





Abstract
The post-translational modification poly(ADP-ribosyl)ation (PARylation) plays key roles in genome maintenance and transcription. Both non-covalent poly(ADP-ribose) binding and covalent PARylation control protein functions, however, it is unknown how the two modes of modification crosstalk mechanistically. Employing the tumor suppressor p53 as a model substrate, this study provides detailed insights into the interplay between non-covalent and covalent PARylation and unravels its functional significance in the regulation of p53. We reveal that the multifunctional C-terminal domain (CTD) of p53 acts as the central hub in the PARylation-dependent regulation of p53. Specifically, p53 bound to auto-PARylated PARP1 via highly specific non–covalent PAR-CTD interaction, which conveyed target specificity for its covalent PARylation by PARP1. Strikingly, fusing the p53-CTD to a protein that is normally not PARylated, renders this a target for covalent PARylation as well. Functional studies revealed that the p53–PAR interaction had substantial implications on molecular and cellular levels. Thus, PAR significantly influenced the complex p53–DNA binding properties and controlled p53 functions, with major implications on the p53-dependent interactome, transcription, and replication-associated recombination. Remarkably, this mechanism potentially also applies to other PARylation targets, since a bioinformatics analysis revealed that CTD-like regions are highly enriched in the PARylated proteome.
INTRODUCTION
Genotoxic stress constantly harms mammalian cells and contributes to severe pathological states, such as cancer, aging, and neurodegenerative diseases. To ensure genome integrity and physiological homeostasis throughout the lifetime of an organism, multiple DNA damage response and repair mechanisms have evolved during evolution. In mammals, two key players in these processes are the ‘caretaker of the genome‘ poly(ADP-ribose) polymerase 1 (PARP1) and the ‘guardian of the genome’ p53.
PARP1 acts as a sensor, transducer and effector within the DNA damage response with a wide spectrum of functions in several DNA repair mechanisms as well as in chromatin remodeling, transcription, energy metabolism, and regulation of cell death. Using NAD+ as a substrate, PARP1 catalyzes the formation of the post-translational PARylation by covalently attaching PAR chains at glutamate, aspartate, lysine, arginine and serine residues of acceptor proteins (1,2). The resulting biopolymer, i.e., poly(ADP-ribose) (PAR), is a highly negatively charged nucleic-acid-like molecule. Apart from covalent PARylation, proteins can ‘read’ PARylation by binding non-covalently to PAR, which has emerged as a key mechanism to regulate protein function, localization, stability, as well as interactions with other macromolecules such as DNA and RNA. Several PAR-binding modules were identified, including the PAR-binding motif (PBM), the PAR-binding zinc finger motif (PBZ), the macrodomain, and the WWE domain (3). The PBM was described as a weakly conserved 20-amino acid (aa) consensus sequence comprising a region rich in basic aa and a core pattern of hydrophobic aa interspersed with basic residues. So far, no defined protein fold was assigned to the PBM, which is present in hundreds of proteins, including p53, XPA, p21, XRCC1, TERT and histones (4,5). Interestingly, PAR binding proteins are very often also acceptors of covalent PARylation, however the molecular relationship and the mechanistic interplay between the two modes of modification are largely unknown.
The tumor suppressor protein p53 also acts as a master regulator in many DNA damage response mechanisms. Inactivation of p53 is an important driving force of carcinogenesis, as p53 is mutated in >50% of human cancers. This renders the TP53 gene the most prominent target for tumorigenic mutations (6). DNA damage triggers p53 stabilization via its phosphorylation and the subsequent release from its suppressor MDM2. In response, p53 acts as a transcription factor and induces the expression of a wide spectrum of genes involved in cell cycle regulation, apoptosis, and DNA repair (7). p53 comprises an N-terminal transactivation domain (TAD), a DNA binding domain (DBD), a tetramerization domain (TD), and a multifunctional C-terminal domain (CTD) (Figure 1A). The CTD, which is highly basic and intrinsically disordered, participates in all aspects of p53 functions, including transcriptional activity, regulation of protein stability, recruitment of co-factors, and its complex binding behavior to DNA (8). Mice carrying a genetic deletion of the CTD exhibit anemia and bone marrow failure, suggesting crucial functions for this region in organismic physiology (9,10). However, the exact role of the CTD in p53 regulation is still controversial. Initial studies reported that the CTD is a negative regulator of sequence-specific DNA binding (11). Later, the CTD was described to have positive regulatory properties, such as facilitating p53 binding to chromatin and long, ‘naked’ DNA stretches (12) as well as sliding along DNA, while searching for cognate binding sites (13,14). Moreover, the CTD controls the stability of p53–DNA complexes by facilitating cooperative contacts between the core DNA binding domains of p53 (15). Most aa of the CTD are targeted by post-translational modifications (PTMs), such as acetylation, ubiquitination, SUMOylation, neddylation or methylation, all of which are vital for p53 activity and stability, depending on the specific cellular conditions (8).
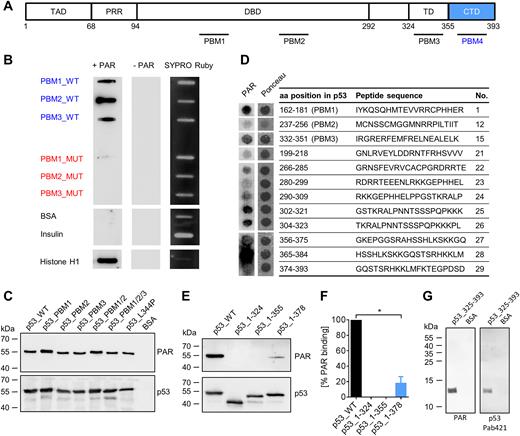
PAR binds non-covalently to the C-terminal domain (CTD) of p53. (A) Schematic representation of the human p53 protein with previously reported (PBM1–3) and novel (PBM4, aa region 363–382) PAR binding motifs (PBMs). (B) PAR overlay assay after slot-blotting of peptides from PBM1–3. Basic amino acids (aa) in wildtype (WT) peptides were exchanged to alanines to generate PBM mutants (peptide sequences are shown in Supplementary Figure S1A). SYPRO Ruby staining served as a loading control. (C) PAR overlay assay after SDS-PAGE of full-length p53 variants with mutated PBMs. BSA served as a negative control. (D) PAR overlay assay of PepSpot peptide array for the identification of potential PAR binding sites in p53. Ponceau S staining served as a loading control. Peptide size: 20 aa. The complete membrane with additional peptides is shown in Supplementary Figure S1B and C. (E) PAR overlay assay after SDS-PAGE of truncated p53 variants. (F) Densitometric quantification of (E). Means ± SEM of 3 independent experiments. The signal of PAR binding to p53_WT was set to 100%. (G) PAR overlay assay after SDS-PAGE of p53_325–393. BSA served as a negative control.
p53 and PARP1 interact at multiple levels; thus, a direct protein-protein interaction was found in vitro and in cells (16,17). Consistently, p53 is a substrate for covalent poly(ADP-ribosyl)ation (PARylation), which has been demonstrated in vitro as well as in cells (18–20). Apart from covalent PARylation, p53 also displays a non-covalent high-affinity interaction with PAR (21,22). Using peptide studies, Malanga et al. reported three potential PBMs within the p53 sequence (22), two of which are located in the DBD (PBM 1&2) and another one in the TD (PBM 3). Interestingly, p53 has a preference for long PAR chains over short ones (21) and PAR was shown to decrease the DNA binding affinity of p53 (22). In addition, it was reported in a murine system that PARylation of p53 blocks the interaction between p53 and the nuclear export receptor Crm1, resulting in nuclear accumulation of p53 (23). Furthermore, p53 deletion in a Parp1 null genetic background accelerates the onset and shortens the latency of mammary tumorigenesis in mice (24). Despite these studies demonstrating a significant relationship of PARylation and p53, many open questions on the molecular and cellular mechanisms of this interaction remain. In particular, similarly to other PARylation substrates, the relationship between non-covalent PAR binding and covalent PARylation of p53 is not understood. Furthermore, it is largely unknown how PAR influences the biochemical properties of p53 and how this translates to a regulation of its cellular functions. Beyond this, it is still enigmatic how PARP1 targets p53 and other proteins for covalent PARylation. Here, we systematically searched for regions in p53 that are essential for non-covalent PAR binding and for its covalent PARylation. We identified a novel region in the p53-CTD that is crucial for non-covalent PAR binding in full-length (fl) p53 and that determines the covalent PARP1-mediated PARylation of p53. Strikingly, fusing the p53-CTD to a protein that is usually not PARylated renders this a target of covalent PARylation as well. These results demonstrate that in the case of p53, PAR binding and covalent PARylation are inherently linked and that the multifunctional CTD of p53 is the center for the regulation of p53 functions by PARylation with implications on protein interactions, transcription and replication-associated recombination.
MATERIALS AND METHODS
Bioinformatic analysis to identify CTD-like regions in PARylated proteins
Protein sequences from the lists of covalently PARylated proteins under genotoxic conditions were retrieved from previous studies (25–27). The IUPred source code (28,29) was modified and used to predict disordered domains in these covalently PARylated proteins, applying the ‘long disorder’ option. A protein region was assumed to be intrinsically disordered, if a disorder tendency of >0.5 was present. The intrinsically disordered fragments were screened for highly positively charged regions. A net charge of at least +4 was chosen in a sliding window of 15 aa as in-silico search pattern. If a directly consecutive region in a protein was found, it was merged with the previous region. To calculate the charge, the amount of basic aa, i.e., arginine or lysine, were subtracted by the amount of acidic aa, i.e., glutamate or aspartate. As control groups, the human proteome (www.uniprot.org, proteome ID UP000005640, reviewed, 20 169 proteins, last modified: 29 January 2017), cytoplasmic proteins from the ‘Eukaryotic Subcellular Localization DataBase’ (eSLDB, gpcr.biocomp.unibo.it/esldb, 776 proteins) (30) or the database of N-glycosylated proteins (N-GlycosylDB, 1118 proteins) from the dbPTM database (dbptm.mbc.nctu.edu.tw) (31) were used.
