype (Fig. 2a, lower panels). Differentiated macrophages exhibited increased expression of antigen-presenting major histocompatibility complex (MHC) class II molecule, HLA-DR, while the monocyte surface marker, CD89, was downregulated as compared to freshly isolated monocytes. To confirm the functional capacity of these macrophages, they were exposed in vitro to two different strains of Escherichia coli (E. coli). Phagocytic activity of macrophages was demonstrated by internalization of enterotoxigenic E. coli (ETEC) incubated for 12 h with MΦ on the same side of the Transwell filter (Fig. 2b). Further evidence of phagocytic activity was provided by the internalization of enteropathogenic E. coli (EPEC) by MΦ seeded on the opposite side of the Transwell filter (Fig. 2c,d). This bacteria-MΦ compartmentalization was chosen to represent their location in the final macrophage-enteroid co-culture model. Macrophage phagocytic activity was associated with morphological changes such as their ability to generate projections that extended through 1.0 μm pore filters to physically interact with EPEC (Fig. 2c, middle and right panels) and its flagella (Fig. 2d). This interaction was facilitated via actin-dependent remodeling (Fig. 2c, lower panels). Together, these results demonstrated the successful generation of a homogeneous population of mature monocyte-derived macrophages that can be seeded underneath 1.0 μm pore filter inserts and respond to compartmentalized (top of the filter added) enteric bacterial pathogens.

(a) Freshly isolated human monocytes (Mo; upper panels) and the derivative differentiated macrophages (MΦ; lower panels) were assessed by flow cytometry for surface expression of immune cell markers including CD14 and CD16 (shown as dot plots) and CD64, CX3CR1, CD89 and HLA-DR (shown as histograms). For Mo, the three main populations were reported as Q1, Q2 and Q3. (b) ETEC incubated with MΦ (schematic representation, left panel). Following overnight infection, bacteria were phagocytosed by MΦ. DNA, blue; ETEC, green; CD14 (MΦ), red. (c,d) MΦ were seeded underneath the filter with EPEC added onto the insert (schematic representation, left panel). MΦ extended projections across the 1.0 μm pore insert filter to reach EPEC, added on the opposite surface of the insert membrane. Dashed lines indicate the position of the 1.0 μm filter. (c) DNA, blue; actin, green; CD14, red; EPEC DNA, arrowheads. (d) EPEC flagella, green; CD14, red.
Establishment of macrophage-enteroid co-cultures and cross communication between macrophages and epithelial cells
A human macrophage-enteroid co-culture was established by culturing human enteroid monolayers and mature human macrophages, separated by a permeable membrane. Enteroids were seeded with the basolateral membrane facing the filter and accessible, through the filter pores, to macrophages seeded underneath the filter (Fig. 3a). Confocal imaging of the macrophage-enteroid co-culture showed fluorescently labeled WGA at the apical surface of DF enteroid monolayers derived from human duodenum and on adherent macrophages facing the basolateral side of the enteroid monolayer (Fig. 3b). The viability of these two cell types was confirmed by propidium iodide (PI) exclusion (Fig. 3b); less than 1% of the total filter-adhered macrophages were PI-positive after 24 h of co-culture (data not shown). The macrophage-enteroid model was successfully expanded to include DF enteroid monolayers developed from multiple sections of the human intestine (i.e. duodenum, jejunum and proximal colon) for the purpose of interrogating tissue specific questions (Fig. 3c). Macrophage-enteroid communication was examined using small intestinal DF enteroid monolayers (developed from duodenum and jejunum). Macrophages co-cultured with DF enteroid monolayers exhibited a distinct morphology: they were enlarged with ruffled membranes (Fig. 3d, right panel). In contrast, macrophages maintained on filters alone were rounded and smaller (Fig. 3d, left panel). The fact that these changes occurred within 24 hours of co-culture, suggests an enteroid-induced effect. Similar to what we described above for the enteroid monolayers alone (Fig. 1g), IL-8, TGF-β1, IFN-γ, and IL-6 were detected in media from macrophage-enteroid co-cultures (Fig. 3e). TNF-α, IL-1β, IL-10 and IL-12p70 were un-detectable (<3 pg) in co-cultures and macrophage monocultures while IL-2, IL-4, and IL-13 were secreted in low quantities likely by macrophages (see Supplementary Table S1). TGF-β1 was again secreted primarily to the basolateral side. The amount produced by the MΦ-enteroid co-culture was not different than the levels produced by the enteroids or MΦ alone, which suggests TGF-β1 could derive from either cell type but without synergism or potentiation (Fig. 3e). Meanwhile, IL-8 was also detected in the macrophage-enteroid co-culture basolateral compartment (Fig. 3e) with levels exceeding those produced by the enteroid alone but significantly lower than those produced by macrophages alone. This 1) suggests that macrophages were the main contributors of IL-8 produced basolaterally in co-culture and 2) demonstrates that epithelial cells lead to the downregulation of IL-8 produced by macrophages. Similar to IL-8, IFN-γ and IL-6 levels were substantially increased in the basolateral compartment of co-cultures upon addition of macrophages, suggesting that they were the main contributors to cytokine production (Fig. 3e). Moreover, the quantity of IFN-γ present in co-culture was almost 3 times lower as compared to level released by MΦ alone.

(a) Schematic representation of human enteroid co-cultured with macrophages (MΦ). (b) Macrophage-enteroid co-culture showed polarized DF duodenal enteroid monolayer indicated by apical WGA labeling (green; left and middle panels) and MΦ facing the basolateral side of the epithelium (middle and right panels), both separated by a permeable membrane shown by dashed lines (middle panel). Propidium iodide (red, white arrow in right panel) did not label substantial number of cells. Nuclei, blue; XZ projection, middle panel; XY projection, left and right panels. (c) Macrophage-enteroid co-cultures were established with duodenal-, jejunal-, and colon-derived enteroid monolayers. Nuclei, blue; actin, white; CD14 (MΦ), red; filter, dashed lines. (d) MΦ cultured 24 h with DF enteroid monolayer (right panel) showed morphological changes when compared to MΦ cultured alone (left panel). Nuclei, blue; WGA, green. (e) IL-8, IFN-γ, and IL-6 secretion was increased in basolateral media of DF enteroid cultured 24 h with MΦ. TGF-β1 secreted in apical and basolateral media of 72 h macrophage-enteroid co-cultures was not significantly modulated by the addition of MΦ. IL-8: *indicates p = 0.0156; **indicates p = 0.0156; ***indicates p = 0.0156; ****indicates p = 0.0469. TGF-β1: *indicates p = 0.0448. IL-6: *indicates p = 0.0226; **indicates p = 0.0184. (f) Both ND and DF enteroid monolayers (left panels) could be cultured with MΦ (right panels). Filter, dashed lines; actin, white; nuclei, blue; CD14 (MΦ), red. (g) The presence of MΦ increased cell height in both ND and DF enteroid monolayers. *Indicates p = 1.06 × 10−12; **indicates p = 4.39 × 10−5. (h) Differentiation of the enteroid monolayers and the addition of MΦ increased TER. *Indicates p = 0.0391; **indicates p = 0.0105. Data are presented as Mean + SEM of at least three independent experiments involving enteroids generated from at least three individual donors.
To further determine whether phagocytic cells could influence intestinal epithelial cell morphology, macrophage-enteroid co-cultures were established by culturing macrophages with either ND or DF intestinal enteroid monolayers (Fig. 3f, right panels). Cell height measurements of fluorescent confocal microscopy images (Fig. 3f,g) revealed a significant increase of epithelial cell height in ND and DF monolayers in the presence of macrophages as compared to ND and DF monolayers alone. Likewise, the TER measured in cultures was significantly greater when MΦ where present (Fig. 3h). These findings suggest a potential role of macrophages in enhancing the maturation of the intestinal epithelium and thickening the physical barrier26.
Taken together, these results demonstrated the feasibility of establishing reproducible and functional macrophage-enteroid co-cultures consisting of human primary cells and revealed the interplay of these cells and its influence on structural, physiological, and immune response features as characterized by changes in maturation, barrier function, and cytokine production.
Bacterial infection promotes molecular and phenotypic responses of macrophage-enteroid co-cultures
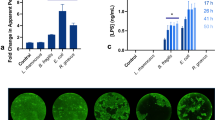
Lastly, we assessed the suitability of the human macrophage-enteroid model to study host-pathogen interactions using ETEC and EPEC strains as model human small intestinal pathogens. Macrophages and DF enteroid monolayers were allowed to equilibrate for 24 h in co-culture, and then mimicking in vivo luminal exposure, ETEC or EPEC (multiplicity of infection (MOI) = 50) were applied to the apical side of enteroid monolayers in the presence or absence of MΦ. Confocal imaging of uninfected or EPEC-infected co-cultures showed the presence of MΦ projections stretching across the permeable filter, in particular upon EPEC infections (Fig. 4a). The quantitative analysis of these macrophage projections demonstrated a significant increase in their numbers as a result of EPEC infection (Fig. 4b, right panel). A similar increase in cell membrane projections was observed when MΦ were exposed to bacteria alone (Fig. 2c,d). EPEC infection was not only associated with increased projections but also with increased number of adherent macrophages (Fig. 4b, left panel) as compared to uninfected (UN) enteroid co-cultures, which suggested that luminal microbial exposure stimulated both retention of macrophages as well as their activation and response. To further localize the MΦ projections, we conducted high-resolution AIRY scan confocal microscopy of EPEC infected macrophage-enteroid co-cultures. The projections were seen extending from underneath the permeable filter and along the lateral membranes of DF enteroid monolayers (Fig. 4c, arrowheads). We further investigated these macrophage morphological changes in macrophage-enteroid co-cultures infected with ETEC. Confocal analyses showed ETEC attached to the apical side of enteroid monolayers (Fig. 4d, arrowheads). Noteworthy, the macrophages seeded underneath the filter in the macrophage-enteroid co-culture physically interacted with ETEC on the apical surface as indicated by yellow pseudocolor merge of red signal, which marks CD14+ macrophages, and green signal, designating ETEC (Fig. 4d, lower right panel, arrowheads). The quantification of bacteria recovered from cultures infected for 16 h demonstrated a significant decrease in viable ETEC number when macrophages where present as compared to DF enteroid monolayer alone (Fig. 4e). Interestingly, the ability of MΦ to kill apical ETEC was already visible after 30 min of infection (data not shown). Also, we noticed a rapid decrease in TER values upon overnight ETEC infection of DF enteroid monolayer, suggesting a loss of barrier function. This seemed to be partially prevented by the presence of macrophages in co-culture (Fig. 4f). Similar to EPEC infection, we found evidence of macrophage projections reaching across enteroid monolayers and trapping ETEC attached on the luminal side of the enteroid monolayer as shown by confocal microscopy and deconvolution 3D reconstruction of ETEC infected co-cultures (Fig. 4g, arrowheads). Finally, overnight infection did not trigger significant changes in production of TGFβ-1, IL-8, IFN-γ, and IL-6 by the co-culture (Fig. 4h).

(a) MΦ projections extended through Transwell filters into DF enteroid monolayers following overnight EPEC infection. Actin, white; nuclei, blue; CD14 (MΦ), red; filter, dashed lines. (b) Adherent MΦ (left panel; *indicates p = 4.42 × 10−5) and number of MΦ projections (right panel; *indicates p = 2.33 × 10−9) increased upon apical overnight EPEC infection of co-cultures. UN, uninfected. (c) High resolution immunofluorescence confocal microscopy showed MΦ projections (arrowheads) going through permeable support and along lateral epithelial membranes upon overnight apical EPEC infection. Actin, white; CD14 (MΦ), red; filter, dashed lines. (d) ETEC adhered to DF enteroid monolayers alone (upper right panel) and co-cultured with MΦ (bottom right panel) following 3 h of infection. Uninfected cultures are represented in left panels. Actin, white; nuclei, blue; ETEC, green; CD14 (MΦ), red; filter, dashed lines. (e) Relative number of viable ETEC collected from the enteroid monolayer following 16 h of infection decreased in the presence of MΦ. *Indicates p = 0.0332. (f) DF enteroid monolayers infected with ETEC for 16 h displayed reduced TER values that were partially restored by the presence of MΦ in the co-culture. (g) Confocal Z-stack image (upper panel) and derivative 3D representation (lower panel) of MΦ extending projections through the membrane to reach the apical surface of DF epithelium infected 3 h with ETEC (arrowheads). Actin, white; CD14 (MΦ), red; nuclei, blue; filter, dashed lines. (h) Overnight ETEC infection did not significantly change the levels of IL-8, TGF-β1, IFN-γ, and IL-6 secreted by the cultures. IL8: *indicates p = 0.0447; **indicates p = 0.0455. Data are presented as the Mean + SEM of three independent experiments involving enteroids generated from three different donors with the exception of IFN-γ and IL-6 production that represent Mean + SEM of two independent experiments with enteroids generated from two different donors.