Protein purification
Human p53, mutants and truncation variants thereof were expressed in Escherichia coli BL21(DE3). Expression was induced at an OD600 of 0.6–0.8 with 20 μM IPTG, followed by an incubation for 12 h at 16°C and 4 h at 10°C. After centrifugation, cells were snap-frozen in liquid nitrogen. Cells were thawed and resuspended in lysis buffer [50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM 2-mercaptoethanol, 10 mM imidazole, 1 mg/ml lysozyme, 1 × EDTA-free protease inhibitor cocktail (Roche)]. Cells were sonicated, followed by DNA digestion with 5 μg/ml DNase I for 30 min. The insoluble fraction was removed by centrifugation at 15,000 g for 30 min. The soluble fraction was filtered and loaded on an ÄKTA chromatography system (GE Healthcare), using a 1-ml HisTrap column (GE Healthcare). Elution was performed with a linear gradient from 10 mM to 500 mM imidazole in a buffer, consisting of 50 mM sodium phosphate pH 8.0, 300 mM NaCl. Elution fractions were dialyzed and thrombin-cleavage was performed overnight for His-tag removal in a buffer, consisting of 20 mM sodium phosphate pH 8, 150 mM NaCl, 10 mM 2-mercaptoethanol. Thereafter, thrombin was inhibited with 0.1 mg/ml Pefabloc (Roche). A second purification step was performed with a HiTrap heparin HP 1-ml column (GE Healthcare), using a linear gradient from 150 mM to 1000 mM NaCl in a buffer, consisting of 20 mM sodium phosphate pH 8.0.
p53_325–393 was expressed and purified as described above, but the second purification step was performed with a HiTrap SP HP 1-ml column (GE Healthcare), instead of a heparin column.
GST-p53_325–393, GST-p53_359–387 or GST were expressed and lyzed as described above, with a modified lysis buffer (50 mM sodium phosphate pH 7.0, 300 mM NaCl, 5 mM 2-mercaptoethanol, 1 mg/ml lysozmye, 1 × protease inhibitor cocktail (Roche)). To the cleared lysate 1 ml of glutathione sepharose 4B (GE Healthcare) was added and incubated rotating for 2 h at 4°C. Beads were washed with 15 ml GST wash buffer 1 (50 mM sodium phosphate pH 7.0, 300 mM NaCl) for 10 min, while rotating. After centrifugation and removal of the supernatant, 7.5 ml GST wash buffer 1 were added to the beads and the slurry was poured into an empty gravity flow column. After washing the column with 5 ml of GST wash buffer 1, 5 ml of GST wash buffer 2 (50 mM sodium phosphate pH 7.0, 150 mM NaCl) were added. Elution was performed with 20 ml of 10 mM glutathione in GST wash buffer 2. Purification of GST-DEK_WT and GST-DEK_PBM was performed as described previously (32). GST-Af1521 was expressed and purified according to a previous study (25). The used plasmid DNA pGEX-4T1::Af1521 was received from Michael L. Nielsen (University of Copenhagen, Denmark). Human PARP1 was expressed in Sf9 cells and purified as described previously (21). Snake venom phosphodiesterase (PDE I) was purchased (Affymetrix) and purified further according to a previous study (33). For the thrombin cleavage assay, the p53 variants p53_28-TCS-29, p53_68-TCS-69, p53_324-TCS-325, as well as p53_WT were covalently PARylated to a larger extent after nickel-affinity chromatography and dialysis. In-vitro PARylation was performed in a solution, consisting of 20 mM sodium phosphate pH 8.0, 150 mM NaCl, 10 mM 2-mercaptoethanol, 10 mM MgCl2, 7.7 μg/ml self-annealed oligonucleotide GGAATTCC, 23 nM rec. PARP1 and 154 μM NAD+ for 1 h at RT. PARylated p53 was purified from this solution with a HiTrap heparin HP 1-ml column, as described above. For determination of PARylation sites by mass spectrometry, p53_WT was purified and PARylated in the same way as described above, with the exception that the His-tag was cleaved during dialysis.
PAR overlay assay
168–403 pmol of custom-synthesized peptides (Genscript) or 5 pmol proteins were blotted on a nitrocellulose membrane (GE Healthcare) either by slot-blotting or by SDS-PAGE and semi-dry blotting, respectively. The membrane was incubated in a solution containing 0.2 μM PAR (synthesized and purified as described previously (21)) in TBST buffer (150 mM NaCl, 10 mM Tris pH 8, 0.05% Tween 20) for 1 h at RT, followed by three washes of 10 min in TBST buffer, containing 1 M NaCl. PAR concentrations refer always to ADP-ribose moieties. The membrane was blocked in TBSMT (TBST with 5% skimmed milk) for 1 h and subsequently PAR was detected using the mouse monoclonal 10H anti-PAR antibody (34) and a HRP-coupled secondary antibody (Dako). Detection of p53 was performed with the mouse monoclonal DO-1 antibody (Merck). As loading control for slot-blotting, SYPRO ruby staining (Thermofisher Scientific) was used directly after blotting. Each PAR overlay experiment was performed at least in 3 independent experiments.
PepSpot peptide arrays
PepSpots peptide arrays (JPT Peptide Technologies) contained custom synthesized peptides with a length of 20 aa covalently linked to a cellulose membrane. Each spot carried approximately 5 nmol of a peptide. A PAR overlay assay was performed as described above with the exception that Alexa488-coupled secondary antibody (Thermofisher Scientific) was used. Fluorescence detection was performed, using a Typhoon FLA 9500 biomolecular imager (GE Healthcare). Ponceau S staining served as loading control.
Electrophoretic mobility shift assay
A 1:2 dilution series of rec. human p53 was prepared in a buffer, consisting of 25 mM Tris-HCl pH 7.4, 150 mM NaCl, 5% (v/v) glycerol and 0.5 mM DTT. The highest p53 concentration was 7 μM, the lowest 27.24 nM. 250 fmol of a double-stranded, Cy5-labeled p53 response element from the p21 promotor (REp21, Oligo_1 + Oligo_2 annealed) was added and incubated for 15 min at RT. 10 × loading dye [250 mM Tris-HCl pH 7.4, 40% (v/v) glycerol, 0.2% (w/v) Orange G] was added to a final volume of 10 μl. Samples were loaded on a 4% native TBE polyacrylamide gel in a Hoefer SE400 gel chamber, containing 0.5 × TBE as running buffer. After 1 h with a voltage of 300 V, fluorescence detection was performed, using a Typhoon FLA 9500 biomolecular imager.
DNA-PAR competition assay
A 1:2 dilution series of non-fractionated, unlabeled PAR was prepared (in concentrations of 200 μM to 1.56 μM). p53_WT (1 μM), p53_1–355 (1.5 μM), p53_PBM4–4 (1 μM) or p53_325–393 (18.7 μM) were added to the PAR solution and incubated for 10 min at RT in a buffer, consisting of 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2% (v/v) glycerol and 0.2 mM DTT. 250 fmol of a double-stranded, Cy5-labeled p53 response element from the p21 promotor (REp21, Oligo_1 + Oligo_2 annealed) or a scrambled variant thereof (scrambled REp21, Oligo_4 + Oligo_5 annealed) were added and incubated for 15 min at RT. 10 × loading dye (250 mM Tris-HCl pH 7.4, 40% (v/v) glycerol, 0.2% (w/v) Orange G) was added to a final volume of 10 μl. Samples were loaded on a 4% native TBE polyacrylamide gel in a Hoefer SE400 gel chamber, containing 0.5 × TBE as running buffer. After applying a voltage of 300 V for 1 h, fluorescence detection was performed, using a Typhoon FLA 9500 biomolecular imager.
Differential scanning fluorimetry
To measure the melting temperature of p53, differential scanning fluorimetry (DSF) was used, according to a previous study (35). The total volume per sample comprised 25 μl. To a buffer, consisting of 50 mM Tris-HCl pH 7.4, 150 mM NaCl and 1 mM DTT, rec. p53 variants were added to a final concentration of 2.3 μM. 50 μM size-fractionated PAR (concentration of ADP-ribose subunits) of different chain length (size-fractionation as described previously (21)) was added and incubated for 10 min at RT. Next, a double-stranded p53 response element from the p21 promotor (REp21, Oligo_2 + Oligo_3 annealed) was added to a final concentration of 2.3 μM (concentration of double-stranded DNA chains), and incubated for 10 min at RT. 2.5 μl of 100 × SYPRO Orange solution (Thermofisher Scientific) was then added, followed by a melt curve analysis with a temperature ramp from 20°C to 95°C at 1°C/min steps, using a quantitative real-time CFX connect thermocycler (Biorad).
In-vitro co-immunoprecipitation (coIP)
Rec. human PARP1 (1 μg) was incubated with 10 μg anti-PARP1 antibody (FI-23) (36) or with mouse IgG control (Santa Cruz) for 1 h at 4°C in 300 μl IP buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate). 10 μl Protein G sepharose beads (Sigma-Aldrich) were added and incubated for 2 h at 4°C, while rotating. Indicated samples were subjected to auto-PARylation of PARP1 by adding 10 mM MgCl2, 7.7 μg/ml self-annealed oligonucleotide GGAATTCC, 1 mM DTT and 100 μM NAD+. Samples were incubated for 5 min at 4°C, while rotating. After centrifugation at 2400 g for 20 s, beads were washed with 500 μl wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% NP-40, 0.05% deoxycholate), followed by a second washing step, using 250 μl IP buffer. After centrifugation and removal of the supernatant, 300 μl IP buffer were added, together with 1 mM DTT and 23 pmol of rec. p53. Incubation took place for 1 h at 4°C while rotating. Beads were washed twice for 5 min in 500 μl IP buffer and once for 5 min in 500 μl wash buffer while rotating. Proteins were eluted from beads by adding 22 μl of 2 × SDS sample buffer, incubating 5 min at 95°C and subjected to SDS-PAGE followed by Western blotting. The rabbit polyclonal anti-PARP1 antibody H250 (Santa Cruz) and the rabbit polyclonal anti-p53 antibody FL-393 (Santa Cruz) were used for immunodetection.