In aggregate, the results show that macrophages facing the basolateral side of the epithelial cells in our co-culture model can sense, capture, and kill luminal pathogens though appendages that extend across the enteroid monolayer without disrupting the epithelial barrier and inducing a significant pro-inflammatory micro-environment.
Discussion
In this study we have described, for the first time, the establishment and characterization of an ex vivo human “mini” intestine model consisting of primary intestinal epithelial cell monolayers derived from stem cell-containing crypts and monocyte-derived macrophages. An important feature of this system is that it more closely recapitulates epithelial cell diversity and functionality13,27,28,29,30 of the human intestine as compared to previously published models, particularly those that employ immortalized cell lines. The macrophage-enteroid co-culture model was successfully used to interrogate epithelial cell-macrophage interactions and innate immune responses to enteric pathogens.
Different from previously described in vitro intestinal models that rely on cancer-derived epithelial cell lines (e.g. Caco-2 and HT-29)7,8,31,32,33,34, the macrophage-enteroid model we established was based exclusively on primary human cells. Also, unlike existing cancer cell models, the enteroids that provide the framework for the macrophage-intestinal epithelium co-culture naturally exhibit the major cell types found within intestinal epithelium in vivo, as shown in Fig. 1f and in previous studies18,22,29,35. They also recreate the phenotypic characteristics of the intestinal segment from which they were derived36. Enteroid monolayers can be generated for experimental needs by proliferation of LGR5+ stem cell-containing ND enteroids that are maintained (bio-banked) frozen, an important practical advantage previously met only by cell lines. Furthermore, the monolayer format, as opposed to the classical Matrigel-embedded 3D cyst culture, allows manipulations both at the apical and basolateral cell surfaces in a compartmentalized manner, thereby broadening options for treatment and collection of specimens (i.e. cells and culture media). It also improves reproducibility in evaluation of outcomes by reducing the variation in cell number and the restricted lumen volume inherent in the cysts cultures20,21,37. An accessible apical membrane, which can only be achieved with the monolayer format, is critical for consistent exposure of the epithelium to bacterial pathogens (e.g. ETEC and EPEC) similar to what occurs in vivo. Although luminal infection can be performed in enteroid cysts, this would require microinjection of bacteria into each enteroid cyst lumen, which is not only impractical, requiring injection of hundreds of enteroids to infect the equivalent number of cells seeded in the enteroid monolayer cultures (~500,000 cells/0.33 cm2 Transwell), but also technically challenging, as the needle puncture to access the cyst lumen can irreversibly damage the enteroid.
The macrophage-enteroid model described consists of epithelial cell monolayers cultured on the top and macrophages attached underneath a permeable Transwell filter. This design allows for the cells to be in close proximity. Both epithelial cells and macrophages remained viable and functionally active in the combined culture. Importantly, the presence of MΦ further stabilized and enhanced the maturation of the enteroid monolayers as evidenced by the increased epithelial cell height and TER (Fig. 3f–h)26. Not only the epithelial cells changed their morphology but also the MΦ experienced visible morphological changes in the co-culture; they appeared noticeably enlarged and exhibited ruffled edges (Fig. 3d). The phenotype of these macrophages is being examined to further understand their potential effector functions. The normal human intestine contains resident anergic macrophages that contribute to maintaining immune tolerance38,39. Following intestinal infection and/or injury, blood-derived monocytes infiltrate the mucosa and differentiate into “responsive” macrophages38. Two major effector macrophage populations known as M1 and M2, and responsible for driving Th1 or Th2 responses (depending on the stimuli, timing, and environment, among other factors), have been described based on animals and in vitro studies40. Studies to elucidate the contribution of the intestinal epithelium in programming macrophages to polarize towards a resident, M1 or M2 phenotype are ongoing using the human enteroid–macrophage co-culture system.
Enteroids and colonoids are reliable human models for the study of viral and bacterial pathogenesis14,20,21,41,42. In the present study, we increased the cellular complexity by integrating macrophages to enteroid monolayers with the purpose of interrogating host-pathogen interactions and innate immune response. This was achieved by exposing a small intestinal monolayer-macrophage co-culture to EPEC and ETEC as representative human enteric pathogens43,44. Macrophages were able to physically sense and interact with E. coli applied to the apical side of the epithelial cells (Fig. 4d,g) and as a result, they exhibited physiological changes (Fig. 4a,c,g) consistent with the activation and deployment of phagocytic machinery and carried out bacterial killing (Fig. 4e). Furthermore, the presence of MΦ tended to prevent the increase of permeability induced by bacterial infection (Fig. 4f). Interestingly, apical E. coli infection improved the adherence of macrophages (Fig. 4b, left panel) and promoted the development of cell membrane projections (Fig. 4b, right panel) that extended across the epithelium and captured the organism (Fig. 4a,c,g). This phenomenon has been largely observed for human dendritic cells co-cultured with epithelial cell lines45,46,47 and it has been hypothesized that human macrophages could have this capacity based on studies in mice48,49,50. However, there was no direct evidence that this would apply to human macrophages. Our results are, to our knowledge, the first demonstration of human macrophage appendages developing and extending across a human primary intestinal epithelial cell barrier to sample luminal bacteria. Another important finding from our study was the successful bacterial attachment to differentiated (as opposed to non-differentiated) enteroid monolayers, which suggests a more efficient or specific recognition of component(s) or molecule(s) from “villus-like” cells by these enteric pathogens. Similarly, we observed a differentiated epithelium-restricted infection in human small intestinal enteroids exposed to rotavirus and human colonoids exposed to enterohemorrhagic E. coli (EHEC)21,41.