In-vitro PARylation assay
Unless stated otherwise, 0.5 μM rec. protein was PARylated in PBS buffer, in presence of 10 mM MgCl2, 7.7 μg/ml self-annealed oligonucleotide GGAATTCC, 1 mM DTT, 23 nM rec. PARP1 and 100 μM NAD+, in a final volume of 10 μl for 1 h at RT. Proteins were separated by 15% SDS-PAGE, followed by semi-dry blotting on a nitrocellulose membrane (GE Healthcare) and immunodetection using the anti-PAR antibody 10H, unless stated otherwise. Each in-vitro PARylation experiment was performed at least in three independent experiments.
Thrombin cleavage assay
Rec. protein (5 pmol, unless stated otherwise) was incubated in a buffer, containing 20 mM sodium phosphate pH 8.0, 100 mM NaCl, 10 mM 2-mercaptoethanol. 1 unit thrombin was added to a final volume of 30 μl and incubation was performed for 16 h. Thrombin cleavage was stopped by addition of SDS sample buffer and samples were subjected to SDS-PAGE, using a 15% acrylamide gel, followed by semi-dry blotting and immunodetection. Each thrombin cleavage experiment was performed at least in three independent experiments.
Analytical size-exclusion chromatography
The tetramerization of p53 variants was analyzed by size exclusion chromatography (SEC). The tetramerization deficient mutant p53_L344P was used as a control. SEC was performed in 50 mM Tris-HCl pH 7.4 and 150 mM NaCl at a flow rate of 0.03 ml/min using a Superose 6 Increase column (GE healthcare). 30 μl of 8 μM protein solution were loaded respectively and elution was analyzed by absorbance measurement at 280 nm. Sizes were calculated by a gel filtration HMW calibration kit (GE Healthcare). To analyze DNA and PAR binding, p53 variants were preincubated with 0.25 μM double-stranded response element from the p21 promotor (REp21, Oligo_2 + Oligo_3 annealed) or 50 μM unfractionated PAR for 10 min and elution was followed by absorbance measurements at 258 nm.
Mass spectrometric identification of PARylation sites
Sample treatment and mass spectrometer analyses were performed as described previously (33). Briefly, PARylated proteins were treated with PDE I overnight at 25°C. The samples were reduced with DTT for 30 min at 56°C and alkylated with chloroacetamide for 60 min at RT. Digestions were performed using trypsin or pepsin for 4 h at 37°C. Digested samples were analyzed by reversed phase liquid chromatography nanospray tandem mass spectrometry (LC–MS/MS) using an Eksigent nano-HPLC equipped with a C18 LC column (5 μm, 100 Å pore size) in a silica capillary (Acclaim PepMap100, Thermofisher Scientific). After sample injection, the column was washed for 5 min with 100% mobile phase A (0.1% formic acid) and peptides were eluted using a linear gradient of 10% mobile phase B to 40% mobile phase B in 35 min, then to 80% B in an additional 5 min, at 300 nl/min. The LTQ-Orbitrap mass spectrometer (Thermofisher Scientific) was operated in a data dependent mode in which each full MS scan (30 000 resolving power) was followed by 5 MS/MS scans where the five most abundant molecular ions were dynamically selected and fragmented by collision-induced dissociation (CID) using a normalized collision energy of 35% in the LTQ ion trap. Dynamic exclusion was allowed. Tandem mass spectra were searched against a suitable protein database using Mascot (Matrix Science) with the specific enzyme cleavage and variable methionine oxidation. For higher-energy collisional dissociation (HCD) measurements, an Orbitrap Fusion Tribrid mass spectrometer (Thermofisher Scientific) interfaced with an Easy-nLC 1100 nanoflow liquid chromatography system (Thermofisher Scientific) was used. In this case, samples were enriched for phosphoribosylated peptides with Sigma PHOS-Select iron affinity gel beads (Sigma-Aldrich).
Cell culture, transient transfection and PARP inhibitor treatment
Human H1299 cells were cultured at 37°C and 5% CO2 in DMEM (Gibco), supplemented with 10% fetal bovine serum (Biochrom), 0.1 u/μl penicillin and 0.1 μg/μl streptomycin (Gibco). K562(HR-EGFP/3’EGFP) cells were cultivated in RPMI 1640 medium (Gibco) supplemented with 13% (v/v) fetal bovine serum (Merck Millipore) and 1.3% (v/v) penicillin–streptomycin–glutamine (Gibco). Transient transfection of H1299 cells was performed with Lipofectamine 2000 (Fisher Scientific) according to the manufacturer's instruction using 3 μl transfection reagent per μg DNA. PARP inhibitor treatment was performed directly afterwards with 10 μM olaparib or veliparib (Selleckchem).
p53 interactome analysis
H1299 cells were transiently transfected with vectors, encoding Strep-tagged p53_WT, p53_PBM4 or with the control vector pcdna3.1 using polyethylenimin (PEI). p53_WT transfected cells were kept in presence or absence of 10 μM olaparib (Selleckchem). As additional control, non-transfected cells were also used. 24 h later, cells were lyzed [1% (v/v) NP-40, 10 mM 2-mercaptoethanol, 1 × protease inhibitor cocktail with EDTA (Roche), 10 μM olaparib, 50 μM gallotannin, in PBS] and treated with 8 μg/ml DNase I for 30 min, while rotating at 4°C. The lysate was centrifuged and the supernatant was subjected to a p53 pulldown, by adding 100 μl Strep-tactin bead slurry (IBA, Germany). The samples were incubated for 2 h while rotating. The samples were centrifuged (from here on always 2400 g, 20 s), supernatant was removed and beads were washed three times for 5 min with wash buffer [0.1% (v/v) NP-40, 10 mM 2-mercaptoethanol, 1 × protease inhibitor cocktail with EDTA, 10 μM olaparib, 50 μM gallotannin, in PBS]. Proteins were eluted from beads by adding 50 μl of a buffer, consisting of 100 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA and 10 mM D-biotin. The eluted samples were subjected to SDS-PAGE and colloidal Coomassie staining. Protein bands were excised from the gel. All samples were reduced with DTT for 30 min at 56°C and alkylated with chloroacetamide for 60 min at RT. Digestions were performed using trypsin for 4 h at 37°C. Analysis was performed on an Orbitrap Fusion Tribrid mass spectrometer (Thermofisher Scientific) interfaced with an Easy-nLC 1100 nanoflow liquid chromatography system (Thermofisher Scientific). Samples were reconstituted in 0.1% formic acid and loaded onto the analytical column (75 μm × 15 cm) and resolved at a flow rate of 300 nl/min using a linear gradient of 5−35% solvent B (0.1% formic acid in acetonitrile) over 45 min. Data-dependent acquisition with full scans in 350−1500 m/z range was carried out using the Orbitrap mass analyzer at a mass resolution of 120 000 at 200 m/z. Most intense precursor ions were selected at top speed data dependent mode with a maximum cycle time of 3 s. Peptides with charge states 2−5 were selected, and dynamic exclusion was set to 30 s. Precursor ions were fragmented using higher-energy collision dissociation (HCD) set to 35%, and MS/MS ions were detected using the ion trap analyzer. Tandem mass spectra were searched against a suitable protein database using Mascot (Matrix Science) with ‘Trypsin/P’ enzyme cleavage, static cysteine alkylation by chloroacetamide and variable methionine oxidation.
Genomic recombination assay
For the determination of the recombination frequency, K562 cells with stably integrated EGFP-based recombination substrate were used [K562(HR-EGFP/3'EGFP)] (37). The principle of this assay is the restoration of a functional EGFP out of two mutated EGFP-variants. For investigation of the replication-associated recombination frequency of K562(HR-EGFP/3'EGFP) cells and corresponding Western blot analysis for protein expression, cells were transiently transfected with pcdna3.1::p53_WT, pcdna3.1::p53_PBM4 expression plasmids or pcdna3.1 empty vector (vector ctr) via electroporation. Afterwards, cells were exposed to 2.5 μM of PARP-inhibitor olaparib or the solvent DMSO. After 72 h, cells were collected via centrifugation and recombination frequency was determined as fraction of green fluorescent cells within the whole living cell population by usage of the diagonal gating method in the FL1/FL2 dot blot (37). Recombination frequencies were measured by quantification of one million cells from EGFP-positive cells within the life cell-population (SSC/FSC gate). Mean values of recombination frequencies of mock-treated p53_WT expressing cells were set to 1 (absolute mean frequency: 7 × 10−6 for mock-treatments).
Luciferase reporter assay
H1299 cells were transfected with DNA vectors encoding p53_WT or p53_PBM4 together with the Cignal p53 reporter mix (Qiagen). The Cignal p53 reporter mix contains a plasmid, encoding firefly luciferase, which is under the control of a minimal CMV promoter and tandem repeats of p53 transcriptional response elements. It also contains a plasmid, encoding renilla luciferase, which is constitutively expressed and used to normalize for transfection efficiencies and cell viability. 10 μM of the PARP inhibitor olaparib or DMSO as solvent control were added to the cells. After 24 h, cells were lyzed with the Dual-Glo luciferase reporter assay (Promega), according to the manufacturer's instructions. Firefly and renilla luciferase luminescence were analyzed in technical triplicates using a Varioscan Flash plate reader (Thermofisher Scientific). The transactivation activity was expressed as relative luminescence, which is the ratio of firefly luciferase luminescence to renilla luciferase luminescence.
Fluorescence recovery after photobleaching (FRAP)
H1299 cells were transfected with DNA vectors, encoding GFP-tagged p53_WT or p53_PBM4. Bleaching experiments were conducted 24 h later. To this end, an LSM880 confocal laser-scanning microscope (Zeiss) equipped with a 63×/1.4 NA oil-immersion objective was used. A rectangular region (width: 20 pixels/2.4 μm) across the nucleus was bleached using a 488 nm Argon-ion laser (100% transmission, pixel dwell time: 8.2 μs). The fluorescence recovery was followed in a time series experiment. Before irradiation 20 images were recorded. Fluorescence intensities within the bleached region and a non-irradiated nuclear region were measured for each time point. To compensate photobleaching and a fast redistribution of unbound proteins during the bleaching step, for each time point a correction factor was calculated. After background subtraction, the fluorescence intensity in the unbleached region was divided by the average fluorescence intensity measured in the unbleached region before the bleaching pulse. For each time point, the background corrected fluorescence intensity was divided by the respective correction factor. The fluorescence intensity was then normalized, setting the initial fluorescence intensity after the bleaching event to ‘0’, and the fully recovered fluorescence intensity to ‘1’.
p21 protein expression analysis
H1299 cells were transfected with DNA vectors, encoding p53_WT or p53_PBM4. Cells were lyzed 24 h later with a buffer consisting of 50 mM Tris-HCl pH 8, 150 mM NaCl, 1% (v/v) NP40 and protease inhibitor cocktail (Roche). Protein-content was determined using a BCA assay kit (Thermofisher Scientific). For expression level analysis using SDS-PAGE, 30 μg or 10 μg of total protein was loaded for detection of p21 or p53, respectively. After Western blotting, p21 was detected using a rabbit polyclonal p21 antibody (C-19, Santa Cruz).
High-throughput RT qPCR
H1299 cells were transfected with DNA vectors, encoding GFP-tagged p53_WT or p53_PBM4. Cells were detached 24 h later using trypsin and 10% of the cells were used to determine transfection efficiency by FACS. The remaining cells were used for RNA isolation followed by high-throughput RT qPCR with Fluidigm dynamic arrays on the BioMark™ System as described previously (38). To consider different transfection efficiencies, the ΔΔCq calculation of the respective p53-dependent genes was adjusted according to Godbey et al. (39).
Statistical information
Unless stated otherwise, Student's t-test was used to calculate statistical significance. Significance was defined at a 5% level and all analyses were performed with GraphPad Prism 6. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
RESULTS
PAR binds non-covalently to the CTD of p53
To investigate how non-covalent PAR binding affects p53’s functions, we sought to generate a PAR-binding-deficient p53 mutant by taking into account the previously published results by Malanga et al. (22). This study reported three potential non-covalent PAR-binding sites within the p53 sequence using synthesized peptides, i.e., PBM1–3 (Figure 1A). Using a well-established PAR overlay assay (40,41), we first verified results by Malanga et al. showing that peptides of the three PAR binding motifs (i.e., PBM1–3_WT) efficiently bind PAR. Mutant peptides derived of PBM1–3_WT comprising exchanges of basic aa to alanines (i.e., PBM1–3_MUT) showed a strongly reduced or abolished PAR binding ability (Figure 1B, peptide sequences in Supplementary Figure S1A). Next, we generated the corresponding recombinant (rec.) fl-p53 mutants using site-directed mutagenesis. Unexpectedly, an SDS-PAGE analysis followed by a PAR overlay assay revealed no reduction in PAR binding of the fl-p53_PBM1–3 mutants (Figure 1C), indicating that another, so far unknown, PAR binding site mediates PAR binding of fl-p53. Using a PepSpot peptide array for screening, we identified a novel site exhibiting very strong PAR binding ability in p53’s C-terminal domain (CTD) (peptide 28, Figure 1D, complete membrane with additional peptides in Supplementary Figure S1B and C). To verify that the CTD interacts with PAR, we generated truncated variants of rec. p53 and tested their PAR binding abilities (Figure 1E and F). In agreement with the PepSpot analysis, the truncated p53 CTD deletion mutant p53_1–355 completely lacked PAR binding, revealing that in the context of the fl-p53 protein, PBMs 1–3 are not relevant for non-covalent PAR interaction. Instead, our data demonstrates that the actual relevant PAR binding site in fl-p53 is located in the CTD, comprising aa 356–393. The mutant p53_1–378, which comprises half of the CTD, exhibited a significant reduction in PAR binding by 82% compared to fl-p53, demonstrating that the main PAR binding capacity of p53 is located at the very C-terminus of aa 379–393. A truncated p53 mutant consisting of the CTD and the TD (p53_325–393) confirmed PAR binding of the CTD (Figure 1G). Since the CTD is highly positively charged (pI = 10.12 for region 356–393), it is plausible that this region can bind to the highly negatively charged PAR. Since basic aa are essential for the interaction with PAR, the PAR binding site can be mapped to aa 363–382. This region resembles the PAR binding motif (PBM) according to Pleschke et al. (4) and is identified by in-silico alignment as a PBM when allowing two aa mismatches (termed ‘PBM4’ from hereon) (Figure 1A). In summary, we have identified the CTD as a novel PAR interaction site within p53, which accounts for the majority of the PAR binding ability of fl-p53.
Generation of a PAR-binding-deficient p53 mutant
To generate a PAR-binding-deficient p53 mutant, a PepSpot array was performed with peptides, in which critical basic aa were substituted to alanines in two overlapping regions from the p53 CTD: aa 356–375 and aa 365–384 (Figure 2A, complete membrane with additional peptides and controls in Supplementary Figure S2A and B). For the region of aa 356–375, 6 aa exchanges were necessary to abolish PAR binding, and at least 7 aa exchanges for the region of aa 365–384. Based on these results, we incorporated 10 (p53_PBM4–10), 7 (p53_PBM4–7), 5 (p53_PBM4–5) or 4 (p53_PBM4–4) aa exchanges within the region of aa 363–382 of fl-p53 and tested those mutants for their PAR binding ability. Since no PAR binding could be detected in any of those mutants (Figure 2B), p53_PBM4–4 was used as a PAR-binding-deficient mutant for subsequent biochemical and functional analyses. Analytical size-exclusion chromatography (SEC) further confirmed a highly reduced PAR binding under native conditions in solution (Supplementary Figure S2C and E). To investigate if p53_PBM4–4 is still functional, the DNA binding to a p53 response element from the p21 promoter (REp21) was analyzed by an electrophoretic mobility shift assay (EMSA) (Figure 2C and D) and by analytical SEC (Supplementary Figure S2D and E). Under the tested conditions, the DNA binding affinity of the p53_PBM4–4 mutant was not significantly altered compared to p53_WT. Furthermore, cellular chromatin binding was comparable between p53_WT and p53_PBM4–4 (Supplementary Figure S2F-H). Fluorescence recovery after photobleaching (FRAP), which is a measure for chromatin binding, further revealed that the cellular mobilities of p53_WT and p53_PBM4–4 are very similar (Figure 2E). Additionally, the tetramerization of p53_PBM4–4 is not adversely affected by the aa exchanges (Figure 2F). In summary, we successfully generated a p53 mutant with strongly reduced PAR binding by exchanging four basic aa within the CTD, again verifying that aa 363–382 of p53 are of critical importance for the non-covalent p53–PAR interaction.
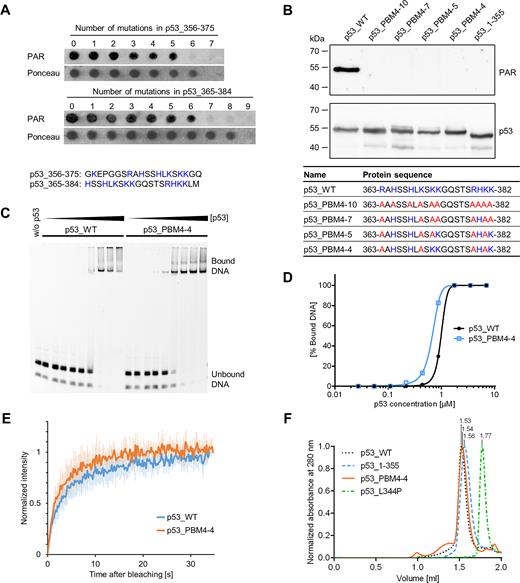
Generation of a PAR-binding-deficient p53 mutant. (A) PAR overlay assay of PepSpot peptide array of CTD mutants with 1–9 aa exchanges. Ponceau S staining served as a loading control. Peptide size: 20 aa. Amino acids highlighted in blue were subjected to exchanges to alanines. The complete membrane with additional peptides and controls is shown in Supplementary Figure S2A and B. (B) PAR overlay assay after SDS-PAGE of full-length p53 with mutated PBM4. Exchanges of 10, 7, 5 or 4 aa were introduced into the CTD, which is shown in the table below. Basic aa (in blue) were subjected to exchanges to alanines (in red). (C) Electrophoretic mobility shift assay (EMSA) with p53_WT and p53_PBM4–4, binding to the fluorescently labeled DNA substrate REp21. (D) Densitometric quantification of (C). (E) FRAP analysis in H1299 cells reconstituted with GFP-tagged p53_WT or p53_PBM4–4. Recovery of GFP was measured after photobleaching. Means ± SD of six cells. (F) Analytical size-exclusion chromatography with p53_WT, p53_1–355, p53_PBM4–4 and a tetramerization-deficient p53 mutant (p53_L344P). Size-calculations are shown in Supplementary Figure S2E.
Influence of non-covalent PAR binding on biochemical properties of p53
The DBD of p53 is known to bind in a sequence-specific manner to p53 response elements, whereas DNA binding abilities of the CTD are thought to be more versatile and sequence-independent (42). As previously reported, non-covalent PAR binding to p53 inhibits p53’s DNA binding ability (22). As these experiments did not distinguish between DBD and CTD-mediated DNA binding, we investigated how non-covalent PAR binding influences the separate DNA-binding abilities of the DBD and the CTD using an in-vitro DNA-PAR competition EMSA. Both p53 fragments, i.e., p53_1–355 and p53_325–393, bound to the fluorescently labeled DNA substrate REp21. However, while DNA binding by p53_325–393 (i.e., comprising the CTD) was highly impaired by PAR and entirely suppressed at higher PAR concentrations, the DNA binding of p53_1–355 (i.e., comprising the DBD) was only mildly influenced (Figure 3A and B). To discriminate between sequence-specific and sequence-independent DNA binding of p53, we compared its DNA binding ability to REp21 or a scrambled version thereof (i.e., scrambled REp21) in the absence or presence of PAR (Supplementary Figure S3). PAR equally efficient outcompeted the binding of p53_325–393 to scrambled REp21 and REp21, reflecting the sequence-independent DNA binding nature of the CTD. Consistent with the finding that the CTD is the center of PAR-dependent regulation, PAR outcompeted the interaction of p53_WT with scrambled REp21 more efficiently as compared to REp21. p53_PBM4–4 showed an intermediate competition behavior between p53_WT and p53_1–355. In summary, these results demonstrate that PAR directly inhibits CTD-mediated sequence-independent DNA binding of p53, however, CTD-bound PAR chains can also alter the DBD-mediated DNA binding in fl-p53.