The analysis of cytokines in the macrophage-enteroid model revealed polarized synthesis of TGF-β1, IL-8, and IFN-γ (Figs 1g and 3e) by the epithelial cells alone or in the presence of macrophages, and IL-6 by the macrophage-enteroid co-culture (Fig. 3e). TGF-β1 and IL-8 were abundantly produced by the epithelium itself and, interestingly, are critical cytokines constitutively produced by the human intestinal mucosa and involved in mononuclear cell recruitment25. TGF-β1 was released primarily to the basolateral compartment regardless of epithelial cell differentiation status, which is consistent with its role of ensuring extracellular matrix maintenance and development of regulatory T cells51. TGF-β1 was produced in similar amounts by the enteroid cells alone, macrophages alone, or both combined, denoting no additional or synergistic effect. Interestingly, the non-differentiated monolayers secreted much higher levels of TGF-β1 than the differentiated tissue, which we suspect could be due to a positive feedback of Wnt3A, a component necessary for sustained growth of LGR5+ stem cells15 that is present in the basal (NDM) but not the differentiated media (DFM). This is in agreement with a recent report that attributed increased expression of TGF-β1 by rat hepatic cells to Wnt stimulation52. In all instances, the TGF-β1 was released in its inactivated form, possibly due to the absence of extracellular matrix known to be a TGF activating factor53.
The production of IL-8 and IFN-γ exhibited a different pattern. They were mainly released from the apical side of the enteroid monolayer, produced more abundantly by differentiated enteroid monolayers and released at remarkably higher levels to the basolateral side of the enteroid-macrophage co-culture (Figs 1g and 3d). Also, the release of IL-6, which was not polarized in enteroid mono-culture, showed a similar pattern in co-culture. Macrophages alone produced higher levels of IL-6, IL-8, and IFN-γ than the enteroids (Fig. 3e) and are presumably the main contributors of these cytokines produced by the co-culture. Interestingly, significantly less IL-8 was released in the basolateral co-culture as compared to macrophage alone. Although not significant, a similar pattern was observed for IFN-γ production (Fig. 3e). The decrease of pro-inflammatory cytokine production upon co-culture suggests a synergism between the cell types promoting the macrophage to acquire an anergic phenotype38. The lack of an increase in pro-inflammatory mediator production upon ETEC infection (Fig. 4h) and the high phagocytic capacity of macrophages (Fig. 4e) further sustain this hypothesis38. The capacity of intestinal cells to promote an anergic polarization of macrophages has been suggested in previous studies that used intestinal cell lines31. Further characterization of co-cultured macrophages, including their phenotype and other effector functions in steady state and during infection is warranted to better understand their functional phenotype and contribution to host homeostasis and immune defenses. The ability of the macrophage-enteroid model to detect changes and compartmentalization of immune mediators including important cytokines known to be produced into the gastrointestinal mucosa further demonstrates its accurate closeness of gut function in vivo.
The results our study may provide insight into how the intestinal mucosa clears pathogenic bacteria following acute infection. In humans, EPEC and ETEC infections cause gastroenteritis which resolves without medication usually within a few days post infection54. It is believed that pathogen clearance occurs from normal loss of infected differentiated intestinal epithelial cells since the turnover of the intestinal epithelium is 5-7 days29. Our findings suggest a supportive role of intestinal macrophages in bacterial clearance (i.e. increase of epithelium proximity as suggested by macrophage retention to the filter and high phagocytic activity [Fig. 4b]) and no increase in secretion of pro-inflammatory cytokines [Fig. 4h]) to promote a broad immune response.
The macrophage-enteroid model is envisioned to provide a modular format in which different (single or multiple) immune cells could be added to the epithelial cell monolayers, allowing for studies to be performed in a reductionist or step-wise approach, and in a controlled manner. The demonstration of successful co-culture of mature macrophages with enteroid monolayers produced from small intestinal and colonic intestinal sections extends its use to examine segment-specific questions (Fig. 3c). This co-culture setting will enable inclusion of epithelial and immune cells from the same subject, supporting personalized treatment studies15. The ability of human macrophage-enteroid to recapitulate cell identity, cytokine signaling, and response to infection support its use as an ex vivo model for studies that require modeling of the human gut. Its multiple improvement features make it unique and far superior than methods using human cell lines considered up to now state of the art for the study of cellular and molecular events occurring at human mucosal surface49,55. More work is currently being performed to include additional immune cell lineage, expand its characterization and identify additional readouts. This modular format also allows for further improvements such as addition of other cell lineages constitutive of the lamina propria (e.g. fibroblasts, nerves).
In conclusion, the macrophage-enteroid model described represents a practical and reliable tool to elucidate human innate immunological processes and cellular communications, and to interrogate host-pathogen interactions with a faithful representation of what would occur in the human intestinal epithelium. Furthermore, this macrophage-enteroid system provides a versatile tool to evaluate host responses to preventive or therapeutic treatments.
Methods
Monocyte isolation and macrophage differentiation
Human peripheral blood was collected in EDTA tubes (BD Vacutainer) from healthy volunteers provided informed consent at the University of Maryland. All experimental protocols were approved by the Institutional Review Board of the University of Maryland (IRB #HP-00040025) and all methods were carried out in accordance with the approved guidelines and regulations. Peripheral blood mononuclear cells (PBMC) were obtained by centrifugation over Ficoll-Paque PREMIUM (GE Healthcare Bio-Sciences AB, USA) and residual red blood cells were lysed by treatment with ACK (Ammonium-Chloride-Potassium) Lysing Buffer (Quality Biological, USA). Monocytes were enriched from PBMC by using the Pan Monocyte Isolation Kit (Miltenyi Biotec, USA), according to the manufacturer’s instructions. For macrophage differentiation, cells were seeded in 6- or 12-well plates, at 106 cells/ml, and incubated for 6 days in RPMI (Gibco/Life Technologies, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone Laboratories, USA), MEM non-essential amino acids 1X, 1 mM sodium pyruvate and 55 μM 2-Mecaptoethanol (Gibco, Life Technologies), Penicillin/Streptomycin (HyClone Laboratories, USA) and 50 ng/ml Macrophage Colony-Stimulating Factor (M-CSF) (BioLegend, USA). Additional M-CSF was added on day 2. Complete media was changed on day 4. Cells were maintained at 37 °C in 5% CO2.