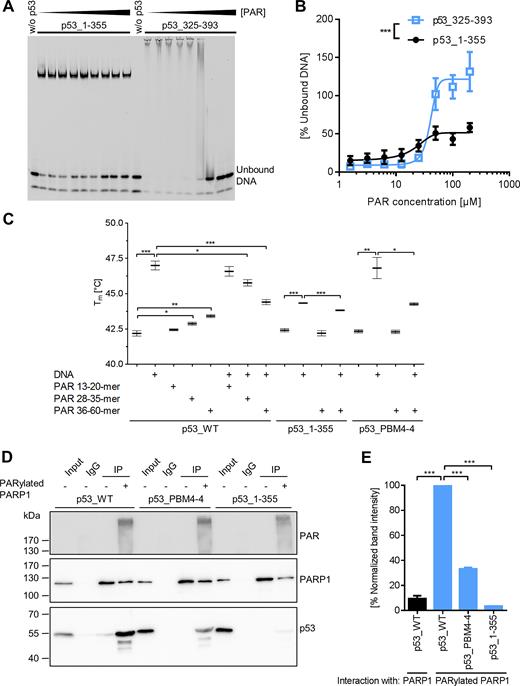
Influence of non-covalent PAR binding on biochemical properties of p53. (A) DNA-PAR competition EMSA. DNA binding of p53_1–355 (1.5 μM) or p53_325–393 (18.7 μM) to a fluorescently labeled DNA substrate REp21 was competed with non-fractionated, unlabeled PAR. Note that DNA binding via p53_1–355 comprising the sequence-specific DBD results in formation of discrete complexes detectable as single bands, whereas p53_325–393 comprising the sequence-independent DNA binding CTD results in formation of heterogeneous complexes detectable as smears. (B) Densitometric quantification of (A). Means ± SEM of >3 independent experiments. Statistical significance was analyzed via two-way ANOVA. (C) Differential scanning fluorimetry (DSF) analysis with p53. Influence of PAR and DNA on the melting temperature of p53 was analyzed. Means ± SEM of 3 independent experiments. Only relevant significances are depicted. (D) Analysis of p53–PARP1 interaction in vitro by coIP. Recombinant (rec.) PARP1 was immobilized on beads and subjected to an auto-PARylation reaction. Rec. p53 was added and the interaction with PARP1 was analyzed using appropriate antibodies. (E) Densitometric quantification of (D). Means ± SEM of three independent experiments.
Previously, it was shown by differential scanning calorimetry that the addition of a p53 response element to the core domain of p53 leads to an increase in its melting temperature (Tm) (43). To test how PAR affects the thermodynamic stability of p53, differential scanning fluorimetry (DSF) was performed (Figure 3C). Indeed, the presence of PAR resulted in an increase of Tm. Furthermore, this increase was PAR chain length dependent being more pronounced with longer PAR chains. This is consistent with previous findings showing increased affinity of longer PAR chains to p53 (21). However, the stabilizing effect of PAR was much weaker than of the DNA substrate REp21, suggesting that the interaction works by different binding mechanisms, potentially by electrostatic interactions. The DNA-PAR competition revealed that longer PAR chains can compete more efficiently with the DNA-p53 binding than short PAR, again being in line with the higher affinity of p53 to long PAR chains. Neither the melting temperature of p53_1–355 alone nor in combination with REp21 was influenced by PAR verifying that PAR binding is mediated by the CTD of p53. Consistent with this notion, the melting temperature of p53_PBM4–4 was not influenced by the presence of PAR itself. However, the increase in Tm mediated by the addition of REp21 could be partly inhibited by addition of PAR, indicating that p53_PBM4–4 still exhibits some residual PAR-binding under certain conditions.
Since in cells it is thought that the majority of PAR molecules are covalently attached to PARP1, we performed in-vitro co-immunoprecipitation (coIP) experiments with auto-PARylated or unmodified PARP1 to investigate how auto-PARylation of PARP1 influences the p53–PARP1 interaction (Figure 3D and E). To this end, rec. PARP1 was immobilized via a PARP1-specific antibody on protein G -coated beads, followed by auto-PARylation, high-stringency washing, and finally co-incubation with rec. p53 variants. About 10-fold more p53 was bound to auto-PARylated PARP1 compared to unmodified PARP1. Consistent with the notion that PAR mediates the p53–PARP1 interaction, the interaction of auto-PARylated PARP1 to p53_PBM4-4 was highly reduced and the interaction to p53_1–355 was almost completely abolished. This reveals that auto-PARylation of PARP1 represents a prerequisite for the efficient interaction with p53 and that PAR functions as a bridge for the p53–PARP1 interaction. The results from Figure 3C–E and Supplementary Figures S2C and S3D suggest that under certain conditions native, tetramerized p53_PBM4–4 is not entirely deficient in PAR-binding, since tetramerization can potentially compensate for the removal of basic aa. Thus, in conclusion, our data indicate that p53_PBM4–4 represents a compromise between reduced PAR binding and still maintained biochemical functionality, e.g., in terms of DNA binding. Keeping this in mind, we used the p53_PBM4–4 mutant in combination with pharmacological PARP inhibition to functionally analyze consequences of non-covalent p53–PAR interaction on a biochemical as well as on a cellular level. In summary, these results show that (i) PAR binding inhibits CTD-mediated sequence-independent DNA binding of p53, (ii) the p53–PAR complex is of lower thermodynamic stability than the p53–DNA complex, despite its high binding affinity, and (iii) the p53–PARP1 interaction can be strongly induced by auto-PARylation of PARP1.
The CTD of p53 is essential for covalent PARylation of p53
To investigate a potential influence of the CTD on the covalent PARylation of p53, truncated p53 variants were tested for their ability to be covalently PARylated using an in-vitro PARylation assay. In this assay, covalent PARylation was tested by mixing rec. p53 variants with rec. PARP1 in the presence of NAD+. Due to covalent PARylation of rec. proteins with different sizes of PAR, the signals in the subsequent Western blot appeared as a diffuse band (‘smear’), by which it can be determined, if a protein is covalently PARylated or not. Strikingly, covalent PARylation was completely abolished in the p53_1–355 variant (Figure 4A), indicating that non-covalent interaction of PAR with the CTD is also essential for the covalent PARylation of p53. Consistently, p53_1–378, which had shown residual non-covalent PAR binding (Figure 1E), exhibited covalent PARylation. To analyze if the CTD is a target for covalent PARylation itself, or if it mediates the interaction to PARP1, we developed a thrombin cleavage assay. To this end, a thrombin cleavage site (TCS) was introduced between aa 324 and 325 (p53_324-TCS-325, Figure 4B), which separates the DBD from the TD. When a PARylation reaction was performed with the p53_324-TCS-325 variant, followed by the thrombin cleavage, a ∼10-kDa down-shift of the PARylated band indicated that the covalent PARylation signal remained at the larger p53_1–324 fragment (Figure 4C, lanes 3 and 4). This demonstrates that covalent PARylation occurs mainly within the region of aa 1–324 of p53. No additional PARylated band was detected at lower molecular weights. To exclude that thrombin itself caused PAR degradation, thrombin was added to PARylated p53_WT lacking a TCS. In this case, no down-shift was observed (Figure 4C, lanes 1 and 2). Previously, covalent PARylation sites were mapped to sites E255, D256 and E268 of murine p53 (23). We exchanged the corresponding aa in human p53 to alanines, but covalent PARylation still occurred (lane ‘p53_EDE/A’ Supplementary Figure S4A). Thus, to map the exact location of covalent PARylation in human p53, we performed high-resolution Orbitrap mass spectrometry (MS) according to Daniels et al. (33). Covalently PARylated p53 was treated with phosphodiesterase I (PDE I) to reduce the PAR chain length, resulting in one remaining protein-bound phosphoribose moiety. Efficient PDE I treatment was controlled by Western blotting for PAR (Supplementary Figure S4B). Using this procedure, we identified several covalent PARylation sites in the TAD of p53 (Figure 4D). Thus, after trypsin digestion, aa E2, D7 or E17 were identified as highly likely targets for covalent PARylation. After pepsin digestion, aa E28 was identified as a covalent PARylation site (Figure 4D, Supplementary Figure S5). We next sought to generate a p53 mutant deficient or impaired for covalent PARylation. To this end, aa E28 alone or in combination with aa E2, D7, E17 or other potential PARylation sites were exchanged to alanines. An in-vitro PARylation assay revealed that covalent PARylation still occurred in these rec. p53 mutants. No decrease in band intensities was observed (Figure 4E). To test if covalent PARylation occurs C-terminally of E28 or outside of the TAD, a TCS was introduced into the p53 sequence between aa 28 and 29 (p53_28-TCS-29) or between aa 68 and 69 (p53_68-TCS-69), respectively. Thrombin cleavage of covalently PARylated p53_28-TCS-29 or p53_68-TCS-69 resulted in a ∼10 or ∼15-kDa down-shift, respectively, of the PARylated bands (Figure 4F), demonstrating that covalent PARylation occurs C-terminally of E28 and C-terminally of the TAD. (N.B. The actual downshift differs slightly from the theoretically calculated one, because of high proline content and additional PARylation of the N-terminus.) We cannot rule out that PARP1 has preferential target sites for covalent PARylation in p53, but our experiments provide substantial evidence that PARP1 modifies various target sites throughout the whole p53 protein with rather broad specificity. To investigate if PARP1 covalently PARylates p53 variants with partial or full deletion of the TAD, we generated N-terminally truncated p53 variants. An in-vitro PARylation assay demonstrated that p53_29–393, as well as p53_69–393 can still be covalently PARylated. Intriguingly, even the p53_325–393 variant can be covalently PARylated, although to a lesser extent (Figure 4G). This suggests that the only denominator that is essential for covalent PARylation of p53 is the CTD and that the selection of the modification site by PARP1 is rather promiscuous.
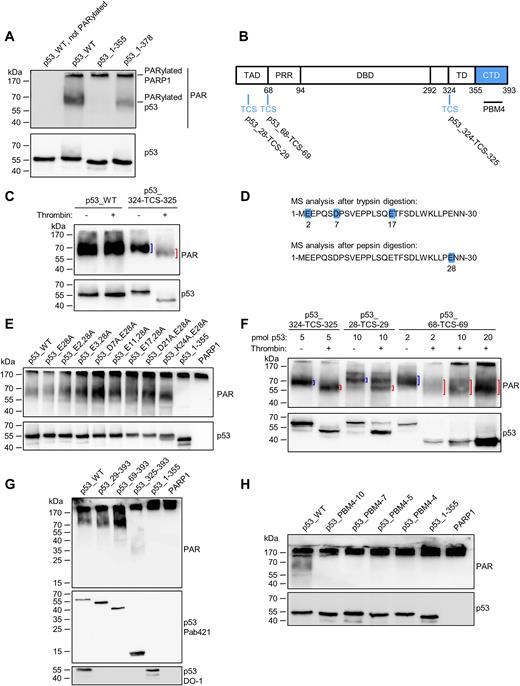
The CTD of p53 is essential for covalent PARylation of p53. (A) In-vitro PARylation assay to test covalent PARylation of C-terminally truncated versions of p53. (B) Schematic representation of the here used human p53 protein variants containing a thrombin cleavage site (TCS). (C) Thrombin cleavage assay with covalently PARylated p53_324-TCS-325. Down-shifted bands are indicated by red brackets, bands of non-thrombin-treated samples by blue brackets. (D) Mass spectrometric identification of covalent PARylation sites in p53_WT. PDE I –treated PARylated p53 was trypsin or pepsin digested. (E) In-vitro PARylation assay with p53 containing mutations at the PARylation sites identified in (D). PARP1: A control PARylation reaction was performed in the absence of p53 variants. (F) Thrombin cleavage assay with covalently PARylated p53_28-TCS-29 or p53_68-TCS-69. (G) In-vitro PARylation assay with N-terminally truncated versions of p53. (H) In-vitro PARylation assay with p53-PBM4 mutants. Exchanges of 10, 7, 5 or 4 aa were introduced into the CTD (protein sequences are shown in Figure 2B).
To examine if non-covalent PAR binding to the CTD is a prerequisite for covalent PARylation, the PAR binding mutants p53_PBM4–10, p53_PBM4–7, p53_PBM4–5 and p53_PBM4–4 were tested for covalent PARylation. Indeed, all of them were deficient for covalent PARylation (Figure 4H), indicating that non-covalent PAR binding of the CTD is necessary for covalent PARylation of p53. Thus, p53_PBM4–4 can be employed as a mutant deficient for both non-covalent as well as covalent PARylation (termed ‘p53_PBM4’ from hereon).
To exclude potential antibody artifacts in the PARylation-deficient mutants or truncations, the in-vitro PARylation assay was performed with radioactive 32P-NAD+ (Supplementary Figure S4C). Consistent with immunochemical results, a subsequent autoradiographic detection showed that p53_PBM4 and p53_1–355 were deficient for covalent PARylation. Additionally, to ensure that only covalent PARylation of p53 is detected in our SDS-PAGE analysis without residual non-covalent PAR–p53 interaction, we added the chaotropic agent urea in a concentration of 8 M to samples after the PARylation reaction, followed by incubation at 95°C for 5 min (Supplementary Figure S4D). No reduction in the PAR signal was observed, verifying that PAR is covalently attached to p53. Furthermore, a PARP inhibitor was added to auto-PARylated PARP1, followed by addition of rec. p53. The absence of a PARylated p53 signal indicated that p53 did not show residual PAR binding, despite the presence of PAR from the auto-modification of PARP1 (Supplementary Figure S4E). Also, when purified PAR was co-incubated with rec. p53, no PAR signals were detected after SDS-PAGE analysis (data not shown).
In summary, these results provide strong evidence that non-covalent PAR binding to the CTD mediates the interaction of p53 with auto-PARylated PARP1, thereby triggering the covalent PARylation of p53. Further, p53 is covalently PARylated throughout the whole protein raising the possibility that PARP1 selects the modification site with rather broad specificity.
Fusing the CTD of p53 to GST renders GST a target for covalent PARylation
Since the interaction of the CTD with auto-PARylated PARP1 appears to be the key factor for targeting p53 for covalent PARylation, we tested if fusing the CTD to a protein that is normally not a substrate for covalent PARylation renders this a target for PARylation. To this end, we chose glutathione S-transferase (GST), which is a widely used negative control for covalent PARylation (44–46). The CTD alone (aa 359–387) or the TD-CTD (aa 325–393) were fused to the C-terminus of GST, separated by a TCS (i.e., GST-p53_359–387 and GST-p53_325–393, respectively). Interestingly, those two fusion proteins were indeed covalently PARylated in an in-vitro PARylation assay, in contrast to GST alone (Figure 5A). Since covalent PARylation potentially can also take place directly within the TD or CTD, we performed a thrombin cleavage assay with PARylated GST-p53_359–387 or GST-p53_325–393. In the case of GST-p53_325–393, a ∼10-kDa down-shift of the PARylated band demonstrated that covalent PARylation mainly occurred indeed within the GST sequence (Figure 5B). To observe the low molecular weight down-shift after thrombin cleavage of PARylated GST-p53_359–387 with suitable sensitivity, we switched to the use of the anti-PAR antibody LP96–10, which recognizes shorter PAR chains than the 10H antibody, thus enabling detection of the PARylation signal in the lower molecular weight range. A ∼4-kDa down-shift of the PARylated protein band after thrombin cleavage clearly demonstrated that covalent PARylation occurred within the GST sequence (Figure 5C). Of note, in cells, GFP, which is usually not covalently PARylated, is targeted for covalent PARylation if it is fused to the extended C-terminal region of p53 (aa 301–393). (Supplementary Figure S4F). In summary, these results demonstrate that fusing the CTD of p53 to a peptide sequence that is usually not PARylated renders it a target for covalent PARylation.
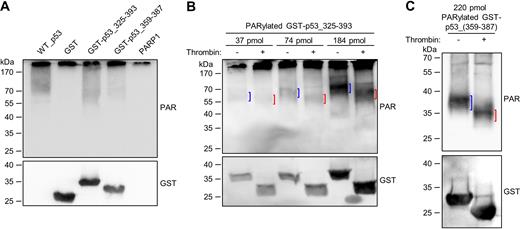
Fusing the CTD of p53 to GST renders GST a target for covalent PARylation. (A) In-vitro PARylation assay with 97 pmol GST, GST-p53_325–393 or GST-p53_359–387. In comparison, covalent PARylation of 5 pmol p53_WT is shown. (B) Thrombin cleavage assay with covalently PARylated GST-p53_325–393. Down-shifted bands are indicated by red brackets, bands of non-thrombin-treated samples by blue brackets. (C) Thrombin cleavage assay with covalently PARylated GST-p53_359–387.
Covalently PARylated proteins are highly enriched in CTD-like regions
Since the CTD targets a protein for covalent PARylation, the question arises if other PARylated proteins also contain regions that resemble p53’s CTD with regards to positive charge density and degree of structural disorder. To identify such regions, we performed a bioinformatics analysis using mass spectrometry data sets for covalently PARylated proteins under genotoxic conditions published by Zhang et al. (27), Jungmichel et al. (26), and Martello et al. (25). First, intrinsically disordered protein regions (IDPRs) were predicted in the PARylated proteins, using the IUpred algorithm (28,29). Next, these IDPRs were screened for presence of highly positively charged regions. Since p53_1–378 and GST-p53_359–387 showed covalent PARylation and a net aa charge of at least +4, this ≥ +4 net charge was chosen as an in-silico search pattern in a sliding window of a total of 15 aa. To calculate the net charge, the amount of the basic aa arginine or lysine was subtracted by the amount of the acidic aa glutamate or aspartate. If a directly consecutive region was identified, it was merged with the previous region. This procedure demonstrated that 68% of the PARylated proteins from Jungmichel et al., 86% from Martello et al. and 84% from Zhang et al. contain highly basic IDPRs (Figure 6A and B, details of identified proteins and sequences in Supplementary Data 1). In comparison, the whole human proteome contains 38% of proteins with highly basic IDPRs. Since PARP1 is mainly active in the nucleus, cytoplasm-localized proteins from the eSLDB database were used as an additional control pool in the analysis (30). 39% of these proteins contain highly basic IDPRs. The proteome of N-glycosylated proteins was used as a non-PARylation-associated control pool, as employed recently also in other studies (20,31). This dataset exhibits only 20% of proteins that contain highly basic IDPRs. These results demonstrate that covalently PARylated proteins are markedly enriched in highly basic IDPRs. Frequently, the PARylated proteins showed very long highly basic IDPRs, with lengths of 40–50 aa and even longer. The highly basic IDPR of p53 is only 24 aa in length, but tetramerization can markedly increase the charge density. To investigate if tetramerization of p53 influences covalent PARylation, we performed the in-vitro PARylation assay in the presence of the tetramerization-deficient mutant p53_L344P (47). PARylation of p53_L344P still occurred, although to a lesser extent than with p53_WT (Supplementary Figure S4A), indicating that tetramerization is not strictly essential, however supports covalent PARylation of p53. To test if similar regulatory mechanisms of non-covalent PAR binding and covalent PARylation exist in proteins other than p53, we analyzed this crosstalk for the DEK protein, which was previously identified as a non-covalent PAR binding protein and a substrate for covalent PARylation (25,26,48). In a recent study, the strongest non-covalent PAR interaction was mapped to aa positions 195–222 (Ganz, Vogel et al., manuscript in preparation). This region was also identified as a highly basic IDPR by our algorithm (i.e., aa 177–227; a second IDPR was identified in the region aa 254–292). Within the PAR binding region of aa 195–222, nine basic aa were exchanged to alanines followed by expression and purification of the recombinant protein. Consistent with results from p53, this mutant showed a reduced non-covalent PAR binding (Figure 6C and D) and covalent PARylation (Figure 6E). To investigate if binding of PAR to other types of PAR binding modules also controls covalent PARylation, we used the in-vitro PARylation assay in the presence of the macrodomain protein Af1521, which lacks highly basic IDPRs (PDB entry 1VHU). The macrodomain binds to PAR non-covalently specifically via the terminal ADP-ribose moiety (49). In this case, we did not detect covalent PARylation of Af1521 in vitro (Supplementary Figure S4A and C), suggesting that not all modes of non-covalent PAR binding render a protein a target for covalent PARylation by PARP1. In summary, these results demonstrate that CTD-like regions are present in most other covalently PARylated proteins, supporting the notion that such highly basic IDPRs may target such proteins for covalent PARylation as well.