Tissue collection and enteroid generation
Human enteroid cultures were established from biopsies obtained after endoscopic or surgical procedures utilizing the methods developed by the laboratory of Dr. Hans Clevers22. De-identified biopsy tissue was obtained from healthy subjects provided informed consent at Johns Hopkins University and all methods were carried out in accordance with approved guidelines and regulations. All experimental protocols were approved by the Johns Hopkins University Institutional Review Board (IRB# NA_00038329). Briefly, enteroids generated from isolated intestinal crypts17 were maintained as cysts embedded in Matrigel (Corning, USA) in 24-well plates and cultured in Wnt3A containing NDM. Once multiple enteroid cultures had been generated, multiple wells were pooled, triturated in Cultrex Organoid Harvesting Solution (Trevigen, USA), and the fragments were collected by centrifugation and resuspended in NDM. Enteroid fragments (100 μl) were added onto 0.4 μm or 1.0 μm pore transparent polyester (PET) membrane 24-well cell culture inserts (Transwell; Corning, USA or Millipore, USA) pre-coated with human collagen IV (30 μg/ml; Sigma-Aldrich, USA). NDM (600 μl) was added to the wells of the receiver plate, and the cultures incubated at 37 °C, 5% CO2. Under these conditions, enteroid cultures reached confluency in 7-14 days. Monolayer differentiation was induced by incubation in Wnt3A-free20 and Rspo-1-free DFM for five days. Monolayer confluency and differentiation were monitored by measuring TER with an ohmmeter (EVOM2; World Precision Instruments, USA). The unit area resistances (ohm*cm2) were calculated according to the growth surface area of the inserts; 0.33 cm2 for both 0.4 and 1.0 μm inserts. Enteroids were derived from segments of the small intestine (duodenum, n = 5; jejunum, n = 1) and proximal colon (n = 1) for Fig. 3c. For all other experiments, results were generated from duodenal and jejunal enteroids derived from intestinal biopsies of healthy subjects.
Enteroid monolayer and macrophage co-culture establishment
ND or DF enteroid monolayers seeded on inserts were inverted and placed into an empty 12-well plate. Fifty microliters of NDM or DFM supplemented with 10 ng/ml of M-CSF and containing 105 monocyte-derived macrophages (MΦ) were added onto the bottom surface of the inserts, and cells were allowed to attach for 2 h at 37 °C in 5% CO2. The inserts remained wet throughout the MФ adherence period. After this incubation, the inserts were turned back to their original position into a 24-well receiver plate and NDM or DFM supplemented with M-CSF (10 ng/ml) was added into the insert (100 μl) and into the well (600 μl). TER measurements indicated as 0 h were collected prior to preparing the co-culture.
Escherichia coli strains and infections
ETEC H-1040756,57 and EPEC E2348/6958 were generously provided by James Kaper (University of Maryland, Baltimore, USA). Both strains were grown from frozen stocks (−80 °C) at 37 °C on Luria broth (LB) agar plates 2-3 days prior to experiments. The day before infections, single colonies were inoculated in 2 ml of Advanced DMEM/F12 tissue culture medium (Gibco/Life Technologies, USA) and grown overnight with vigorous shaking at 37 °C. The day of the infections, the E. coli cell density of the overnight culture(s) was adjusted to 109 CFU/ml in Advanced DMEM/F12, and 5 μL (5 × 106) was added to MΦ, monolayers or 24 h co-cultures to achieve a multiplicity of infection of 50 relative to MΦ (105). E. coli infections were allowed to progress for 30 min, 3 h, overnight (14 h–16 h), or 24 h.
ETEC killing assays
The ability of MΦ in co-culture to kill ETEC was measured by bacterial killing assay. Enteroid monolayers alone or co-cultured with MΦ for 24 h were infected apically with ETEC (infection doses ranged from 5 × 106 to 10 × 106 CFU) for 30 min or overnight. After infection, the monolayers were washed 3 times with sterile PBS (phosphate buffered saline; 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.3) and lysed in a solution of 0.5% sodium deoxycholate (Sigma-Aldrich) by applying vigorous vortexing. The lysate was serially diluted in sterile PBS and spread onto Luria Broth agar plates. CFU were counted after overnight incubation at 37 °C. Results for 16 h infection were obtained from three different small intestinal lines (n = 3). Data are presented as the relative change in the number of viable bacteria observed in ETEC-infected DF macrophage-enteroid monolayer co-cultures compared to DF enteroid monolayers alone exposed to ETEC.
Immunofluorescence staining
Human enteroid monolayers and Mɸ were fixed in aqueous 4% paraformaldehyde (PFA; Electron Microscopy Sciences, USA) for at least 30 min at room temperature (RT) and then washed with PBS for 10 min at RT. Permeabilization and blocking were carried out simultaneously in a solution of 15% FBS, 2% BSA, and 0.1% saponin (all Sigma-Aldrich, USA) in PBS for 30 min at RT. Cells were rinsed with PBS and incubated overnight at 4 °C with primary antibodies diluted 1:100 in PBS containing 15% FBS and 2% BSA. Primary antibodies included Rabbit anti-Muc2 (Sigma-Aldrich, USA), Rabbit anti-phospho-Ezrin (Abcam, USA), Rabbit anti-lysozyme (DAKO, USA), Mouse anti-CD14 (Abcam, USA), Mouse anti-chromogranin A (The Clinical for Proteomic Technologies for Cancer; National Cancer Institute, NIH, USA) and rabbit sera containing anti-colonization factor 1 (CFA/I) for ETEC and anti-H6 flagellin for EPEC (provided by James Kaper). Because EPEC flagella are lost soon after adhesion to Caco-2 monolayers59, EPEC were also visualized by DNA staining in the absence of H6 staining (Fig. 2c). Stained cells were then washed 3 times for 10 min each with PBS followed by secondary antibodies diluted 1:100 in PBS. Secondary antibodies included Goat anti-Rabbit-Alexa-488, Goat anti-Mouse-Alexa-488, Goat anti-Rabbit-Alexa-568, Goat anti-Mouse-Alexa-568 (all from Molecular Probes/Invitrogen, USA). Probes included wheat germ agglutinin (WGA-488) and phalloidin (568 or 633) (Life Technologies). Hoechst (Vector Laboratories, USA) for nuclear/DNA labeling was used at a 1:1000 dilution in PBS. After incubation, cells were washed 3 times for 10 min each and mounted in ProLong Gold (Vector Laboratories, USA) overnight at 4 °C.
Confocal Microscopy
Confocal imaging was carried out at the Fluorescence Imaging Core of the Hopkins Basic Research Digestive Disease Development Center using an LSM-510 META laser scanning confocal microscope (Zeiss, Germany) running ZEN 2012 (black edition) imaging software (Zeiss, Germany). All images were captured with a 40X oil objective. For qualitative analysis, image settings were adjusted to optimize signal. For quantitative analysis, the same settings were used to image across samples (e.g. for enumeration of MΦ or MΦ projections). Deconvolved rendering of confocal fluorescent image data were obtained using Volocity 3D Image Analysis Software (v6.1, PerkinElmer; USA) (Fig. 4g, bottom panel). The optical section (0.159 μm) shown in Fig. 4c was generated on an LSM-880 with AIRY scan detector and processor running ZEN 2.1 (black edition) under a 63X oil objective. Images were minimally manipulated using either ZEN 2012 or Photoshop (version CS3, Adobe) to emphasize macrophages and bacteria staining. Signal processing was applied equally across the entire image.
Flow cytometry
The following human specific monoclonal antibodies were obtained from BD PharMingen, USA: 2331/FUN-1 (anti-CD86, FITC-conjugated), 10.1 (anti-CD64, FITC-conjugated), M5E2 (anti-CD14, APC-conjugated), G46-6 (anti-HLA-DR APC- and V450-conjugated), L307.4 (anti-CD80, PE-Cy7-conjugated), and 3G8 (anti-CD16, PE-Cy7-conjugated). K0124E1 (anti-human CX3CR1, PE-conjugated), 10.1 (anti-CD64, PerCP/Cy5.5-conjugated), and A59 (anti-human CD89, PE-Cy7-conjugated) were obtained from BioLegend, USA. Appropriate isotype-matched control antibodies were purchased from the same manufacturers and used at the same concentration as their respective specific antibody. Cells were blocked in 2% normal mouse serum (Invitrogen, Frederick, MD, USA) for 15 min at 4 °C. After washing, 105 to 106 cells were resuspended in 100 μl of PBS containing 0.5% BSA and 2 mM EDTA (all Sigma-Aldrich) and were incubated with antibodies for 30 min at 4 °C. Cells were then washed 3 times in staining buffer and either analyzed or fixed in 4% PFA for 15 min at 4 °C. Fixed cells were maintained up to 3 days at 4 °C before acquisition. Cells were analyzed in a Guava 8HT using Guava ExpressPro software (Millipore USA) or BD LSR Fortessa using FACSDIVA software (BD Biosciences, USA). Data were analyzed using FlowJo software (v10.1). Fluorescence minus one (FMO) or alternatively beads (OneComp Beads, eBiosciences) were used for compensation. Cells were counted using Guava Viacount Flex Reagent (Millipore, USA) according to the manufacturer’s instruction and analyzed on a Guava 8HT using Guava ViaCount software (Millipore, USA).
Cytokine and chemokine measurements
Cytokine and chemokines were measured by electrochemiluminescence microarray using commercial assays (Meso Scale Diagnostic, Rockville, MD) following the manufacturer’s instructions. IL-8, IL-1β, IL-2, IL-4, IL-10, IL-13, IL-12p70, TNF-α, IFN-γ, IL-6, and TGF-β1 were reported as pg contained in the total volume of culture supernatant obtained from the apical and basolateral compartments of the enteroid or enteroid-MΦ co-culture, or from the MΦ cultured alone at the specified times.
Statistics
Statistical significances were calculated using the Student’s t-test or alternatively Mann–Whitney–Wilcoxon test in absence of normal distribution of the values. TER and cell height values from ND or DF monolayers, cytokine/chemokine values from different conditions, and apical and basolateral compartments from ND or DF were considered as paired for p calculation. Cytokine/chemokine production by ND and DF enteroids, number of MΦ, and MΦ projection values were considered as unpaired for p calculation. Unequal variance was considered for ETEC CFU value comparisons. Statistical significances were assessed to compare groups including minimum n = 3 replicates. Exact p values are indicated in Figure legends.
Additional Information
How to cite this article: Noel, G. et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci. Rep. 7, 45270; doi: 10.1038/srep45270 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
03 May 2017
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has been fixed in the paper.
03 May 2017
Scientific Reports 7: Article number: 45270; published online: 27 March 2017; updated: 03 May 2017 The previous PDF version of this Article contained a typographical error in the Results section under the subheading ‘Generation of mature human macrophages to establish a macrophage-enteroid co-culture system’.
References
Peterson, L. W. & Artis, D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14, 141–153, doi: 10.1038/nri3608 (2014).
Duell, B. L., Cripps, A. W., Schembri, M. A. & Ulett, G. C. Epithelial Cell Coculture Models for Studying Infectious Diseases: Benefits and Limitations. Journal of Biomedicine and Biotechnology 2011, 852419, doi: 10.1155/2011/852419 (2011).
Gibbons, D. L. & Spencer, J. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol 4, 148–157 (2011).
Tsilingiri, K. et al. Probiotic and postbiotic activity in health and disease: comparison on a novel polarised ex-vivo organ culture model. Gut 61, 1007–1015, doi: 10.1136/gutjnl-2011-300971 (2012).
Bimczok, D. et al. Human gastric epithelial cells contribute to gastric immune regulation by providing retinoic acid to dendritic cells. Mucosal Immunol 8, 533–544, doi: 10.1038/mi.2014.86 (2015).