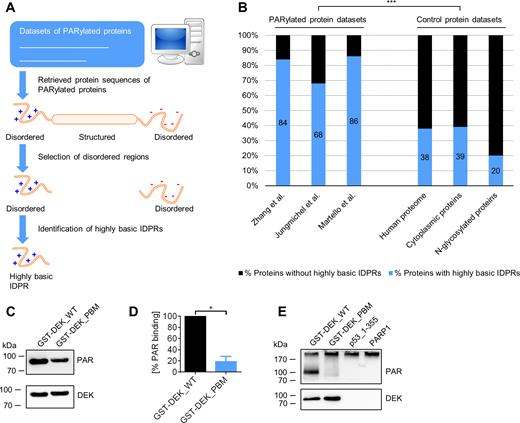
CTD-like regions are present in the majority of covalently PARylated proteins. (A) Schematic representation of the bioinformatics analysis to search for CTD-like regions in PARylated proteins. Protein sequences from data sets of covalently PARylated proteins (25–27) were retrieved and analyzed for presence of predicted intrinsically disordered protein regions (IDPRs). In a next step, screening for highly basic IDPRs was performed, which represent CTD-like regions. (B) Bioinformatics analysis for the identification of CTD-like regions in covalently PARylated proteins. Data from cultured cells under genotoxic stress conditions used from Zhang et al. (27), Jungmichel et al. (26) and Martello et al. (25). P < 0.0001 for any pairwise comparison between a PARylated protein dataset and a control protein dataset; chi-square test. (C) PAR overlay assay after SDS-PAGE of full-length GST-tagged DEK with 9 aa substitutions (GST-DEK_PBM) in a highly basic IDPR PAR binding motif. (D) Densitometric quantification of (C). Means ± SEM of three independent experiments. The signal of PAR binding to DEK_WT was set to 100%. (E) In-vitro PARylation assay with DEK mutant from (D) using anti-PAR anibody LP96–10.
Influence of PAR on cellular functions of p53
To investigate if and how PAR affects cellular functions of p53, pharmacological PARP inhibition and/or the p53_PBM4 mutant were employed to analyze (i) the transactivation activity of p53, (ii) p53-dependent replication-associated recombination and (iii) the PAR-mediated p53 interactome. To this end, p53-deficient human cells were reconstituted with p53_WT or p53_PBM4 by transfection and treated with the PARP inhibitor (PARPi) olaparib or left untreated. Since the transfection procedure itself produces considerable amount of genotoxic stress (50,51) and high levels of p53, we analyzed cellular effects without inducing DNA damage using additional genotoxins. By using protein purification of His-tagged p53 from transfected cells, we confirmed that under those experimental conditions covalent PARylation of p53 occurs in cells (Supplementary Figure S6).
The transactivation activity was determined by a luciferase reporter assay, using a firefly luciferase gene driven by tandem repeats of p53 response elements. Notably, p53_PBM4 showed a significant 37%-reduction in relative luminescence compared to p53_WT (Figure 7A). PARPi treatment in p53_WT-transfected cells reduced the relative luminescence also significantly by 19%, while PARPi in p53_PBM4-transfected cells did not change the relative luminescence, indicating an epistatic effect. To examine the influence of PARylation on gene expression in a chromatin environment, we analyzed CDKN1A (encoding for p21) mRNA transcript levels and p21 protein levels (Figure 7B–D). These results demonstrated that PAR enhances the transactivation activity of p53 also in an endogenous chromosomal encoded gene. mRNA levels of the p53-dependent target genes SLC30A1 and RRM2B showed comparable results (Supplementary Figure S7). MDM2 mRNA levels exhibited a dependence on olaparib, whereas PLK3 showed a p53_PBM4-dependent effect. These results indicate that aa exchanges in PBM4 can also result in PAR independent effects, which is not surprising considering the broad modification of the CTD by other post-translational modifications and its versatile role in the regulation of many p53 functions. Other potential p53-dependent target genes from a previously published gene set (38) showed no dependence on PARP inhibition or p53_PBM4.
A previous report demonstrated that two NLS sequences of low activity are located in the CTD (52). To exclude that the transactivation function is influenced due to altered subcellular localization of p53, high-content fluorescence microscopy with GFP-tagged p53 variants was performed. The ratio of nuclear to cytoplasmic p53-GFP intensity was not significantly different for p53_WT (with or without PARPi) or for p53_PBM4 (Supplementary Figure S8), indicating that PARPi treatment and the aa exchanges in the CTD of the p53_PBM4 mutant did not influence p53’s nuclear localization in the experimental system used in this study.
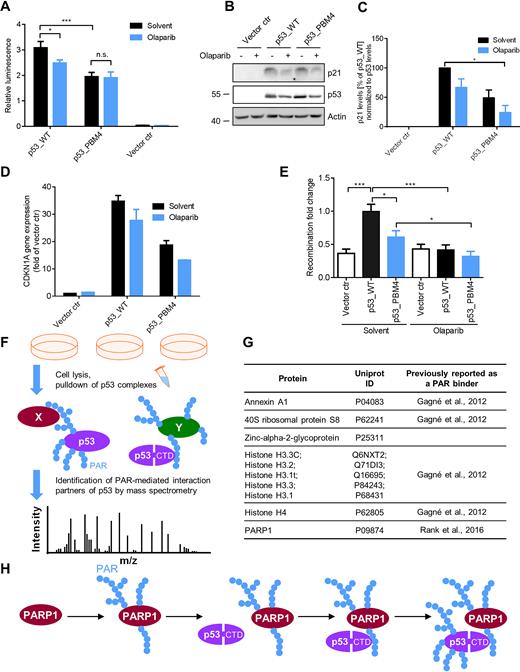
Influence of PAR on cellular functions of p53. (A): Analysis of p53 transcriptional activity using a luciferase reporter assay. H1299 cells were transfected with p53_WT or p53_PBM4, ± PARP inhibitor. Relative luminescence: Ratio of firefly to renilla luciferase luminescence. Renilla luciferase luminescence was used for the normalization of transfection efficiency. Means ± SEM of six independent experiments. (B) p21 protein expression level of experimental system in (A). Actin served as loading control. (C) Densitometric quantification of (B). Means ± SEM of 3 independent experiments. p21 levels were normalized to p53 levels and p53_WT without olaparib was set to 100%. (D) CDKN1A gene expression of experimental system in (A) using RT-qPCR. Means ± half range of 2 independent experiments. (E) Determination of relative replication-associated recombination frequencies of K562(HR-EGFP/3'EGFP) cells, transfected with p53_WT or p53_PBM4, ± PARPi. Means ± SEM of 12 independent measurements. Statistical significance was analyzed via two-tailed Mann–Whitney U test. (F) Schematic representation of the proteomics experiment to identify PAR-mediated interaction partners of p53. H1299 cells were reconstituted with p53_WT ± PARPi or p53_PBM4, followed by pulldown of p53 complexes and MS analysis. (G) Identified PAR-mediated interaction partners of p53 by the proteomics experiment described in F. (H) Mechanistic model of targeting p53 for covalent PARylation by PARP1. PARP1 is activated upon DNA single-strand break detection and subsequently auto-PARylates itself. p53 binds via the CTD non-covalently to PAR chains from auto-PARylated PARP1 and is put into spatial proximity of the catalytic center of PARP1, followed by covalent PARylation of p53 by PARP1. In consequence, the spatio-temporal function of p53 are affected, such as DNA binding properties, transcriptional function, replication-associated recombination and specific protein-protein interactions.
Since p53 stimulates replication-associated recombination, which is assumed to protect replicating DNA (53), we analyzed recombination using a chromosomally integrated recombination reporter substrate (Supplementary Figure S9A) (37,54). Notably, p53_PBM4 showed a significantly reduced stimulation of the recombination rate compared to p53_WT (Figure 7E and Supplementary Figure S9B). Furthermore, p53_WT in presence of PARPi displayed a complete loss of recombination stimulation. These results suggest that PAR is of critical importance for the p53-mediated recombination stimulation, however, additional PAR-dependent factors, which are not directly related to the non-covalent p53–PAR interaction, appear to be also involved in the regulation of replication-associated recombination.
To identify PAR-mediated p53 interaction partners, a p53 pulldown combined with a proteomics analysis was performed, using a high-stringency approach. Thus, an interaction of p53 with another protein was considered to be PAR-mediated, if at least a 4-fold difference in abundance was present between p53_WT and p53_PBM4, as well as between p53_WT and p53_WT treated with PARPi. The fact that PARP1 was identified as a PAR-mediated interaction partner confirms our above coIP results in a cellular environment. Furthermore, Annexin A1, 40S ribosomal protein S8, zinc-alpha-2-glycoprotein, histone H4 and histone H3 variants were identified as PAR-mediated interaction partners (Figure 7F and G and Supplementary Data 2, Supplementary Figure S9C). With the exception of zinc-alpha-2-glycoprotein, all of these proteins were previously identified as PAR binding factors (55). Our results now show that these PAR-protein interactions bridge the interaction with p53 in the cell, thereby demonstrating that the interactome of a given protein, i.e., p53, is directly PAR dependent.
In summary, these results demonstrate that PAR acts as a significant regulator of p53 cellular functions regarding transactivation function, p53-mediated replication-associated recombination, and p53 protein interactions.