Bisping, G. et al. Patients with inflammatory bowel disease (IBD) reveal increased induction capacity of intracellular interferon-gamma (IFN-gamma) in peripheral CD8+ lymphocytes co-cultured with intestinal epithelial cells. Clin Exp Immunol 123, 15–22 (2001).
Leonard, F., Collnot, E.-M. & Lehr, C.-M. A Three-Dimensional Coculture of Enterocytes, Monocytes and Dendritic Cells To Model Inflamed Intestinal Mucosa in Vitro. Molecular Pharmaceutics 7, 2103–2119, doi: 10.1021/mp1000795 (2010).
McKay, D. M., Croitoru, K. & Perdue, M. H. T cell-monocyte interactions regulate epithelial physiology in a coculture model of inflammation. Am J Physiol 270, C418–428 (1996).
Parkos, C. A., Delp, C., Arnaout, M. A. & Madara, J. L. Neutrophil migration across a cultured intestinal epithelium. Dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. Journal of Clinical Investigation 88, 1605–1612 (1991).
Nanthakumar, N. N., Fusunyan, R. D., Sanderson, I. & Walker, W. A. Inflammation in the developing human intestine: A possible pathophysiologic contribution to necrotizing enterocolitis. Proc Natl Acad Sci USA 97, 6043–6048 (2000).
Trapecar, M., Goropevsek, A., Gorenjak, M., Gradisnik, L. & Slak Rupnik, M. A Co-Culture Model of the Developing Small Intestine Offers New Insight in the Early Immunomodulation of Enterocytes and Macrophages by italic Lactobacillus spp. italic through STAT1 and NF-kB p65 Translocation. PLoS ONE 9, e86297, doi: 10.1371/journal.pone.0086297 (2014).
Bermudez-Brito, M., Plaza-Diaz, J., Fontana, L., Munoz-Quezada, S. & Gil, A. In vitro cell and tissue models for studying host-microbe interactions: a review. Br J Nutr 109 Suppl 2, S27–34, doi: 10.1017/S0007114512004023 (2013).
Foulke-Abel, J. et al. Human Enteroids as a Model of Upper Small Intestinal Ion Transport Physiology and Pathophysiology. Gastroenterology 150, 638–649 e638, doi: 10.1053/j.gastro.2015.11.047 (2016).
In, J. G. et al. Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol 13, 633–642, doi: 10.1038/nrgastro.2016.142 (2016).
Sato, T. & Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194, doi: 10.1126/science.1234852 (2013).
Zachos, N. C. et al. Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. J Biol Chem 291, 3759–3766, doi: 10.1074/jbc.R114.635995 (2016).
Stelzner, M. et al. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol 302, G1359–1363, doi: 10.1152/ajpgi.00493.2011 (2012).
Barker, N. & Clevers, H. Lineage tracing in the intestinal epithelium. Curr Protoc Stem Cell Biol Chapter 5, Unit5A 4, doi: 10.1002/9780470151808.sc05a04s13 (2010).
Mukherjee, S. & Hooper, L. V. Antimicrobial defense of the intestine. Immunity 42, 28–39, doi: 10.1016/j.immuni.2014.12.028 (2015).
Foulke-Abel, J. et al. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 239, 1124–1134, doi: 10.1177/1535370214529398 (2014).
In, J. et al. Enterohemorrhagic Escherichia coli Reduces Mucus and Intermicrovillar Bridges in Human Stem Cell-Derived Colonoids. Cellular and Molecular Gastroenterology and Hepatology 2, 48–62.e43, doi: 10.1016/j.jcmgh.2015.10.001 (2016).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772, doi: 10.1053/j.gastro.2011.07.050 (2011).
Schuijers, J. et al. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 16, 158–170, doi: 10.1016/j.stem.2014.12.006 (2015).
Durand, A. et al. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc Natl Acad Sci USA 109, 8965–8970, doi: 10.1073/pnas.1201652109 (2012).
Smythies, L. E. et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol 80, 492–499, doi: 10.1189/jlb.1005566 (2006).
Tabbaa, M. G., Axon, A. T. R. & Dixon, M. F. Enterocyte dimensions in patients with abnormal intestinal permeability. European Journal of Gastroenterology and Hepatology 6, 607–610 (1994).
Grant, C. N., Nellis, E. D. & Chahine, A. A. A case of small intestinal cast causing SBO in complicated intestinal graft-versus-host disease. Pediatr Surg Int 30, 685–688, doi: 10.1007/s00383-014-3515-9 (2014).
Mizutani, T. et al. Real-time analysis of P-glycoprotein-mediated drug transport across primary intestinal epithelium three-dimensionally cultured in vitro. Biochem Biophys Res Commun 419, 238–243, doi: 10.1016/j.bbrc.2012.01.155 (2012).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265, doi: 10.1038/nature07935 (2009).
Walker, N. M. et al. Cellular chloride and bicarbonate retention alters intracellular pH regulation in Cftr KO crypt epithelium. Am J Physiol Gastrointest Liver Physiol 310, G70–80, doi: 10.1152/ajpgi.00236.2015 (2016).
Spottl, T. et al. A new organotypic model to study cell interactions in the intestinal mucosa. Eur J Gastroenterol Hepatol 18, 901–909 (2006).
Barrila, J. et al. Organotypic 3D cell culture models: using the rotating wall vessel to study host-pathogen interactions. Nat Rev Microbiol 8, 791–801, doi: 10.1038/nrmicro2423 (2010).
Carvalho, H. M., Teel, L. D., Goping, G. & O’Brien, A. D. A three-dimensional tissue culture model for the study of attach and efface lesion formation by enteropathogenic and enterohaemorrhagic Escherichia coli. Cell Microbiol 7, 1771–1781, doi: 10.1111/j.1462-5822.2004.00594.x (2005).
Nickerson, C. A. et al. Three-dimensional tissue assemblies: novel models for the study of Salmonella enterica serovar Typhimurium pathogenesis. Infect Immun 69, 7106–7120, doi: 10.1128/IAI.69.11.7106-7120.2001 (2001).
Barker, N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 15, 19–33, doi: 10.1038/nrm3721 (2014).
Middendorp, S. et al. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells 32, 1083–1091, doi: 10.1002/stem.1655 (2014).
VanDussen, K. L. et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64, 911–920, doi: 10.1136/gutjnl-2013-306651 (2015).
Smith, P. D. et al. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol 4, 31–42, doi: 10.1038/mi.2010.66 (2011).
Smythies, L. E. et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem 285, 19593–19604, doi: 10.1074/jbc.M109.069955 (2010).
Italiani, P. & Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol 5, 514, doi: 10.3389/fimmu.2014.00514 (2014).
Saxena, K. et al. Human Intestinal Enteroids: a New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. J Virol 90, 43–56, doi: 10.1128/JVI.01930-15 (2015).
Zhang, Y. G., Wu, S., Xia, Y. & Sun, J. Salmonella-infected crypt-derived intestinal organoid culture system for host-bacterial interactions. Physiol Rep 2, doi: 10.14814/phy2.12147 (2014).
Gonzales-Siles, L. & Sjoling, A. The different ecological niches of enterotoxigenic Escherichia coli. Environ Microbiol 18, 741–751, doi: 10.1111/1462-2920.13106 (2016).
Humphries, R. M. & Armstrong, G. D. Sticky situation: localized adherence of enteropathogenic Escherichia coli to the small intestine epithelium. Future Microbiol 5, 1645–1661, doi: 10.2217/fmb.10.124 (2010).
Rescigno, M., Rotta, G., Valzasina, B. & Ricciardi-Castagnoli, P. Dendritic cells shuttle microbes across gut epithelial monolayers. Immunobiology 204, 572–581, doi: 10.1078/0171-2985-00094 (2001).
Strisciuglio, C. et al. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. J Crohns Colitis 7, 534–541, doi: 10.1016/j.crohns.2012.08.009 (2012).
Yin, Y. et al. CpG DNA assists the whole inactivated H9N2 influenza virus in crossing the intestinal epithelial barriers via transepithelial uptake of dendritic cell dendrites. Mucosal Immunol 8, 799–814, doi: 10.1038/mi.2014.110 (2014).
Chieppa, M., Rescigno, M., Huang, A. Y. & Germain, R. N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med 203, 2841–2852, doi: 10.1084/jem.20061884 (2006).
Kim, H. J., Li, H., Collins, J. J. & Ingber, D. E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci USA 113, E7–15, doi: 10.1073/pnas.1522193112 (2016).
Mazzini, E., Massimiliano, L., Penna, G. & Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity 40, 248–261, doi: 10.1016/j.immuni.2013.12.012 (2014).
Travis, M. A. et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361–365, doi: 10.1038/nature06110 (2007).
Wang, Y. P. et al. Effects of Wnt3a on proliferation, activation and the expression of TGFb/Smad in rat hepatic stellate cells. Zhonghua Gan Zang Bing Za Zhi 21, 111–115 (2013).
Shi, M. et al. Latent TGF-beta structure and activation. Nature 474, 343–349, doi: 10.1038/nature10152 (2011).
Petruccelli, B. P., Kollaritsch, H. & Taylor, D. N. Treatment of travelers’ diarrhea, 92–100 (BC Decker, 1997).
Mills, M. & Estes, M. K. Physiologically relevant human tissue models for infectious diseases. Drug Discov Today 21, 1540–1552, doi: 10.1016/j.drudis.2016.06.020 (2016).
Evans, D. G., Silver, R. P., Evans, D. J. Jr., Chase, D. G. & Gorbach, S. L. Plasmid-controlled colonization factor associated with virulence in Esherichia coli enterotoxigenic for humans. Infect Immun 12, 656–667 (1975).
Evans, D. J. Jr. & Evans, D. G. Three characteristics associated with enterotoxigenic Escherichia coli isolated from man. Infect Immun 8, 322–328 (1973).
Levine, M. M. et al. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet 1, 1119–1122 (1978).
Cleary, J. et al. Enteropathogenic Escherichia coli (EPEC) adhesion to intestinal epithelial cells: role of bundle-forming pili (BFP), EspA filaments and intimin. Microbiology 150, 527–538, doi: 10.1099/mic.0.26740-0 (2004).
Acknowledgements
The authors wish to acknowledge the Integrated Physiology Core and the Imaging Core of the Hopkins Conte Digestive Disease Basic and Translational Research Core Center. They also want to thank Robin Barnes, a Clinical Research Nurse at University of Maryland for her assistance in obtaining peripheral blood samples and Dr. James Kaper (University of Maryland School of Medicine) for kindly providing EPEC and ETEC strains and antibodies. In addition, the authors wish to thank Alma Arnold and Glenn Doherty from Carl Zeiss Microscopy (Thornwood, NY) for capture and AIRY scan processing of image data presented in Fig. 4c. This work was funded by Grand Challenge Exploration award OPP1118529 and National Institutes of Health awards P30-DK089502 and P01-AI125181.
Author information
Authors and Affiliations
Contributions
G.N., N.B., J.S., M.P. and N.Z. designed the experiments. G.N., N.B. and J.S. conducted the experiments and analyzed the data. G.N., N.B. and J.S. compiled the data. G.N., N.B., J.S., M.P. and N.Z. wrote the manuscript. M.D. and O.K. participated in scientific discussion and manuscript editing. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Noel, G., Baetz, N., Staab, J. et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep 7, 45270 (2017). https://doi.org/10.1038/srep45270
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45270
This article is cited by
-
Advancements in understanding bacterial enteritis pathogenesis through organoids
Molecular Biology Reports (2024)
-
Towards Full Thickness Small Intestinal Models: Incorporation of Stromal Cells
Tissue Engineering and Regenerative Medicine (2024)
-
Novel 3D Flipwell system that models gut mucosal microenvironment for studying interactions between gut microbiota, epithelia and immunity
Scientific Reports (2023)
-
Interrogation of the mammalian gut–brain axis using LC–MS/MS-based targeted metabolomics with in vitro bacterial and organoid cultures and in vivo gnotobiotic mouse models
Nature Protocols (2023)
-
Bioengineering translational models of lymphoid tissues
Nature Reviews Bioengineering (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