DISCUSSION
This study reveals that the previously reported high-affinity interaction of PAR with p53 is mediated by its CTD. The non-covalent PAR binding mediates the interaction of p53 with auto-PARylated PARP1, which then targets p53 for covalent PARylation. In consequence, PARylation regulates several essential cellular functions of p53, such as its transcriptional activity, replication-associated recombination, and specific protein interactions, revealing the CTD as the center for regulation of p53 by PARylation (Figure 7H). By this, the current study gives a prime example of how the two modes of PAR modification, i.e., non-covalent PAR binding and covalent PARylation, crosstalk in case of one of the most important tumor suppressor proteins, i.e., p53, with significant functional consequences on the cellular level.
The CTD plays a multifunctional role in nearly every aspect of p53 regulation, such as protein stability, recruitment of co-factors, and the complex binding behavior of p53 to DNA (8). Apart from its sequence-independent interaction with double-stranded DNA, various other DNA structures act as CTD binding partners, such as single-stranded DNA, insertion/deletion mismatches, recombination intermediates, as well as DNA single-strand and double-strand breaks. Additionally, interaction with RNA was demonstrated (8,56,57). Due to this variety of different binding partners, which are based on electrostatic interactions, the CTD was previously suggested to be a sequence-independent DNA binding domain (42). The different functions of the CTD are regulated by a host of post-translational modifications, such as phosphorylation, ubiquitination, methylation, and acetylation. While a role of PARylation was previously proposed in the regulation of p53 (22,23), its molecular details and functional significance are only poorly understood. Thus, Malanga et al. suggested that non-covalent p53–PAR interaction may take place in p53’s DBD and TD (i.e., PBMs 1–3) (22). We reproduced these results, which were based on peptide studies. However, interestingly PBMs 1–3 were of no or only minor relevance for PAR binding in fl-p53, potentially because these binding sites are not accessible in the full-length protein. Instead, we revealed that the actual PAR binding ability resides in the CTD of p53. The reason of why Malanga et al. did not identify the CTD as a PAR binding region may be attributed to the fact that in some cases peptides are not efficiently bound to the membrane during the PAR overlay assay, because of solubility issues.
Due to the similar chemical characteristics compared to DNA and RNA, a high-affinity electrostatic interaction of PAR with the CTD is plausible. The ability to interact with different types of nucleic acids, i.e., DNA, RNA, PAR, may be necessary to regulate the binding of p53 to the different nucleic acid targets and thereby control its specific cellular functions. Given that PAR contains two negatively charged phosphate groups per ADP-ribose moiety, the charge density and thus the affinity is probably at least as high as for RNA or DNA. Since upon DNA damage, cellular PAR levels can rise >100-fold (58) and the local PAR content at the DNA damage site can reach high peak values, PAR potentially has the ability to outcompete DNA and RNA binding (59). In this way, PAR may serve as an important and highly dynamic factor to regulate the binding of p53 to other nucleic acids in a spatio-temporal manner. We demonstrated that PAR inhibited mainly the sequence-independent, CTD-mediated DNA binding of p53. In contrast, the sequence-specific DNA binding via the DBD was only moderately affected. It was previously demonstrated that p53 binds to a high degree at non-specific DNA target sites in cells. Only about 2–5% of p53-bound DNA target sites have a p53 consensus motif (60,61). Thus, non-covalent PAR binding to the CTD could possibly decrease the sequence-independent DNA binding by competition, leading to an increased specific DNA binding after PAR formation.
Intriguingly, our results show that the PAR-binding CTD is also essential for covalent PARylation of p53. Together with the finding that p53 binds more efficiently to auto-PARylated than unmodified PARP1, this strongly indicates that non-covalent p53–PAR interaction provides the target specificity for its covalent PARylation by PARP1 (Figure 7H). In general, the connection between non-covalent PAR binding and covalent PARylation is understood incompletely. Previous work showed that the PARP1 interaction to many proteins, such as CHD2 (62) or DNMT1 (63) is PAR-mediated. This indicates that the covalent PARylation of these proteins by PARP1 may be PAR-mediated, as well. This is supported by the identification of CHD2 as a target for covalent PARylation (27). Consistently, many PAR-binding proteins are also targeted by covalent PARylation (3). In support of this notion, we showed that in addition to p53, covalent PARylation of DEK is dependent on non-covalent PAR-DEK interaction (Figure 6C–E). We propose that a combination of intrinsic disorder and high positive charge confers non-covalent PAR binding and subsequently leads to PARP1-mediated covalent PARylation of p53, DEK, and potentially other proteins. For example, the proteins FUS, EWS and TAF15, which bind non-covalently to PAR by their long, intrinsically disordered and highly positively charged RGG repeats (64), are also a target for covalent PARylation (26). The same was demonstrated for the short N-terminal tails from histones, which also comprise highly basic IDPRs (44). On the contrary, covalent PARylation was not detected in case of the PAR-binding macrodomain (this and previous studies (26)). A possible explanation can be that IDPRs provide the ability to slide along the PAR chain towards PARP1 to reach spatial proximity for covalent PARylation, which is supported by studies showing that IDPRs slide along DNA in a sequence-independent manner (13,14,65). In contrast, the macrodomain binds to the terminal ADP-ribose moiety of the PAR chain by a specific fold and is probably too distant from the catalytic center of auto-PARylated PARP1 for covalent PARylation. Recently, thousands of covalently PARylated proteins were identified by mass spectrometry. Interestingly, the majority of these proteins have regions that are similar to the CTD of p53 in the way that they are also highly basic and disordered. These regions could account for non-covalent PAR binding and for covalent PARylation of proteins, offering a possible explanation of how PARP1 can select its thousands of targets for covalent PARylation.
In general, protein kinases recognize their target sites for phosphorylation at a specific aa consensus sequence (66). In comparison, our results strongly suggest that PARP1 does not require a certain aa consensus sequence for the selection of covalent PARylation sites. Instead, PARP1 targets a protein, if it exhibits a PAR interaction domain like the CTD and suitable acceptor aa. The high variety of different acceptor aa, such as glutamate, aspartate, lysine and serine supports this notion (2,33). Furthermore, PARP1 can even covalently modify DNA strand break termini in DNA fragments, at least in vitro (67). In consequence, the selectivity of PARP1 towards specific aa substrates is under current discussion in the field (3,68). In principle, such a broad specificity might be necessary to render the response upon certain PARP1-activating stimuli as wide and adaptive as possible. Importantly, however, in a cellular context, PARP1 specificity could be regulated by additional co-factors, as was recently demonstrated for HPF1, which is necessary for serine-specific PARylation (2).
This study demonstrates that PARylation participates in the transactivation function of p53 in cells, which is consistent with previous studies (17,69). Additionally, we show that this regulation is dependent on PBM4. A possible mechanism may be that p53 needs to dissociate from DNA for the start of transcription, which may be mediated by PARylation of p53. Treating cells with a PARPi prevents the dissociation of p53 and TAFs (TATA binding protein associated factors) from the p21 promotor (70). Our data implicate that covalent PARylation of p53 can also occur with DNA-bound p53, since DBD-mediated DNA binding was only moderately influenced by the presence of PAR. Therefore, auto-PARylated PARP1 can bind to p53, followed by covalent PARylation and dissociation of p53 from the DNA. Moreover, PARylated p53 might enhance the recruitment of transcriptional co-factors to the promoter. Indeed, it was previously demonstrated that PAR can affect protein-protein interaction and assemble multi-protein complexes (55). Here we specifically show that PAR regulates protein interactions of p53 in the cell. Thus, PAR mediates the p53 interaction to Annexin A1, 40S ribosomal protein S8, zinc-alpha-2-glycoprotein, histone H4, histone H3 variants or PARP1. Of these, histone H3 and H4 (71), PARP1 (17) and Annexin A1 (72) were previously identified as interaction partners of p53. It will be interesting to disentangle in future studies to what extent the cellular effects on p53 function are dependent on non-covalent PAR binding or/and covalent PARylation.
The CTD is a target for various PTMs, such as acetylation, ubiquitination, methylation, and neddylation, mainly modifying its lysines, including those mutated in p53_PBM4. Thus, it is conceivable that a modification or an interplay of different modifications can influence and regulate non-covalent PAR binding as well as covalent PARylation of p53. Interestingly, while mice carrying a genetic deletion of the CTD in their TP53 gene exhibit anemia and bone marrow failure (9,73), mice with aa exchanges of all lysines to arginines within the CTD show only a mild phenotype of γ-irradiation hypersensitivity (74). This raises the hypothesis that in such a mutant, the p53-CTD is still able to bind PAR and to trigger covalent PARylation due to unaltered positive charge, which could explain the absence of a severe phenotype.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Martin Scheffner for the plasmids pET14b::p53_WT, pET11a::p53_WT, pGEX2TK::p53_325–393 and pcdna3.1::HA-p53_WT; and Michael L. Nielsen for the plasmid pGEX-4T1::Af1521. Further, we thank the proteomics and the FACS facilities of the University of Konstanz for their support of the mass spectrometric analyses and FACS measurements, respectively; Desiree Schütz, Alexandra Semmler and Samy Aliyazdi for associated contributions during their Masters’ or Bachelor's theses work; Marcel Leist for access to the Cellomics instrument; and Andreas Marx and Martin Scheffner for helpful discussions on this project.
Author contributions: Conceptualization, A.F., A.M., A.B.; Methodology, A.F., A.K., S.H., G.A., M.H., M.S.; Formal Analysis and Software, G.A.; Investigation, A.F., A.K., S.H., G.A., L.R., M.H., M.S., J.F., S.V., P.R., M.G.; Writing - Original Draft, A.F., A.M.; Writing - Review & Editing, A.F., A.M., A.K., S.H., G.A., L.R., M.H., M.S., J.F., S.V., P.R., M.G., E.F.M., A.H., L.W., K.H., A.B.; Supervision, Project Administration and Funding Acquisition, A.M., A.B.
FUNDING
German Research Foundation (DFG) through the Konstanz Research School Chemical Biology (KoRS-CB); Research Training Group 1331 [RTG1331]; Collaborative Research Center 969 [CRC969, project B04]; Zukunftskolleg of the University of Konstanz as well as the International Graduate School in Molecular Medicine, Ulm. Funding for open access charge: University of Konstanz.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments