Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease
 Alan Chait
Alan Chait  Laura J. den Hartigh*
Laura J. den Hartigh*- Division of Metabolism, Endocrinology and Nutrition, Department of Medicine, University of Washington, Seattle, WA, United States
Adipose tissue plays essential roles in maintaining lipid and glucose homeostasis. To date several types of adipose tissue have been identified, namely white, brown, and beige, that reside in various specific anatomical locations throughout the body. The cellular composition, secretome, and location of these adipose depots define their function in health and metabolic disease. In obesity, adipose tissue becomes dysfunctional, promoting a pro-inflammatory, hyperlipidemic and insulin resistant environment that contributes to type 2 diabetes mellitus (T2DM). Concurrently, similar features that result from adipose tissue dysfunction also promote cardiovascular disease (CVD) by mechanisms that can be augmented by T2DM. The mechanisms by which dysfunctional adipose tissue simultaneously promote T2DM and CVD, focusing on adipose tissue depot-specific adipokines, inflammatory profiles, and metabolism, will be the focus of this review. The impact that various T2DM and CVD treatment strategies have on adipose tissue function and body weight also will be discussed.
Introduction
Obesity has now reached epidemic proportions, with over 60% of the US population classified as overweight or obese (defined by a body mass index ≥ 25 or 30 kg/m2, respectively) (1). The incidence of type 2 diabetes mellitus (T2DM) has also risen in parallel to the obesity epidemic, and thus is considered a major co-morbidity associated with obesity (2, 3). Recent epidemiological evidence has shown that 85% of type 2 diabetic adults are also obese (4), and it has been projected that more than 300 million people worldwide will have T2D as a consequence of obesity by 2025 (5). While much recent research has aimed to delineate the precise cause(s) of obesity-associated T2DM, the primary mechanism is believed to be insulin resistance that derives from white adipose tissue, liver, and/or skeletal muscle, accompanied by impaired insulin secretion by pancreatic β-cells (6). Furthermore, both obesity and T2DM increase the risk of cardiovascular disease (CVD), increasing morbidity and mortality by greater than 2-fold (7–10). The distribution of adipose tissue is of great importance with regards to these co-morbidities. Insulin resistance often occurs when fat accumulates in intra-abdominal depots and is associated with a constellation of CVD risk factors, in what is known as the metabolic syndrome (11). Simply measuring body weight, waist circumference, or calculating BMI does not portray a clear picture of body composition nor fat distribution. Thus, other indices have become more useful for assessing body fat distribution, such as waist-to-hip ratios, as well as methods for assessing body composition, including anthropometry, dual-energy X-ray absorptiometry (DEXA), and computed tomography (CT) scanning. A clear picture of body fat distribution in obese subjects is critical for determining how susceptible they are or will be to developing diabetes and/or cardiovascular disease. In this comprehensive review, the complex and interrelated associations between obesity, diabetes, and CVD will be explored in greater detail.
Types of Adipose Tissue
Adipose tissue can be classified by morphology into white, brown, or beige subsets. In addition, white adipose tissue (WAT) can be broadly classified by location, largely defined as subcutaneous (located under the skin) and visceral/omental (located intra-abdominally, adjacent to internal organs). Adipose tissue is comprised of many different cell types, which coordinately secrete numerous cytokines, chemokines, and hormones. Approximately one third of the cells within adipose tissue are adipocytes, with the rest represented by fibroblasts, endothelial cells, macrophages, stromal cells, immune cells, and pre-adipocytes. In most lean, healthy individuals, WAT is confined to defined depots. But in certain conditions such as obesity and lipodystrophy, WAT mass can increase ectopically in areas that may influence the susceptibility to comorbidities such as diabetes and atherosclerosis. Such ectopic WAT areas are mostly located within the visceral cavity, and include intrahepatic (discussed in the section on Ectopic Fat below), epicardial (epiWAT, between the heart and the pericardium), perivascular (PVAT, surrounding major blood vessels), mesenteric fat (MWAT, contiguous with digestive organs in the viscera), omental fat (OWAT, an apron of fat that stretches over the intestines, liver, and stomach), and retroperitoneal fat (RWAT, surrounding the kidneys). The latter three depots (MWAT, OWAT, and RWAT) will be classified together herein as “visceral fat” (12). In addition to WAT depots, brown adipose tissue (BAT) represents a distinct type of adipose tissue that is characterized by its morphology and function, with concentrated mitochondria giving it a characteristic brown appearance. Beige fat represents a third new classification of adipose tissue, in which brown adipocytes appear within classical WAT depots. Each of these adipose depots will be discussed in more detail below.
Adipose Tissue Distribution
Subcutaneous Fat
Primarily localized to upper and lower body depots in humans, subcutaneous WAT is the most prominent WAT depot in lean, healthy subjects, making up ~80% of all adipose tissue (13). Thus, more than any other depot, subcutaneous WAT represents a physiological buffer for excess energy intake during times of limited energy expenditure. Subcutaneous WAT acts as a metabolic “sink” for excess lipid storage (14). When this storage capacity is exceeded, either due to an inability to generate sufficient new adipocytes (limited hyperplasia) or an inability to further expand existing adipocytes (limited hypertrophy), fat begins to accumulate ectopically in areas outside the subcutaneous WAT (see sections on Ectopic and Visceral Fat below). Additionally, subcutaneous WAT functions as an insulator to prevent heat loss, as a barrier against dermal infection, and as a protective cushion against physical external stress (15).
Subcutaneous WAT likely arises from adipocyte precursor cells that are distinct from adipocytes that arise ectopically, for example in visceral fat (16). Elegant work by Kahn et al. has demonstrated that pre-adipocytes isolated from mouse and human subcutaneous WAT expresses developmental genes that are present prior to the development of WAT in a pattern that is maintained throughout adulthood, suggesting a cell-autonomous function (16). Thus, WAT distribution has a strong heritable component (17).
The beneficial effects of subcutaneous WAT to glucose metabolism have been demonstrated in numerous ways. However, subcutaneous WAT can be further subdivided into “upper” and “lower” regions, located primarily in the trunk and gluteo-femoral regions, respectively. Upper subcutaneous WAT is often lumped together with visceral WAT, classified together as “abdominal fat.” The distinction between upper and lower subcutaneous WAT and how they contribute to metabolic health will be discussed in later sections.
Epicardial Fat
Epicardial adipocytes share embryonic origins with mesenteric and omental adipocytes (18). epiWAT (also termed pericardial WAT) is in close proximity to the myocardium, enabling a shared microcirculation between epiWAT and certain areas of the heart (19). Due to its proximity to the heart, epiWAT is thought to be approximately twice as metabolically active as other WAT depots, with higher levels of fatty acid uptake and fatty acid release due to lipolysis (20). As a metabolically active WAT depot, epiWAT secretes several adipokines and vasoactive substances such as adiponectin, resistin, vascular endothelial growth factor (VEGF), and inflammatory cytokines and chemokines that impact the adjacent myocardium (21). In fact, due to the complete lack of a fibrous fascial layer between epiWAT and the myocardium, diffusion of fatty acids and other bioactive hormones from epiWAT to myocytes and coronary vessels is easily facilitated (22). Most humans possess a small amount of epiWAT, which provides fatty acids through lipolysis of its triglyceride stores for energy use by the heart. However, obese humans possess an enlarged epiWAT depot, which is clinically related to features of the metabolic syndrome (discussed in later sections).
Perivascular Fat
Fat that surrounds blood vessels is termed perivascular fat (PVAT). It has now been recognized that PVAT has characteristics that resemble both BAT and WAT, and is considered to be an active participant in vascular homeostasis (23). PVAT produces many bioactive molecules that influence vascular reactivity, including adipokines (e.g., leptin, adiponectin, omentin, visfatin, resistin, and apelin), cytokines/chemokines [e.g., interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and monocyte chemotactic protein-1 (MCP-1)], and vasoactive molecules (e.g., nitric oxide, prostacyclin, and angiotensin II) (24). Thus, PVAT can directly contribute to vascular tone, in addition to playing a supportive role in maintaining vessel structure. It has been suggested that PVAT in the thoracic aorta resembles BAT, while PVAT in the abdominal aorta exhibits properties of both BAT and WAT (24). Thus, if PVAT becomes dysfunctional in the setting of obesity, it can pivot from providing an atheroprotective role to promoting atherosclerosis. This concept will be evaluated further in later sections.
Visceral Fat
Fat localized within the visceral compartment has been classified as omental, mesenteric, and retroperitoneal. Lean, healthy individuals do not have large amounts of visceral fat, which largely falls into the category of ectopic fat. Visceral fat is highly metabolically active and is constantly releasing free fatty acids (FFA) into the portal circulation. As such, visceral fat content contributes to various features of the metabolic syndrome, such as hyperinsulinemia, systemic inflammation, dyslipidemia, and atherosclerosis (25), to be discussed in more detail in later sections pertaining to obesity.
Brown Fat
BAT is localized to distinct anatomical regions that have been well-characterized in rodents (26). By taking up circulating fatty acids, BAT functions to generate heat by uncoupling chemical energy production (ATP) via oxidative phosphorylation into heat production (non-shivering thermogenesis), thereby contributing to the clearance of plasma triglycerides and the mitigation of ectopic lipid storage (27). While originally believed to be a depot exclusive to hibernating and small mammals, and present to some degree in human infants, adult humans have recently been shown to have functional and inducible levels of BAT that respond to cold and sympathetic nervous system activation (28–30). Such BAT represents between 1 and 2% of total fat stores in humans, and is localized primarily in the cervical, axillary, and paraspinal regions (26, 31, 32). Similarly to WAT, BAT synthesizes and secretes “batokines” such as fibroblast growth factors (FGFs) including FGF21, neuregulin 4, VEGF, and cytokines such as IL-6 (33). Given the relatively small amount of BAT present in humans, the endocrine potential of batokines is relatively unknown, but it is clear that factors secreted from BAT exert paracrine and autocrine functions. While the relative BAT mass in humans and rodents is small compared to other adipose depots, its relative contribution to metabolic health may be higher.
In rodents and other small mammals, the primary BAT depots are located in the interscapular space and supraclavicular regions, among many others (26, 34). With prolonged stimulation, i.e., cold exposure, the size and activity of these BAT depots will increase, a term called BAT recruitment. BAT recruitment is associated with enhanced proliferation and differentiation of BAT precursor cells.
Beige Fat
In addition to WAT and BAT, a third fat type has been described, termed “browned,” “beige,” or “brite” (brown-in-white) fat. As the name suggests, beige fat has been described as the presence of brown adipocytes within classic WAT depots. While beige fat shares some features of classical BAT such as systemic triglyceride-lowering, beige fat is thought to be physiologically distinct from BAT, with differential expression of certain genes involved in metabolism, inflammation, and transcription (35, 36). Moreover, human BAT exhibits similar morphology and function as both rodent BAT and beige tissue (30, 37–39), complicating comparisons between the two species. In rodents, subcutaneous WAT is the most susceptible depot to browning, while in humans it is visceral WAT (40). It is generally believed that the majority of WAT depots can develop browning under particular conditions, but more work is needed in this area. There is a growing list of physiological stressors that can promote the browning of WAT, including cold exposure, exercise, bariatric surgery, cancer cachexia, severe burns, as well as pharmacological and dietary components such as conjugated linoleic acid, short-chain fatty acids, capsaicin, non-caffeinated green tea extract, thiazolidinediones (TZDs), and β-adrenergic receptors (41–52).
There is some debate regarding the origins of beige adipocytes, as well as their impact on energy homeostasis. Beige adipocytes may arise from de novo adipogenesis from specific progenitor cells when initially stimulated by cold exposure (36, 53), but then may lie “dormant” until stimulated again (54). This theory suggests that dormant beige adipocytes can become quickly and readily activated when needed, reminiscent of an immune response. This newly defined relative flux between “dormant” and “active” beige cells may be what has been previously termed “transdifferentiation” of white-to-beige adipocytes (54). Beige adipocytes were initially thought to arise from transdifferentiation from white adipocytes, with the ability to de-differentiate back into white adipocytes (55, 56). Additional studies in vitro suggest that this is likely not the case (57). The identity of committed beige adipocyte precursors has not been fully elucidated, but there is evidence from isolated WAT stromal cells that beige adipocyte precursors are distinct from white adipocyte precursors (36, 39, 58). It has been suggested that strategies that increase the number of beige adipocytes in mouse WAT also protect them from diet-induced obesity (59–63).
Normal Adipose Tissue Function
White Adipose Tissue: Energy Storage and Distribution
Adipose tissue is an essential organ for the regulation of energy homeostasis. Primarily tasked with storing excess energy as triglycerides, adipocytes undergo hyperplasia to increase the number of adipocytes and hypertrophy to increase the size of each adipocyte, allowing adipose tissue to expand in times of nutrient excess. As needed, i.e., during fasting and exercise, triglycerides stored in adipose tissue are mobilized to provide fatty acids for energy utilization by the rest of the body. Stored triglycerides are therefore in a constant state of flux, whereby energy storage and energy mobilization are determined largely by hormonal fluctuations. Thus, adipose tissue functions as an energy balance “hub” that integrates and services the energy requirements of diverse organ systems, such as the liver, skeletal and heart muscle, pancreas, and brain (64).
In healthy lean individuals, the majority of adipose tissue resides in subcutaneous depots, where it serves a thermoregulatory function, and from which stored triglycerides can be readily mobilized when needed (65). Conditions that favor adipose tissue expansion, if endured chronically, will eventually exceed the storage capacity of defined adipose tissue depots, leading to the ectopic deposition of triglycerides in other tissues, including intra-abdominal depots (discussed in more detail in later sections).
Non-shivering Thermogenesis
BAT plays an important role in thermoregulation in mammals, including adult humans (66). BAT tissue is rich in mitochondria and uniquely expresses uncoupling protein-1 (UCP-1), which enables heat production by uncoupling ATP synthesis. BAT-mediated thermogenesis has garnered substantial attention recently, as increasing BAT mass or activity could be an effective strategy to combat obesity. While the primary function of WAT is to manage energy storage, brown adipocytes efficiently burn fatty acids released from WAT during adaptive thermogenesis (67). BAT plays an active role in metabolism in animals and humans (28); therefore, strategies that increase BAT mass and/or activity could promote fat loss in obese populations. In addition, beige fat could also contribute to fat catabolism, potentially reducing WAT stores. Human brown adipogenesis occurs in response to chronic or repeated cold stimulation, or in response to pharmacologic compounds such as beta adrenergic receptor (β-AR) agonists (68, 69). However, these browning-inducing methods mediated by the sympathetic nervous system are not practical as a weight loss strategy for several reasons: (1) the browning effects of cold exposure are rapidly reversible, (2) repeated cold exposure is too time- and energy-consuming to be a practical therapeutic, and (3) β-ARs promote adverse cardiometabolic events. Therefore, mechanisms of WAT browning that are long lasting and act independently from the sympathetic nervous system are highly sought after. A new mechanism of WAT browning that does not involve the sympathetic nervous system (SNS) has recently been described. Adipose tissue resident macrophages can secrete norepinephrine (NE), the neurotransmitter that is also secreted by sympathetic neurons to activate BAT and WAT browning (70). Several follow up studies have suggested that eosinophils, type 2 cytokines, and alternatively activated macrophages play critical roles in supporting WAT browning with concomitant increased energy expenditure and weight loss (71–79). However, the notion that immune cells can influence WAT browning has recently been challenged, using different murine and in vitro approaches (80). As such, there is some discordance regarding the role of macrophages in WAT browning, necessitating further studies.
Secretion of Hormones and Adipokines
Originally classified as a simple energy storage organ, adipose tissue is now known to function as a major endocrine system that secretes adipokines, growth factors, cytokines, and chemokines (81). The secretion pattern of adipokines appears to vary by adipose tissue depot and is dependent on the energy status of the adipose depot, leading to variable paracrine/autocrine effects of adipokines within particular depots. Adipokines are important mediators of various metabolic processes such as fatty acid oxidation, de-novo lipogenesis, gluconeogenesis, glucose uptake, insulin signaling, and energy expenditure in metabolically active tissues such as the liver, skeletal muscle, and brain (81). The various adipokines secreted from adipose tissue and their functions will be described in more detail below. The discussion will be limited to adipokines that are known to be produced to a large extent by adipocytes, in addition to other cell types within adipose tissue such as immune cells.
Leptin
Discovered in 1994, leptin is a peptide hormone that is expressed exclusively by adipocytes and is essential for body weight regulation. Leptin, adiponectin, and omentin (the latter two will be described below) are the only generally accepted adipokines with true endocrine function, meaning they are released from adipose tissue and exert effects on distant target organs. Leptin is encoded by the obesity gene (ob). Leptin-deficient (ob/ob) mice become spontaneously obese due to unrestricted food intake, highlighting the importance of this adipokine in suppressing appetite through the central nervous system (82). Rodents and humans that lack either leptin or the leptin receptor (LEPR) are not only extremely obese, but are also hyperglycemic and extremely insulin resistant (83). In lean and obese animals and humans, circulating leptin levels positively correlate with adiposity (84). Prolonged fasting is associated with a sharp drop in plasma leptin levels, which drives food intake (85). While leptin is expressed in all adipose depots, including BAT, its expression is highest in subcutaneous WAT (86).
Adiponectin
As one of the first adipokines discovered in the mid-1990s (87–90), adiponectin is a well-described insulin-sensitizing hormone that impacts a wide range of tissues. Adiponectin is a distinctly unique adipokine, as its expression and circulating levels are inversely proportional to adiposity levels, in stark contrast to leptin. Adiponectin expression levels vary between sexes, with higher levels observed in females than males (91–93), and between adipose tissue depots, with higher expression in subcutaneous than visceral WAT (94, 95). The insulin sensitivity-promoting properties of adiponectin are well-known, and are exemplified by the development of insulin resistance in adiponectin-deficient mice (96), and the preservation of insulin sensitivity in adiponectin-overexpressing mice (97). Adiponectin signals through two related receptors, ADIPOR1 and ADIPOR2, followed by docking of the adaptor protein APPL1 (98). The resulting signaling pathway, mediated through peroxisome proliferator-activated receptor alpha (PPARα), leads to metabolic improvements involving decreased hepatic gluconeogenesis, increased liver and skeletal muscle fatty acid oxidation, increased glucose uptake in skeletal muscle and WAT, and decreased WAT inflammation (99). Thus, adiponectin receptors are highly expressed in skeletal muscle, liver, and adipose tissue. In addition, adiponectin receptors are expressed in the pancreas, where adiponectin functions to mitigate β-cell loss by neutralizing inflammatory and lipotoxic ceramides and diacylglycerols (100). In addition to β-cells, adiponectin has also been shown to exhibit strong anti-inflammatory effects on other cell types such as macrophages and fibrogenic cells (99, 101, 102). Taken together, adiponectin plays a protective role in mitigating features of the metabolic syndrome.
Resistin
Resistin is a polypeptide that is secreted by obese adipose tissue. It was originally described as an adipocyte-specific hormone, but it is now thought to originate from macrophages residing in inflamed adipose tissue in mice (103) and from circulating monocytes and tissue macrophages in humans (104, 105). Human resistin is only 59% homologous to mouse resistin (106), which has raised some controversy over the pathogenic role of resistin, and limits comparisons between animal models and human disease (107). Resistin is so named due to its ability to “resist,” or interfere with insulin action (108), based on initial studies in mouse models. Evidence for this comes from an initial study in which it was observed that plasma resistin levels are elevated in a diet-induced obese mouse model, that blocking resistin action using a neutralizing antibody improves insulin sensitivity, and that recombinant resistin administration to healthy mice promotes insulin resistance (108). These initial studies led to the suggestion that resistin plays an important role in modulating insulin resistance in the context of obesity, and it has been shown to correlate with insulin resistance in mice and humans (109). Plasma resistin levels have been shown to be increased in obese animal models and humans (110–113) and to decrease with weight loss in humans (114). Conversely, some studies have shown that adipose tissue-derived resistin is suppressed in obesity (115–117), inciting the controversy over what role resistin plays in obesity that persists today. Evidence suggests that visceral fat is the largest contributor to circulating resistin levels (113), supporting the case for an association between resistin and insulin resistance. Moreover, resistin is believed to be an active participant in propagating inflammatory responses. Resistin can upregulate inflammatory cytokines such as TNFα and IL-6 in monocytes and macrophages in a nuclear factor kappa-B (NFκB)-dependent manner (118), and is positively associated with circulating inflammatory markers such as C-reactive protein (CRP) and TNFα (107). Thus, while resistin is an established adipokine and has been shown in some cases to be associated with adverse health conditions such as obesity and insulin resistance, a clear role for resistin is still under active investigation.
Omentin
Initially described as an adipokine secreted from omental WAT (119), it is now generally accepted that omentin is also expressed in other WAT depots such as epicardial fat, and that it derives specifically from the stromal vascular fraction of WAT (119, 120). Omentin is a true endocrine hormone that circulates in the blood (121, 122). Omentin levels are reduced in subjects with obesity (123) and T2DM (124, 125), leading investigators to speculate that omentin may be involved in glucose homeostasis. Indeed, studies using in vitro models showed that omentin enhances insulin-stimulated glucose uptake in human adipocytes by activating Akt signaling pathways (119), and studies in humans show a significant negative correlation between serum omentin levels as well as adipose omentin mRNA levels with insulin resistance (124, 126, 127). Omentin levels have been shown to gradually increase in response to weight loss (128, 129). Additional studies suggest that omentin has anti-inflammatory properties. Omentin blunts cytokine expression in endothelial cells (130), vascular smooth muscle cells (131, 132), macrophages (133), cardiomyocytes (134), and adipose tissue itself (135), and is negatively associated with systemic inflammatory markers such as TNF and IL-6 (136). Thus, omentin is considered to be a biomarker for metabolic health that may function to blunt obesity-related cytokine effects (137).
Fibroblast Growth Factor 21 (FGF21)
FGF21 is an endocrine hormone that is involved in the regulation of lipid, glucose, and energy homeostasis (138). FGF21 has received a lot of attention for its insulin-sensitizing and weight loss-inducing effects when administered pharmacologically (139). The liver is the primary source of circulating FGF21, induced by metabolically stressful conditions such as fasting, a ketogenic diet, protein restriction, and bariatric surgery (140), while the brain and adipose tissue are primary FGF21 targets (141, 142). Other tissues are known to also secrete FGF21, including the pancreas and skeletal muscle (143, 144). However, under certain metabolic conditions such as obesity, WAT and BAT may also produce FGF21 (145). This is supported by several studies showing that BMI and adiposity positively correlate with circulating FGF21 levels in mice and humans (145–149). It is clear that FGF21 levels become elevated as obesity develops in mice and humans, and are positively correlated with BMI, adiposity, and FGF21 expression levels in adipose tissue (145–149). While many studies have shown that adipose tissue expresses FGF21 in rodents (145, 150–154), there is still some debate about whether FGF21 is readily expressed in human adipose tissue. There are a handful of studies that suggest that adipose tissue FGF21 mRNA expression is below detection levels (155) or not expressed by adipose tissue (156). However, numerous additional studies have found detectable FGF21 mRNA expression in visceral WAT (157, 158), subcutaneous WAT (145, 157, 158), epicardial WAT (159), cervical adipose tissue (160, 161), and PVAT (162, 163), with the latter two depots containing both WAT and BAT. FGF21 protein has also been detected in adipose tissue by Western blot and immunohistochemistry (162). It is not clear why some but not all groups have been able to detect FGF21 expression in human adipose tissue, but could depend on the metabolic and/or nutritional status of the subjects sampled (e.g., whether subjects were fasting or fed).
Some studies suggest that adipose-derived FGF21 is a marker of metabolic stress, as it has been shown to correlate with features of the metabolic syndrome (145, 164, 165). Regardless, a clearly-defined function of adipose-derived FGF21 has not yet been established, nor whether adipose-derived FGF21 promotes primarily local effects or contributes to the circulating FGF21 pool under particular metabolic conditions. Elegant studies using tissue-specific Fgf21 KO mice show that adipocyte-derived Fgf21 is not involved in obesity-associated insulin resistance, and that adipose-derived Fgf21 doesn't circulate, instead acting in a paracrine fashion (140). However, the mice used in that study were fasted for 24 h, introducing a metabolic stress that would likely only induce liver-derived Fgf21 that may have masked any contribution from adipose-derived Fgf21. In later studies, a thermogenic role for adipose-derived Fgf21 has been described, in which the browning of WAT was shown to require adipocyte-Fgf21 (141, 166). Thus, it is possible that hepatic- and adipose-derived FGF21 are induced by different stimuli, and that more studies are required to conclusively define a role for adipose-derived FGF21.
Obesity
Obesity results when energy intake chronically exceeds energy expenditure. Many factors are involved, including genetic, epigenetic, hormonal, and lifestyle factors that are beyond the scope of this review. Adipocyte number is believed to be tightly regulated and determined during childhood (167). However, during the development of obesity, adipose tissue can expand by either hypertrophy (an increase in adipocyte size) or hyperplasia (an increase in adipocyte number due to the recruitment of new adipocytes). Obesity is characterized by dysfunctional adipose tissue, in which adipocytes initially become hypertrophic during periods of caloric excess and secrete adipokines that result in the recruitment of additional pre-adipocytes, which differentiate into mature adipocytes as compensatory protection against some of the adverse metabolic consequences of obesity (168). This concept is supported by observations in AdipoChaser mice, a model for tracking adipogenesis (169). AdipoChaser mice fed a high fat diet display evidence of hypertrophy of visceral WAT within 1 month, while hyperplasia occurs after 2 months. Importantly, subcutaneous WAT does not undergo hyperplasia, and hypertrophy lags behind the visceral compartment, with evidence of subcutaneous WAT hypertrophy after 2 months of high fat feeding (170). However, when the capacity for adipocyte recruitment and hypertrophy is overwhelmed, fat accumulates in ectopic sites such as visceral depots, the liver, skeletal muscle, and pancreatic beta cells. These changes are accompanied by inflammation, insulin resistance and other features of the metabolic syndrome, and have been termed metabolically unhealthy obesity (MUHO) (171, 172). In contrast to MUHO, some people accumulate fat mainly in subcutaneous depots, a condition that has been termed metabolically healthy obesity (MHO). MHO is not accompanied to any great extent by insulin resistance, adipose tissue and systemic inflammation, and other features of the metabolic syndrome such as dyslipidemia and hypertension (173–176). Thus, the distribution of fat accumulation is a major determinant of metabolic complications associated with obesity, which can increase the risk of CVD. Various features that contribute to dysfunctional WAT in obesity will be discussed in the sections that follow.
Metabolically Healthy Obesity (MHO)
A sub-group of obese individuals remain insulin-sensitive, and exhibit normal metabolic and hormonal profiles despite having a BMI that would characterize them as obese (177, 178). Such individuals have been classified as having “metabolically healthy obesity” (MHO), and appear to be distinct from those with “metabolically unhealthy obesity” (MUHO) in that they remain insulin sensitive and do not have much adipose tissue inflammation or other features of the metabolic syndrome (179, 180). Therefore, MHO individuals have a lower risk for developing T2DM and cardiovascular disease (174). MHO is sometimes defined as having 2 or less features of the metabolic syndrome or based on homeostatic model assessment of insulin resistance (HOMA-IR) measures, but consensus on a precise definition does not exist (176). Thus, some individuals classified as having MHO rather fall somewhere between metabolically healthy and unhealthy. Moreover, individuals with so-called MHO can progress to develop features of the metabolic syndrome with time (181–184). Because CVD outcomes in general relate to the number of metabolic abnormalities present in individuals with MUHO (185–188), there is less CVD in individuals with MHO than those with the metabolic syndrome. In addition, while MHO individuals are so defined due to a healthier cardiometabolic profile than those with MUHO, the true clinical benefits of MHO remain in question, as the cardiometabolic profile and insulin sensitivity of MHO individuals typically does not improve significantly with weight loss (179, 189–192). Nevertheless, evidence from animal models and cultured adipocytes do suggest that the preservation of the capacity for subcutaneous WAT expansion mitigates extensive visceral and hepatic fat accumulation, potentially driving the MHO phenotype (76, 97, 193).
Metabolically Unhealthy Obesity (MUHO)
Visceral Adiposity and the Metabolic Syndrome
Other obese individuals tend to accumulate fat mainly intra-abdominally in visceral depots, which is also known as central obesity. Visceral adiposity is associated with insulin resistance, a predisposition to diabetes, local and systemic inflammation, dyslipidemia [characterized by hypertriglyceridemia, a preponderance of small, dense low-density lipoprotein (LDL) particles and reduced high-density lipoprotein (HDL)-cholesterol levels], insulin resistance, dysglycemia [a broad term that refers to an abnormality in blood sugar stability], adipose tissue and systemic inflammation, hypertension, a thrombogenic profile and non-alcoholic fatty liver disease (NAFLD) (194). This constellation of CVD risk factors associated with visceral obesity is widely known as the metabolic syndrome and is a hallmark of MUHO, illustrated in Figure 1. Visceral obesity and the metabolic syndrome are associated with an increased risk of developing CVD, which is exacerbated when overt diabetes develops as a result of insulin secretion failing to adequately compensate for insulin resistance. Interestingly, even normal weight individuals who accumulate fat intra-abdominally have these metabolic abnormalities (195, 196), including an increased risk of CVD. Asians and Asian-Americans are particularly prone to accumulate intra-abdominal fat and have features of the metabolic syndrome despite having normal weights and BMI values by Western standards (196), raising the question of whether different normal values should apply to individuals of Asian ancestry. Moreover, this raises the question of the validity of body weight or body mass index (BMI -weight in kg/height in m2) as an index of obesity or adiposity, since these measures do not differentiate the 2 major types of obesity. Measures such as waist circumference, waist/hip ratio and weight to height ratio have been used. These indexes are notable for their inclusion of upper subcutaneous WAT, which some consider to contribute as much, if not more, to metabolic syndrome than visceral WAT alone (197). CT scanning at the level of the umbilicus has been found to be useful but is expensive and not practical other than for research purposes at present. Lower body subcutaneous WAT does not correlate with risk factors for the metabolic syndrome, potentially due to a slower FFA turnover, higher levels of adipocyte hyperplasia, and lower levels of inflammation (198–201).
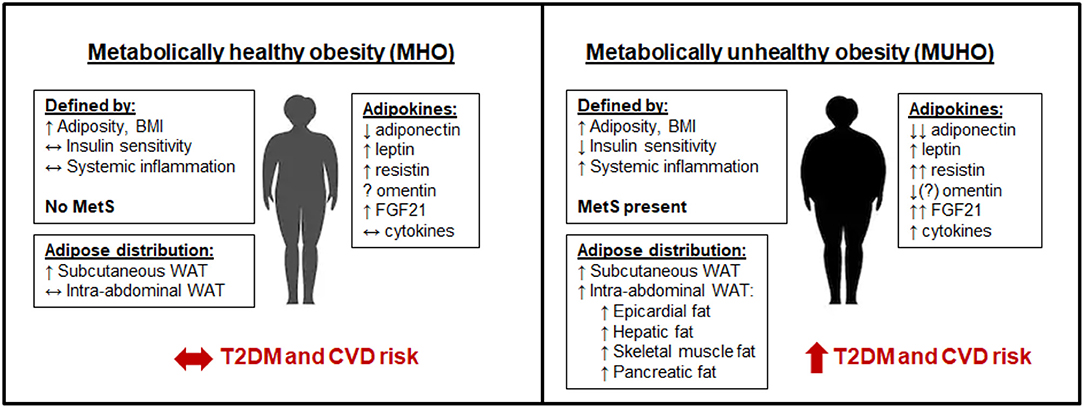
Figure 1. Metabolically healthy obesity (MHO) vs. metabolically unhealthy obesity (MUHO). In comparison with lean metabolically healthy subjects, those with MHO have increased adiposity and BMI, but with reduced systemic inflammation and retained insulin sensitivity, thus defining them as not having metabolic syndrome (MetS). MHO subjects have elevated subcutaneous white adipose tissue (WAT) levels, without excessive accumulation of visceral fat. Their adipokine profile is similar to lean subjects, but with increased leptin, resistin, and FGF21, and decreased adiponectin, which limits their risk of developing type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD) in the short term. By contrast, those with MUHO exhibit elevated insulin resistance and systemic inflammation in addition to increased adiposity and BMI over lean controls, contributing to MetS. MUHO individuals have excess subcutaneous and intra-abdominal adipose tissue, with increased hepatic fat and fat distributed amongst other visceral organs. This leads to a dysfunctional adipokine profile, characterized by reduced adiponectin and omentin, with further elevated leptin, resistin, FGF21, and cytokines when compared to lean controls. Thus, MUHO subjects are at risk for developing T2DM and CVD.
Notable differences in the adipokine profile between MHO and MUHO subjects have been reported, which could contribute to their respective risks for T2DM and CVD. Leptin has been shown to be higher in MUHO than MHO obese Chinese children in one study (202), but was not found to differ between adult groups in several other studies (203–205). By contrast, adiponectin has consistently been shown to be higher in subjects with MHO than in those with MUHO, despite both populations having lower adiponectin than metabolically healthy lean controls (203, 205–209). Resistin and FGF21 levels tend to be highest in the MUHO population (148, 208). Data on whether omentin levels differ between MHO and MUHO has been inconsistent, with one study suggesting that MUHO subjects have higher omentin levels than MHO subjects (210), and other suggesting the opposite, that omentin levels are negatively correlated with the metabolic syndrome (122, 211). Cytokines such as TNFα and IL-6 as well as the chemokines SAA and MCP-1 have been shown to be elevated in MUHO (208). These adipokine differences between subjects with MHO and MUHO are depicted in Figure 1.
White Adipose Tissue Inflammation
Macrophages and inflammation
Adipose tissue expansion in obesity is accompanied by inflammatory changes within adipose tissue, contributing to chronic low-grade systemic inflammation that is characterized as mildly elevated levels of circulating cytokines, chemokines, and acute phase reactants. In mice fed a high fat diet, obesity is associated with the induction of a large number of inflammatory pathways, constituting as many as 59% of total pathways that are differentially regulated (212). Expansion of adipose tissue depots during weight gain is accompanied by an infiltration of new inflammatory cells, the major one initially being macrophages. Reported to represent ~5–10% of total cells within lean adipose tissue, macrophages in obese adipose tissue represent up to 60% of all cells present (213). These pro-inflammatory cells are recruited in response to chemokines such as monocyte chemotactic protein-1 (MCP-1) produced by hypertrophic adipocytes (213, 214). Studies in mice have demonstrated that most macrophages in obese adipose tissue are derived from circulating monocytes (213), although a small percentage appear to derive from proliferation of resident tissue macrophages (215). Resident macrophages that are present in normal adipose tissue express markers of “alternatively activated,” or M2 macrophages such as the mannose receptor (CD206), macrophage galactose type C-type Lectin/CD301a/CLEC10A (MGL1), and arginase-1 (ARG1). These anti-inflammatory macrophages are believed to be responsible for maintaining tissue homeostasis (216). It remains unclear whether the derivation of adipose tissue macrophages is the same in human obesity.
Macrophage accumulation occurs to a greater extent in visceral than in subcutaneous adipose depots in both rodents and humans (217–220). Macrophages are seen in crown-like clusters, where they are thought to represent an immune response to dead and dying adipocytes (219). These recruited macrophages demonstrate a phenotypic switch from being anti- to pro-inflammatory, and develop some features similar to “classically activated,” or “metabolically-activated” macrophages (MMe) (221–223). However, use of genetic markers show that these cells have significant differences from classical M1 macrophages and alternate nomenclatures have been suggested for these pro-inflammatory cells. Morris and Lumeng have divided adipose tissue macrophages into several populations based on cell surface markers and expression profiling (224). Using a proteomics approach, Kratz et al. showed that markers of classical activation were absent on ATMs from obese humans. Stimulation of macrophages with glucose, insulin, and palmitate resulted in the production of a “metabolically activated” MMe phenotype distinct from classical activation. Such markers of metabolic activation were expressed by pro-inflammatory macrophages in adipose tissue from obese humans and mice and correlated with the extent of adiposity (225).
Other immune cells
In addition to macrophages, T-cells also are present in normal adipose tissue and demonstrate phenotypic change during weight gain. Both CD4+ and CD8+ T cells are found in adipose tissue and are increased in the obese state. Th2 cytokines (e.g., IL-4 and IL-13) are responsible for generating “alternatively activated” (M2) macrophages in lean adipose tissue. With weight gain in mice there is a shift away from a predominance of TH2 T cells present in lean adipose tissue and toward more TH1 and cytotoxic T cells as well as a reduction in regulatory T cells (Tregs) (226). Interferon γ (IFNγ)–expressing Th1 polarized T cells appear to promote adipose tissue inflammation and increased IFN-γ activity has been reported in adipose tissue in both mice and humans (227, 228). T-cell activation involves peptide antigen presentation via major histocompatibility complex (MHC) class II (CD4+) or MHC class I (CD8+). A subset of T cells called natural killer T (NKT) cells respond to lipid or glycolipid antigens (229–231). The number of invariant NKT (iNKT) numbers has been observed to be reduced in adipose tissue and livers from obese mice and humans (232–235). B-cells and mast cells also are increased in adipose tissue in the obese state (227, 236, 237). Use of specific cell surface markers has also demonstrated the presence of dendritic cells in adipose tissue, and studies indicate that dendritic cells are independent contributors to adipose tissue inflammation during obesity (238, 239).
Chronic inflammation in obesity
Adipose tissue inflammation in obesity differs from typical inflammatory responses employed in host defense in that it is chronic, sterile, low grade, and affects the metabolic control of nutrient flow in adipose tissue, liver, muscle and pancreas, and has been termed “meta-inflammation.” One way it affects nutrient flow is by causing insulin resistance. There is good evidence to support the notion that the systemic inflammation that is associated with obesity and contributes to insulin resistance begins with adipose tissue inflammation. The regulation of hepatic C-reactive protein (CRP) and serum amyloid A (SAA) is likely in response to IL-6 secretion from visceral adipose tissue that directly targets the liver via the portal circulation (240–244). CRP is a prominent biomarker for insulin resistance and CVD (245–247), and SAA antagonizes insulin action in adipocytes, thus contributing to systemic insulin resistance (248). SAA also has been associated with CVD in some rodent and human models (218, 249–253). In summary, the discovery of elevated secretion of inflammatory cytokines by obese adipose tissue provides evidence that obesity directly mediates systemic inflammation, which contributes to insulin resistance and CVD (discussed further in later sections).
Cytokines and Chemokines
Obesity is associated with elevated circulating levels of IL-6 and TNFα, which are subsequently decreased with weight loss (254, 255). Adipose tissue is a major source of these cytokines (256) as well as the chemokine MCP-1, which is important for recruitment of inflammatory cells such as macrophages to expanding adipose tissue (257). While such inflammatory mediators that originate from adipose tissue could technically be classified as adipokines, they are also produced by the majority of cell types in the body and will therefore be described in further detail in this section. It should be noted that cytokine and chemokine production is limited in lean adipose tissue and in subjects with MHO. Many cell types synthesize and secrete these cytokines and chemokines, including several that make up the adipose tissue milieu such as monocytes, macrophages, dendritic cells, B cells, and T cells. As such, they play a prominent role in adipose tissue pathophysiology associated with obesity.
IL-6
Much research has been devoted to the role that adipose-derived IL-6 plays in the etiology of obesity. The expansion of adipose tissue is accompanied by excessive adipocyte lipolysis and subsequently elevated FFA levels, which promotes adipocyte IL-6 secretion (258, 259). Omental fat produces 2 to 3-fold higher levels of IL-6 than subcutaneous fat (260), providing a potential mechanism for the higher contribution of omental WAT to insulin resistance (261). Most studies in vitro and in mice suggest that adipose-derived IL-6 promotes hepatic insulin resistance and glucose intolerance (259, 262, 263), while some indicate that in certain contexts IL-6 signaling in WAT and liver may be protective against metabolic disease (264, 265). For example, mice with genetic disruption of the IL-6 receptor specifically in the liver exhibit exacerbated hepatic inflammation and impaired glucose tolerance (264), suggesting that IL-6 may also function to limit hepatic inflammation. Thus, the context in which IL-6 signaling is studied is critically important for the interpretation of its function.
TNFα
In addition to its secretion from inflammatory cells such as monocytes and macrophages, TNFα was first described as an adipokine in 1993 (266). As with IL-6, TNFα levels positively correlate with adiposity, BMI, insulin levels, and insulin resistance (267, 268). While adipocytes themselves can secrete TNFα, the majority of TNFα secreted from adipose tissue is derived from immune cells in the stromal vascular fraction, and that obesity-associated increases in TNFα largely reflect the infiltration of pro-inflammatory macrophages within expending adipose tissue (213). One mechanism by which adipose-derived TNFα may promote insulin resistance is by directly activating hormone sensitive lipase (HSL), thereby increasing FFA release from adipocytes which promotes insulin resistance in the liver and skeletal muscle (269). Another mechanism is via autocrine activation of insulin receptor substrate-1 (IRS-1), which prevents insulin from interacting with its receptor (270).
MCP-1
Monocyte chemotactic protein-1 (MCP-1) is a potent chemotactic factor that promotes monocyte and macrophage recruitment into sites of inflammation during tissue injury and infection. It is secreted by adipocytes during the development of obesity and leads to infiltration of monocytes, which differentiate to become adipose tissue macrophages. The macrophages in turn secrete additional MCP-1 leading to further recruitment of inflammatory cells (271, 272). Body mass index and adiposity strongly correlate with adipose CCL2 (the gene encoding MCP-1) expression levels, and MCP-1 decreases following weight loss in humans (273). Ob/ob mice, a commonly used mouse strain that spontaneously develops obesity due to leptin deficiency-induced hyperphasia, as well as diet-induced obese mice, display elevated levels of plasma Mcp-1 and Ccl2 adipose tissue expression (213, 274, 275). In addition, mice engineered to express elevated levels of Ccl2 specifically from adipocytes exhibit increased macrophage recruitment into adipose tissue, and subsequently increased insulin resistance, effects that were not observed in diet-induced obese mice that were deficient in Ccl2 (274). Potential mechanisms by which adipose-derived MCP-1 could increase insulin resistance include changes in liver mRNA expression of genes involved in lipid and glucose metabolism in response to elevated FFA (274), or more likely due to increased recruitment of macrophages into adipose tissue (described in the section on “Obesity and insulin resistance”). Evidence suggests that human visceral WAT secretes higher levels of MCP-1 than subcutaneous WAT (276). These studies and others have prompted the suggestion that MCP-1 could be a viable therapeutic target for the treatment of obesity and associated insulin resistance.
Serum amyloid A (SAA)
While well-described as an acute phase protein secreted by the liver in response to pro-inflammatory cytokines, SAA is also expressed in adipocytes and macrophages and correlates with adiposity (244, 277–281). There are 4 subtypes of SAA: SAA1–4. SAA1 and SAA2 are highly upregulated in response to inflammation, while SAA4 is largely constitutively expressed. SAA3 is a pseudogene in humans, replaced by SAA1 and SAA2 in extra-hepatic tissues. While the best defined cell source of SAA1 and SAA2 is hepatocytes, SAA1 and SAA2 are also expressed from adipocytes and macrophages under inflammatory conditions in metabolic diseases such as obesity, insulin resistance, and cardiovascular disease (250). SAA3 expression is increased during hypertrophy of cultured mouse adipocytes (214) and in gonadal fat in obese mice (218, 282). Inducible forms of SAA also are expressed in both subcutaneous (277) and omental WAT (283) from obese humans. Thus, the increased adipocyte size and number that accompanies obesity is also associated with elevated adipose tissue-derived SAA levels, likely in part due to increased hepatic secretion in response to cytokines produced in adipose tissue.
Ectopic Fat
In obesity, white adipose tissue may become dysfunctional and unable to properly expand to store excess ingested energy, triggering storage of triglycerides in sites where the primary function is not fat storage. Ectopic fat that is localized to major glucose regulatory organs such as the liver, skeletal muscle, and pancreas is commonly regarded as being “lipotoxic,” since this ectopic fat can interfere with normal insulin signaling and promote insulin resistance and increase the risk for T2D (284, 285). Excessive amounts of visceral fat also is considered to be a form of ectopic fat, and as noted earlier, is associated with features of the metabolic syndrome and an increased risk of T2DM and cardiovascular complications (286). In animal models as well as in humans, it has been shown that the accumulation of lipotoxic diacylglycerols (DAGs) and ceramide, as occurs with visceral obesity, leads to impaired insulin signaling and reduced glucose uptake in skeletal muscle and liver (287–290). More specific mechanisms by which ectopic fat accumulation in particular tissues promotes insulin resistance will be explained in the following sections.
Hepatic lipid accumulation and inflammation
Several studies have reported an inverse relationship between hepatic lipid content and whole-body insulin sensitivity (291–293). The liver is a major target for the excessively produced inflammatory cytokines and FFAs released from obese WAT (294) (see later). It has been estimated that nearly 60% of ectopic hepatic triglycerides in obese NAFLD patients derive from FFA released from adipose tissue (295). FFA-derived triglycerides accumulate in the cytoplasm of hepatocytes in the form of lipid droplets. While the lipid droplets may not be lipotoxic per se, various intermediate lipid moieties generated during triglyceride synthesis (e.g., DAGs and ceramide) have been shown to promote lipotoxicity and enhance hepatic insulin resistance (296), likely by inhibiting insulin signaling pathways (297, 298). Selective upregulation of ceramide degradation pathways in the liver has been shown to reverse hepatic lipid accumulation and improve glucose tolerance in diet-induced obese mice (299). Moreover, obesity-associated reductions in adiponectin have also been shown to contribute to hepatic steatosis, presumably by blunting hepatic fatty acid oxidation, a process regulated by adiponectin (300–302).
It also has been suggested that adipose tissue inflammation contributes to hepatic lipid accumulation. Kanda et al. showed that overexpressing Ccl2 from adipocytes in mice led to macrophage accumulation in adipose tissue and subsequent hepatic steatosis and hepatic insulin resistance, without an obese phenotype (274). Similarly, mice in which Ccl2 had been deleted showed resistance to high fat diet-induced insulin resistance and hepatic steatosis, an effect that was accompanied by reduced expression of TNFα in adipose tissue (274). Additional evidence to support the notion that adipose tissue inflammation promotes hepatic steatosis derives from studies showing that adipose-derived cytokines promote lipolysis of WAT stores (303, 304), thus increasing circulating FFA levels.
Kupffer cells are liver-resident macrophages, and reportedly comprise 80–90% of all tissue-resident macrophages in the body (305). In the healthy liver, the role of Kupffer cells is to phagocytose pathogens and toxins and to maintain tissue homeostasis and repair, akin to an M2 macrophage (306, 307). In contrast with adipose tissue macrophages, hepatic Kupffer cell numbers do not increase with adiposity, but instead become “activated,” akin to M1 or MMe macrophages (308). The primary stimuli for Kupffer cell activation likely derive from dysfunctional adipose tissue, including FFA, cytokines, and adipokines (309). Adipokine imbalance such as the hypoadiponectinemia that results from visceral adipose tissue expansion fails to suppress hepatic inflammation and oxidative stress, contributing to Kupffer cell activation. Thus, signals from dysfunctional obese adipose tissue propagate hepatic inflammation by activating resident Kupffer cells, which then themselves secrete pro-inflammatory cytokines, further amplifying systemic inflammation (310).
Ectopic fat in skeletal muscle
Lipids also can be stored within skeletal muscle when the capacity for fat storage by WAT is exceeded (311). Lipids can be stored either between muscle fibers (as adipocytes, or extramyocellular lipids), or within muscle cells (cytosolic triglycerides, or intramyocellular lipids) (312). Pre-adipocytes have been identified within skeletal muscle, providing evidence that distinct adipocyte cells may reside between skeletal muscle fibers (313). There is an association between ectopic skeletal muscle fat and insulin resistance that is largely dependent on BMI, but this association persists when BMI is statistically accounted for (314–316). It remains to be determined whether skeletal muscle fat is simply a marker of metabolic dysfunction or if it plays an active role in mediating insulin resistance. Ectopic skeletal muscle fat, as with ectopic fat in other areas, has the potential to impair insulin action in skeletal muscle through the inhibition of insulin signaling by lipotoxic DAGs and ceramide (317, 318). Several large clinical trials including SECRET and CARDIA have recently suggested that skeletal muscle fat could play a direct role in increasing cardiometabolic risk (319–322). However, while ectopic fat in skeletal muscles is often associated with metabolic disease, highly trained athletes have been reported to have comparable amounts of skeletal muscle fat as subjects with T2DM, yet their tissue remains highly insulin sensitive (323). This phenomenon has been called “the athlete's paradox” and is likely due to the high energy demands of skeletal muscle in extremely fit athletes.
Ectopic fat in the heart
Obesity and T2DM are both independently associated with fat accumulation in the heart (324), rendering ectopic fat in the heart as a strong predictor of CVD (325, 326), particularly in subjects with T2DM (327). Similar to the liver, excess circulating FFA can also lead to increased triglyceride deposition in the heart. Cardiac tissue mainly utilizes FFA for metabolism, but when delivered in excess of basal myocardial fatty acid oxidation rates can also lead to the accumulation of lipotoxic products (328). In addition to ectopic cardiac myocyte lipid storage, excess FFA can be stored in epiWAT, pericardial fat (between the visceral and parietal pericardia), or PVAT (329). PVAT in particular has a major impact on vascular homeostasis. As a source of several vasoactive mediators, PVAT influences vascular contractility. Healthy PVAT is thought to be a largely anti-inflammatory tissue (330), with characteristics akin to BAT in the areas surrounding the thoracic aorta in particular (331). However, in the setting of obesity, dysfunctional PVAT releases predominantly vasoconstrictive and proinflammatory mediators that negatively influence vascular homeostasis (332–334). Similarly, epiWAT is a source of bioactive molecules that negatively impact cardiac rhythm and perpetuate an atherogenic environment in obesity (335). Patients with T2DM express higher levels of the LDL and very low-density lipoprotein (VLDL) receptors in epiWAT than non-diabetic control subjects (336), suggesting that altered lipid metabolism in epiWAT could be associated with T2DM.
Ectopic fat in the pancreas
Mounting evidence suggests that excessive fat in the pancreas is associated with an increased risk of metabolic disorders, with reports that nearly 2/3 of the obese population has excessive pancreatic fat (337). Recent studies have connected ectopic pancreatic fat with β-cell dysfunction and T2DM (338–340), which in turn is associated with an increased risk of CVD. Therefore, lipotoxic lipid intermediates may also play a role in increasing the risk of CVD by elevating levels of pancreatic fat, thus leading to T2DM (341). In contrast to skeletal muscle, ectopic pancreatic fat is characterized mostly by adipocyte infiltration rather than intracellular lipid accumulation (342). The accumulation of fat in the pancreas also has been reported to accelerate acute pancreatitis due to increased levels of lipolysis and inflammation (343, 344).
Brown and Beige Adipose Tissue, Inflammation, and the Metabolic Syndrome
Compared with healthy lean controls, obese subjects display reduced BAT content, identified as tissue that actively takes up 2-[18F]fluoro-2-deoxyglucose (FDG) (345). This reduction in active BAT mass appears to be more prevalent in visceral obesity (346, 347). Concurrently, individuals with detectable BAT activity display lower blood glucose, triglyceride and FFA levels, lower glycated hemoglobin (Hb1Ac) levels, and higher HDL cholesterol levels than people with no detectable BAT (348, 349). As discussed in other sections, BAT acts as an important “sink” for excess blood glucose and FFA disposal. Thus, loss of BAT function in association with obesity could contribute to the development of insulin resistance and hyperlipidemia. It has been shown that while cold exposure can activate BAT to a certain degree in obese subjects and those with T2DM, the levels of BAT activation achieved are substantially lower than in healthy lean subjects (350, 351). While BAT is largely resistant to the development of mild obesity-induced local inflammation, BAT inflammation becomes quite pronounced with stronger obesogenic insults (352). Such inflammation can directly upset the thermogenic potential of BAT by impairing its ability to take up glucose (described in more detail in later sections) (353, 354). Whether individuals who inherently possess less active BAT are more prone to obesity and facets of the metabolic syndrome or whether these pathological conditions themselves reduce BAT activity requires further investigation. Regardless, it is still widely believed that strategies that augment BAT or beige activity could represent viable therapeutics to combat metabolic syndrome (355, 356). Efforts to enhance BAT activation in humans consist of intermittent regular cold exposure, introduction of β3-adrenergic receptor agonists, and exercise (29, 357). However, robust reductions in body weight in humans have not yet been shown to be clinically significant when BAT is activated (358), necessitating further mechanistic studies to elucidate whether BAT activation is a viable target for metabolic improvement in humans.
Whether BAT undergoes similar immune cell changes as WAT under obesogenic conditions is still not clear. In one study, BAT isolated from mice made obese by 13 weeks of high fat diet feeding displayed lower mRNA expression of inflammatory genes, lower immunostaining for macrophage markers F4/80 and CD68, and lower macrophage content by FACS analysis (331). However, subsequent studies have shown that BAT becomes inflamed in obese mice, with increased mRNA expression levels of inflammatory markers Tnf and Emr1 (the gene that encodes the macrophage marker F4/80) (359–361). Such BAT inflammation reportedly lowers the thermogenic potential of this tissue (359), presumably due to increased local insulin resistance (360, 362), which could reduce the glucose and fatty acid oxidizing capacity of BAT.
Similar to BAT, beige adipocyte quantity and functionality appear to be sensitive to local inflammation. A study in which IkB kinase (IKK, an enzyme that is required for NFκB activation and subsequent inflammatory cytokine transcription) was inactivated in mice, not only blunted adipose tissue inflammation and body weight gain, but enhanced WAT browning (363). Similarly, inhibiting a major intracellular mediator of toll-like receptor 4 (TLR4) signaling, interferon regulatory factor 3 (IRF3), blunted WAT inflammation and augmented WAT browning (364). Moreover, it has been shown that the immune cell infiltration of subcutaneous WAT that accompanies obesity directly interferes with the differentiation and/or recruitment of beige adipocytes (365). Thus, accumulating evidence suggests that obesity-associated inflammation hinders the thermogenic and insulin sensitizing effects of both BAT and beige adipocytes.
Obesity and Insulin Resistance
Abundant evidence indicates that adiposity and adipose tissue inflammation are associated with insulin resistance, which refers to a reduced response to binding of insulin to its receptor in peripheral tissues such as adipose tissue and skeletal muscle. This differs from glucose effectiveness, which is uptake of glucose by peripheral tissues in an insulin-independent manner. Insulin inhibits hepatic glucose output and stimulates lipogenesis in the liver, both of which are reduced in the presence of insulin resistance. Such desensitization of insulin signaling pathways also inhibits glucose uptake in peripheral tissues and stimulates lipolysis in adipose tissue. To compensate for reduced insulin sensitivity, insulin secretion is increased in order to maintain euglycemia. If the pancreatic beta cells are unable to secrete sufficient insulin to compensate for the reduced insulin sensitivity (termed beta cell dysfunction), hyperglycemia will ensue, leading to glucose intolerance and eventually T2DM (366). While the precise mechanisms that lead to beta cell dysfunction are not completely understood, ectopic fat accumulation may contribute, as discussed earlier. Nonetheless, ample evidence suggests that excess adiposity and adipose tissue inflammation contribute to insulin resistance [reviewed in (64, 367)]. Many studies have demonstrated that excess adiposity is correlated with insulin resistance in humans. Cross-sectional studies in men of European, Asian Indian, and American descent have shown that total, visceral, and subcutaneous adiposity, BMI, and waist circumference are all negatively associated with insulin sensitivity (368, 369). As noted earlier, adiposity, especially visceral adiposity, is characterized by adipose tissue inflammation.
Several hypotheses have been put forth to account for the relationship between adipose tissue inflammation and insulin resistance. These include production of pro-inflammatory cytokines by adipocytes and adipose tissue macrophages (discussed previously in the section on WAT Inflammation), excess FFA, decreased adiponectin, increased resistin and retinol binding protein, ceramide accumulation, and ectopic fat accumulation in liver and skeletal muscle (367).
Free Fatty Acids
It was initially hypothesized that excess adiposity promoted insulin resistance due to the accelerated release of FFA by obese adipocytes, which inhibit insulin signaling in liver and muscle due to excessive lipotoxicity and/or ectopic fat storage in these tissues (64), and also contribute directly to beta cell dysfunction (366). It has been shown that adipose tissue mass correlates with circulating FFA in obese humans, with a tendency for individuals with visceral adiposity to have higher FFA turnover (370–372). It has also been reported that individuals with T2DM tend to have elevated FFA levels over non-diabetic controls (373), an effect found to correlate more strongly with insulin sensitivity rather than obesity (374). Consistent with this, one study reported that FFA levels were lower in MHO subjects than those with MUHO (375). In addition to dysregulated energy metabolism, disruption of the endocrine function of obese adipose tissue has now been shown to contribute to insulin resistance, described in more detail below.
Adipokines
Adipocytes in obesity simultaneously secrete lower levels of adiponectin and elevated levels of cytokines and chemokines, such as TNFα, IL-6, MCP-1, and SAA. Not only is there evidence that such inflammatory cytokines contribute directly to insulin resistance in hepatocytes and myocytes (366), they also directly inhibit adiponectin production from adipocytes (376).
There is evidence that hypoadiponectinemia plays a role in obesity-associated T2DM (377–380). Subjects with T2DM exhibit reduced circulating adiponectin levels (379, 380); similarly, MHO subjects have higher circulating adiponectin than those with MUHO (206). Obese mice that are deficient in leptin (LepOb/Ob mice) that are engineered to overexpress adiponectin are protected from obesity-associated insulin resistance, despite having elevated adiposity (97). This may be explained by the nature of adipose tissue expansion in these transgenic mice, which had smaller, less inflamed adipocytes and less liver fat content. Similarly, administration of recombinant adiponectin improved glucose tolerance and insulin sensitivity in obese high fat diet-fed or Leprdb/db mice (377).
As discussed in earlier sections, FGF21 is a hormone produced by the liver as well as adipocytes that exerts insulin-sensitizing effects. However, recent evidence has paradoxically suggested an association between serum FGF21 levels and obesity-associated metabolic syndrome (145, 381). FGF21 levels have been reported to be 2-fold higher in MUHO when compared to MHO (148). Moreover, subjects with T2DM were reported to have significantly higher plasma levels of FGF21 than insulin-sensitive controls, with FGF21 levels positively correlated with BMI, HOMA-IR, and Matsuda index, suggesting a strong correlation with insulin resistance (157). Plasma FGF21 levels also correlated strongly with visceral, epicardial, hepatic, and skeletal muscle ectopic fat levels, measured using 64-slice multidetector CT scanning (157). Given that FGF21 has been shown to improve insulin sensitivity and promote negative energy balance (382, 383), some have suggested that obesity and associated metabolic syndrome represent an “FGF21-resistant” state (146). This conclusion was reached based on some observations that circulating FGF21 levels are increased in obesity, with lower FGF21 receptor expression levels on target tissues such as adipose tissue (146, 384). However, this notion has been challenged by evidence that obese subjects are equally responsive to pharmacological administration of FGF21 (384, 385). Thus, it has now been proposed that obesity-associated FGF21 is increased as a compensatory mechanism to preserve insulin sensitivity (386). As such, a clear role for adipocyte-derived FGF21 in obesity and associated metabolic syndrome is still lacking.
Adipose Tissue Plasticity
Evidence suggests that ineffective adipose expansion promotes local inflammation and an insulin resistant phenotype (387). However, sufficient adipogenesis and hyperplasia (i.e., the ability to distribute fat among newly differentiated adipocytes without the need for significant hypertrophy) mitigates such inflammation and subsequent insulin resistance (388). Thus, strategies to increase the recruitment of adipocyte progenitor cells to expand adipose tissue by increasing adipose cell numbers could be protective against the metabolic consequences of obesity.
A key structural and functional component of adipose tissue is made up of extracellular matrix (ECM) molecules, including collagen and proteoglycans such as versican and biglycan, among others (389). Adipose tissue makes large quantities of ECM during active remodeling, as would occur during WAT expansion in obesity (390–392). Obese animal models and humans with obesity and/or T2DM exhibit large increases in visceral WAT ECM content, which can contribute to the local inflammatory milieu (393, 394). To date, most studies of WAT ECM function have centered around collagen, which can form a scaffold that constrains adipocyte expansion due to mechanical stress (391, 392, 395). Targeting ECM components to release adipocytes from such constraints due to excessive ECM production could potentially alleviate the ectopic accumulation of fat that drives the metabolic syndrome.
Visceral Adipose Tissue vs. Hepatic Lipid Accumulation?
While the majority of adipose tissue in humans is localized subcutaneously (396), the volume of visceral adipose tissue is believed to be a strong predictor of insulin resistance (397), independent from subcutaneous fat quantity (397, 398). The association between insulin resistance and visceral adipose mass is particularly striking in certain ethnic populations, with T2DM rates of 46.6% in Filipino, 14.7% rates in African American, and 9.8% rates in Caucasian populations (398), suggesting a strong genetic component. While visceral adiposity is positively associated with insulin resistance, there is evidence to suggest that it may not be a causal factor. Other conditions associated with visceral adiposity, such as hepatic fat content, may instead drive insulin resistance (292, 399). Some clinical studies have dissociated the glucose metabolic effects of visceral adiposity from hepatic lipid accumulation. In one such study, significant differences in insulin sensitivity in the liver, skeletal muscle, and adipose tissue were reported in obese human subjects who differed in hepatic lipid content, with no such differences observed in obese subjects who differed in visceral adiposity (291). Similarly, in a study in which obese subjects were matched for liver fat content, no differences in indices of glucose metabolism were noted (293). Insulin-sensitive MHO individuals tend to have lower visceral and intrahepatic fat accumulation than their MUHO counterparts (203, 400, 401), providing further evidence that these fat depots contribute to insulin resistance. Collectively, while visceral adiposity and hepatic fat content are both strongly associated with whole-body and tissue-specific insulin resistance, hepatic lipid accumulation may play a more direct role in negatively modulating glucose homeostasis.
Subcutaneous Adipose Tissue
Many studies have suggested that fat distribution is strongly associated with insulin resistance, with visceral adiposity being the strongest predictor of insulin resistance (198, 402, 403). While the detrimental effects of visceral and hepatic lipid accumulation on glucose metabolism are clear, it is also becoming increasingly appreciated that lower body subcutaneous adiposity may be metabolically protective (404–406). Large-volume liposuction of subcutaneous WAT has shown little to no metabolic benefit in human trials (407). Gluteofemoral adipose mass is positively associated with insulin sensitivity in humans, coupled with a slower rate of lipolysis and subsequent FFA release, lower levels of inflammatory cells and cytokines, and elevated adipokines such as leptin and adiponectin (404). Evidence from animal models has suggested that transplantation of subcutaneous WAT into the visceral cavity of recipient mice promotes less body weight and adiposity gain than transplantation with visceral WAT, resulting in greater insulin sensitivity in the liver and endogenous WAT (408). Taken together, a growing body of evidence suggests that adipose tissue and ectopic lipid distribution contribute to whole-body glucose homeostasis.
Brown and Beige Adipose Tissue
With the purported potential to improve glucose homeostasis, interest in BAT and beige adipose tissue as therapeutic targets has increased in recent years. Studies in rodents in which BAT is transplanted into diseased mouse models have shown that transplanted BAT improves insulin sensitivity, glucose metabolism, and obesity (409–411), likely mediated by batokine effects. While the predominant energy source that contributes to brown adipocyte heat production derives from fatty acids (412) (~90%), with only ~10% of energy derived from glucose, BAT is still regarded as having a strong impact on glucose homeostasis. As a highly metabolically active organ, BAT contributes to glucose clearance by taking up relatively large amounts of glucose from the circulation, thus reducing insulin secretion by pancreatic β-cells (413). Indeed, individuals that possess detectable BAT have lower fasting glucose concentrations than those without active BAT (414). Glucose disposal through activated BAT occurs by both insulin-dependent and insulin-independent mechanisms (415). For example, the cold exposure-mediated influx of glucose into active BAT has been suggested to be an insulin-independent process (416–418). However, as the insulin receptor is highly expressed in BAT tissue, it is considered to be one of the most sensitive insulin target tissues and thus an important organ for glucose disposal (413). BAT activation further enhances insulin signaling in BAT itself by augmenting insulin-independent glucose uptake associated with thermogenesis and glucose uptake due to insulin signaling. Thus, strategies that activate BAT and beige adipose tissue have the capacity to improve insulin resistance by clearing excess glucose (419–421).
Links Between Obesity, Insulin Resistance, and CVD
Obesity as a Risk Factor for CVD
Several pathologic conditions, including hypercholesterolemia and systemic inflammation, are hypothesized to drive atherosclerotic CVD. With a primary function of sequestering lipotoxic lipids and the known potential for chronic inflammation, obese adipose tissue has emerged as a potential player in the regulation of these atherogenic factors. Obesity has been officially classified as an independent risk factor for CVD by the American Heart Association since 1995, meaning that obesity treatment is likely to lower the incidence of CVD (422). As alluded to in previous sections, people with MHO are at a lower risk of experiencing cardiovascular events than people with MUHO (423), yet those without obesity are at a considerably lower risk for future events. Thus, even a moderate level of weight loss, if sustainable, could potentially lower the risk of adverse CVD events (120). However, some studies have shown that individuals with established CVD and heart failure with moderate degrees of obesity present a more favorable prognosis than those who are normal or underweight, a situation that has been termed the “obesity paradox” (424, 425). Possible reasons include confounding factors such as smoking and the presence of co-morbidities that are associated with lower body weights, or the use of BMI rather than measures of visceral obesity for most studies on the obesity paradox. Despite the obesity paradox in those with established CVD, the following sections will provide information regarding potential links between obesity T2DM and CVD. The various features of adipose tissue depots, including ectopic fat, and how they contribute to T2DM and CVD are summarized in Figure 2. Notably, there are many similarities between adipose depot characteristics that contribute to both T2DM and CVD.
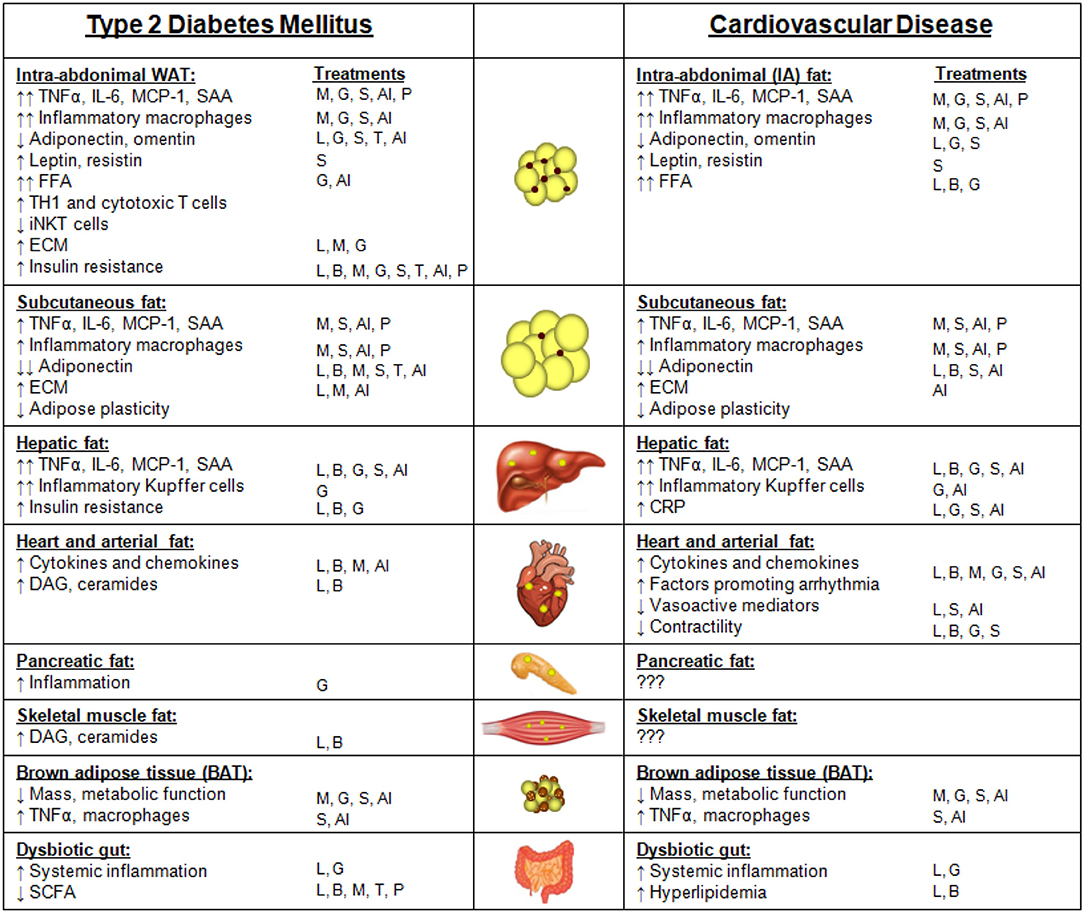
Figure 2. Adipose depots and ectopic fat sites and their features that contribute to type 2 diabetes mellitus (T2DM) or cardiovascular disease (CVD). Features of intra-abdominal white adipose tissue (WAT), subcutaneous fat, hepatic fat, heart and arterial fat (inclusive of epicardial, pericardial, and perivascular fat), pancreatic fat, skeletal muscle fat, brown adipose tissue, and a dysbiotic gut that contribute to either T2DM or CVD. Arrows indicate changes in comparison with subjects without T2DM or CVD. The T2DM treatment strategies that have been reported to improve each adipose depot feature are listed under “treatments.” Treatments: weight loss due to lifestyle changes (L); weight loss due to bariatric surgery (B); metformin (M); GLP-1 receptor agonists (G); SGLT-2 inhibitors (S); thiazolidinediones (TZDs, T); anti-inflammatory approaches (AI); microbiome modulation with pre- or pro-biotics (P).
Adipose Depot-Specific Links With CVD
Visceral and Subcutaneous White Adipose Tissue
The accumulation of visceral fat in obesity is associated with the metabolic syndrome, its associated CVD risk factors, and an increased risk for clinical CVD (426). This distribution of WAT has been shown to have the greatest effect on CVD risk and mortality among patients with normal body weight (427). The risk of CVD in the metabolic syndrome has been considered to result from the presence of multiple CVD risk factors such as dyslipidemia (hypertriglyceridemia, an excess of small, dense LDL particles and reduced HDL-cholesterol levels), hypertension, dysglycemia, and a thrombogenic profile that have been reviewed elsewhere (428–430). However, there are several additional potential mechanisms by which visceral WAT might contribute directly to CVD that involve FFA, insulin resistance, and inflammation. Visceral WAT has higher lipolytic activity than subcutaneous WAT due to its having fewer insulin receptors, and thus is a significant source of FFA. Visceral-derived FFA can directly impact the liver via the portal vein, facilitating FFA uptake by the liver and subsequent hepatic insulin resistance. Similarly, excess FFA from visceral fat might directly impair lipid metabolism and lead to dyslipidemia, which increases CVD risk. In obese diabetic subjects, plasma FFA levels have been shown to be elevated compared to BMI-matched non-diabetic subjects (373), supporting the notion that insulin resistance further elevates circulating FFA levels. Moreover, the incidence of T2DM is nearly doubled in patients with the highest levels of FFA (90th percentile) when compared with subjects with the lowest FFA levels (10th percentile) (431). In one study, obese T2DM subjects who had undergone overnight fasting during pharmacological inhibition of lipolysis exhibited improved insulin sensitivity and glucose tolerance (432), providing further evidence for an inhibitory effect of FFA on insulin sensitivity.
The adipokine profile of visceral WAT also contributes substantially to its association with CVD risk. Obese visceral WAT primarily secretes inflammatory cytokines such as resistin, TNFα, IL-6, IL-1β, MCP-1, and SAA, with reduced levels of adiponectin (433). Plasma adiponectin levels are decreased in patients with CVD (434). Adiponectin is believed to contribute to CVD protection by several mechanisms, including the reduction of lipid levels, repressing expression of inflammatory mediators such as VCAM, ICAM, E-selectin, TNFα, and IL-6, and by acting directly on the heart to improve ischemic injury by activating AMPK and subsequently increasing energy supply to the heart (435–438). Adiponectin also stimulates endothelial nitric oxide synthase (eNOS), which maintains healthy vascular tone (439, 440). Thereby, adiponectin would play a protective role in the development of CVD. Conversely, leptin levels are positively associated with acute myocardial infarction, stroke, coronary heart disease, chronic heart failure, and left cardiac hypertrophy (441–445), although the reasons for this remain largely unknown. Leptin receptors are expressed in the heart, indicative of an important impact of direct leptin signaling (446). Resistin is positively associated with systemic inflammatory markers (447), upregulates endothelial expression levels of VCAM-1 and endothelin-1 (448) and promotes the proliferation of smooth muscle cells (449). Resistin also associates positively with coronary artery calcification levels, and negatively with HDL cholesterol (450). Thus, adipose-derived resistin levels could be used to predict the severity of coronary atherosclerosis (450). Similarly, cytokines and chemokines such as those secreted from obese visceral WAT can induce expression of endothelial adhesion molecules (451), recruit macrophages (452), increase thrombosis (453), and reduce vasoreactivity (454), and are positively associated with cardiovascular events (249, 455). While visceral WAT-derived cytokines are associated with these CVD-inducing processes, it is important to note that the direct contribution from visceral WAT is not currently known, as these are also secreted from other tissues.
As discussed in previous sections, in addition to cytokines and exclusive adipokines, WAT is also a source of FGF21. While the liver is considered to be the major source, adipocytes have also been shown to produce FGF21 to varying degrees in response to various stimuli. In addition to its associations with obesity and T2DM, FGF21 levels have also been associated with increased risk for CVD (456–460). Subjects with CVD that also had diabetes exhibited even higher levels of FGF21 (459), suggesting an important role in diabetes-accelerated atherosclerosis. In particular, FGF21 levels have been shown to positively correlate with hypertension and triglyceride levels, and to negatively correlate with HDL-cholesterol levels (461). One study by Lee et al. suggested that plasma FGF21 levels are associated pericardial fat accumulation (462), which suggests that ectopic fat could be a source of FGF21 in metabolic disease. Further studies are needed to discern whether adipocyte- or hepatic-derived FGF21 contribute to these effects. In stark contrast to these effects of physiological FGF21, pharmacological administration of FGF21 in humans and non-human primates reduces blood glucose, insulin, triglycerides, and LDL cholesterol, and increases HDL cholesterol (142, 463, 464). Thus, there is a disconnect between the physiological and pharmacological effects of FGF21 that requires further study.
Epicardial, Perivascular, and Brown Adipose Tissues
It is becoming increasingly clear that adipose tissue expansion contributes directly to obesity-associated cardiovascular disease risk (465). Obesity is accompanied by not only excess visceral adiposity, but also by excess epicardial and perivascular WAT (466). Due to their proximity to the heart, coronary arteries, and other major arterial blood vessels that are prone to atherosclerosis, it is not surprising that epiWAT and PVAT are important regulators of cardiac and vascular. The respective sizes of these adipose depots are associated with risk factors for the metabolic syndrome, including elevated visceral fat content, blood glucose, hypertension, systemic inflammation, insulin resistance, circulating LDL levels, mean arterial pressure, and atherosclerosis (19, 467–471), as well as adverse cardiovascular events (472–475). The mechanisms behind these associations include increased secretion of pro-inflammatory cytokines, vasoactive factors, and vascular growth factors (476–478); increased release of lipotoxic FFA (479, 480); increased macrophage content (481); increased oxidative stress (482); and decreased secretion of adiponectin (483), which are triggered by obesity. In a prospective cohort of patients with aortic stenosis, a positive association between epiWAT volume and left ventricular mass was found (484), suggesting that in addition to changes in adipokine secretion, epiWAT could negatively influence cardiac function by placing a restrictive burden on the heart. Mechanisms by which PVAT influences CVD are more nuanced and complex. As an adipose depot that features some characteristics of both WAT and BAT, and with different functions depending on the anatomical location (i.e., thoracic vs. abdominal aortic PVAT), PVAT can play either a cardioprotective or a pathological role (24). As obesity progresses, PVAT can become dysfunctional in that it more resembles WAT, and contributes to a pro-inflammatory and lipotoxic microenvironment that promotes atherosclerosis (485). Similarly to healthy PVAT, BAT provides atheroprotection by serving as a protective “buffer” for the vasculature against lipotoxic FFA (486). However, BAT can become dysfunctional as obesity progresses, undergoing a phenotypic “whitening” switch that promotes atherosclerosis (487). Thus, while PVAT and BAT play atheroprotective roles in healthy individuals, obesity promotes dysfunction of these depots, blunting this protective effect against CVD.
Strategies for Reducing T2DM and/or CVD Risk That Impact Adipose Tissue
Strategies for weight loss are multi-faceted, including combinations of diet and lifestyle modifications, pharmaceutical therapy, and various forms of bariatric surgery (488). While there is some debate over this, it is generally believed that small degrees of weight loss in MUHO obese populations can have a dramatic impact on cardiometabolic health (489, 490); thus, strategies that improve obesity are likely to also decrease risk factors for CVD. Similarly, CVD treatment strategies are centered around a combination of pharmaceutical use and lifestyle modifications, which also impact adipose tissue. In this section, we will describe the effects that various CVD treatment strategies have on adipose tissue metabolism and inflammation. How these treatment strategies impact the contributions of particular adipose depot features to T2DM and CVD are listed in Figure 2.
Weight Loss
Lifestyle Modifications: Dietary Changes
As most patients with T2DM and/or CVD are overweight or obese, weight loss often is the first strategy to reduce the severity of T2DM and/or CVD. Traditional methods prescribed for weight loss include restricting food intake and increasing energy expenditure. Despite a large number of fad diets that dictate particular proportions of dietary fat, protein, and carbohydrates to facilitate weight loss [summarized in (488, 489)], the simple fact remains that for weight loss to occur, energy balance must be negative. Thus, energy intake must be less than energy expended, which includes resting energy expenditure, physical activity, and the thermic effect of food.
It has been previously reported that for every kilogram of body weight lost due to dietary restrictions, visceral adiposity is reduced by around 2–3% (491). Subsequently, additional studies have shown that modest weight loss due to dietary changes in people with overweight or obesity is due to roughly equivalent fat lost from subcutaneous and visceral depots, while the addition of exercise leads to more weight loss from subcutaneous fat as well as loss of ectopic skeletal muscle fat (492–495). The loss of visceral fat is associated with reduced CVD risk factors, including reduced systemic inflammation, total cholesterol, LDL cholesterol, and triglycerides (493, 496), as well as reduced fasting glucose and insulin levels (496, 497). A post-hoc analysis from the Look AHEAD study showed that weight loss of ~10% in overweight or obese subjects with T2DM yielded a 21% lower risk of the primary outcome (including CVD-related death, non-fatal acute myocardial infarction, non-fatal stroke, or hospital admission for angina) (498). As the subjects recruited for the Look AHEAD trial had T2DM, this and other post-hoc analyses suggest that weight loss in T2DM subjects also lowers the risk of CVD events (499, 500).
Lifestyle Modifications: Including Exercise
It is well established that aerobic exercise increases fuel mobilization from adipose tissue by increasing lipolysis and subsequent FFA mobilization, which ultimately decreases adiposity and adipocyte size (501–504). Such enhanced fuel mobilization is thought to be highest for visceral WAT (505). Several studies have shown that a high level of fitness (defined by a high activity level with maximal oxygen uptake) negatively associates with visceral adiposity (506–508), even in subjects with obesity and/or T2DM, suggesting that aerobic exercise contributes to a favorable adipose distribution profile that reduces the risk of metabolic syndrome. Hepatic fat is also mobilized and decreased following intense aerobic exercise (509). Studies in mice suggest that not only visceral fat mass is lost with regular exercise, but subcutaneous and brown fat mass are also diminished (510). As expected with fat loss, exercise is coincident with reduced plasma and adipose tissue leptin levels (511–516). The effects of exercise-induced fat loss on adiponectin levels are less clear, with some studies showing no changes in circulating adiponectin levels (517–519), some showing increased plasma adiponectin (520–522), and others showing increased subcutaneous WAT expression of adiponectin mRNA (523–525). A meta-analysis showed that pediatric subjects with obesity exhibit reduced resistin levels following aerobic exercise (526). Little is known about the impact of exercise on FGF21 in obese humans, but one study suggested that aerobic exercise training in obese women reduced circulating FGF21 levels (527). By contrast, studies in rodents have shown that circulating FGF21 levels are not altered by exercise in obese animals (528). Collectively, such exercise-induced changes to WAT distribution and adipokine secretion likely facilitate the observed improvements in insulin sensitivity and CVD risk factors observed with exercise.
While many studies have reported that exercise training increases subcutaneous WAT browning in rodent models of obesity (529–532), there is limited data to support this in humans. Many studies have shown that there is no effect of aerobic exercise training to recruit beige adipocytes in humans (533). However, one study compared subcutaneous WAT from lean, sedentary young men with age- and weight-matched endurance-trained men and reported no differences in beige markers such as UCP1, PGC1A, or CIDEA (534). Another study found evidence of subcutaneous WAT browning (i.e., increased UCP1 and CPT1B expression) in overweight sedentary individuals that had undertaken a 12-week bicycle training program (535). There is some debate about what role brown or beige adipose tissue would play in exercise, if it indeed occurs. It is known that BAT and beige activity is increased when thermogenesis is required, and exercise is a highly thermogenic activity that raises core body temperature, so it is not immediately clear why exercise would increase BAT and/or beige activity. Exercise is known to activate the sympathetic nervous system, which also activates BAT to quickly release stored energy, so it is possible that BAT activation is secondary to exercise-induced sympathetic activation (536). Nevertheless, further studies are needed to determine what role if any BAT and/or beige adipocytes play in mediating the metabolically beneficial effects of exercise.
Loss of adipose tissue mediated by dietary changes, exercise, liposuction, or bariatric surgery (discussed in the section on Bariatric Surgery) is accompanied by decreased markers of adipose tissue and systemic inflammation (537, 538). Weight loss achieved through calorie restriction and/or exercise resulted in decreased systemic IL-6, CRP, TNFα, MCP-1, soluble intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) (273, 539–542). Fat loss by liposuction yielded similar changes in systemic inflammatory markers in one study (543), but did not improve plasma cytokine levels in another (407). The removal of visceral fat from Zucker diabetic fatty rats resulted in dramatic reductions in systemic cytokines (544); this suggests that removing visceral fat, rather than the subcutaneous fat that is routinely removed during liposuction, is more advantageous in terms of resolving inflammation. Many studies also have shown that weight loss following bariatric surgery leads to reductions in systemic inflammatory markers (545), with notable reductions in adipose tissue inflammatory cytokine and macrophage expression (546–548). However, some similar studies do not show improvements in adipose tissue inflammation following various weight loss modalities, such as bariatric surgery or very low-calorie diets (549–551). It has been suggested that pronounced weight loss over time can lead to improvements in adipose tissue inflammation that were not observed in the same subjects following acute moderate weight loss (552). This implies that adipose tissue inflammation during the initial stages of weight loss could be required for the pronounced adipose tissue remodeling required for fat loss (284, 553).
Medications Indicated for the Treatment of T2DM That Lead to Weight Loss
Metformin
Metformin is the most commonly prescribed medication to treat T2DM, particularly in subjects with obesity (554). Metformin has been proposed to lower blood glucose levels through suppression of gluconeogenesis in the liver, activation of AMP-activated protein kinase (AMPK), inhibition of the mitochondrial respiratory chain (complex 1), and by unknown mechanisms in the gut (555, 556). Thus, the precise mechanisms by which metformin lower blood glucose are complex and still evolving. While some diabetes medications have adverse effects on body weight, patients taking metformin often lose a small amount of weight [reviewed in (557)]. Studies in T2DM suggest that metformin may reduce body fat stores and promote a more metabolically healthy fat distribution (558–560). The effect of metformin on adiposity may be partially due to reported nausea and anorexic effects of the drug (561–563). Metformin has been shown to decrease visceral WAT mass, potentially by promoting fatty acid oxidation and/or adaptive thermogenesis (564). With much recent attention focused on BAT as a potential target for obesity treatment, it has recently been shown that BAT is an important effector organ in the glucose-lowering effects of metformin (565). Some studies have reported increases in omentin following metformin therapy, which could be due to visceral fat loss (566). Metformin also reduces hepatic steatosis through inhibition of ApoA5 and steroyl-CoA desaturase-1 (SCD1) which combine to limit de novo lipid synthesis, which is partially mediated by its actions on AMPK and liver X receptor (LXR) activity (567, 568). It also has been suggested that metformin reduces ECM remodeling that is dysregulated in obesity (see previous section on adipose tissue plasticity), and reduces lipogenesis (564).
In addition to the increasingly recognized anti-obesity effects of metformin, its ability to improve CVD risk is also becoming apparent (569). A recent meta-analysis suggested that metformin could contribute to a 16% decrease in all-cause mortality, but may also contribute to a 48% increased risk of stroke (570). The mechanism may include improvements in the lipid profile, such as mild reductions in plasma VLDL cholesterol and triglycerides with slight elevations in HDL cholesterol (571). In addition, metformin has been shown to have anti-inflammatory properties, reported to reduce circulating CRP and MCP-1, reduce NFκB activity, and to reduce advanced glycation end products (AGE) (572–576).
GLP-1 receptor agonists
Glucagon-like peptide-1 (GLP-1) is a peptide hormone that is continuously secreted at low levels during fasting by intestinal L cells. Consumption of a meal enhances GLP-1 secretion, which functions to reduce plasma glucose levels by stimulating insulin secretion from pancreatic beta cells. In addition, GLP-1 receptors are abundant in brain areas that control food intake regulation, such as the hypothalamus, where GLP-1 functions to reduce the drive to eat (577, 578). Thus, several GLP-1 receptor agonists have been developed to mimic the glucose-lowering and anorexic effects of GLP-1 to treat obesity and T2DM.
Liraglutide, a GLP-1 receptor agonist, has shown efficacy in not only glucose control, but also in promoting weight loss and reduced waist circumference based on results from the Liraglutide Effect and Action in Diabetes (LEAD) study (579–581). Liraglutide has also been shown to reduce total adiposity, and specifically visceral fat mass (582, 583). While initially described as being devoid of GLP-1 receptors (584), it has now been confirmed that adipocytes express the GLP-1 receptor (585, 586). Adipose tissue may therefore be an additional target for GLP-1 receptor agonists to promote adipose remodeling by unknown mechanisms. In addition to its effects on body weight and glucose metabolism, GLP-1 receptor agonists may also provide protection against CVD (587). The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial showed that liraglutide lowered the risk of myocardial infarction and non-fatal stroke among patients with T2DM that had high CVD risk (587). GLP-1 receptor agonist treatment has been shown to protect against atherosclerosis in animal models and in humans, potentially by lowering plasma lipids and by reducing circulating CRP and soluble ICAM-1 levels (588–590). Liraglutide, when administered in combination with metformin as indicated for the treatment of T2DM, has been shown to reduce epicardial WAT volume with simultaneous increased omentin expression (591). Thus, liraglutide may provide cardioprotection through reduced levels of ectopic fat, lipids, and inflammation.
SGLT-2 inhibitors
Inhibitors of the sodium-glucose cotransporter 2 (SGLT-2) have been shown to reduce blood glucose levels in subjects with T2DM by enhancing urinary glucose excretion (592). The SGLT-2 inhibitor empagliflozin, alone and in combination with the GLP-1 receptor agonist liraglutide, has been shown to reduce CVD risk (593), as well as cardiovascular death to a greater extent than statins alone (488). Empagliflozin also is associated with decreased hypertension, reduced arterial stiffness, and decreased vascular resistance (594, 595). In both rodents and humans with non-alcoholic fatty liver disease, SGLT-2 inhibitors have been shown to reduce ectopic liver fat by blunting de novo hepatic lipogenesis (596–599), with reduced alanine transaminase (ALT) and aspartate transaminase (AST) levels (600), two markers of hepatic metabolic stress. Furthermore, empagliflozin is associated with weight loss in humans when administered in combination with other therapeutics, such as metformin, thiazolidinediones, and sulfonylureas (601–603). In rodents, SGLT-2 inhibitors have been shown to suppress high fat diet-induced weight gain and to markedly reduce obesity-induced inflammation in WAT, potentially by increasing fat oxidation and the recruitment of beige adipose tissue (604, 605). Thus, in addition to correcting hyperglycemia, SGLT-2 inhibitors can also impact adipose tissue physiology; whether this is through direct or indirect mechanisms remains to be elucidated.
Bariatric surgery
Bariatric surgical techniques, including Roux-en-Y gastric bypass (RYGB) and sleeve gastrectomy, are widely acknowledged to be the most effective treatment strategies for obesity, achieving relatively low levels of obesity remission (606). Within the first year of surgery, some patients experience the loss of around half of their adipose tissue mass (607), often with roughly equivalent losses from subcutaneous and visceral WAT (608, 609). As weight loss progresses, studies have shown that later weight loss is largely from visceral depots (610–612), an effect that correlates with the degree of diabetes remission (609). It has also been reported that ectopic skeletal muscle and pancreatic fat are reduced following bariatric surgery (610, 613, 614), which could contribute to improved glucose metabolism. Studies in humans have reported that subcutaneous adipocytes become smaller following bariatric surgery, resembling adipocytes from lean individuals, but that total adipocyte number remains unchanged (615, 616). Little is known regarding the size and number of visceral adipocytes, which are extremely difficult to sample from humans. As expected with reduced adipocyte size, leptin levels have been shown to decrease following bariatric surgery, while adiponectin has been shown to increase in some studies (617, 618), but not in others (549, 550). Whether changes in adipokine secretion are important for the sustained metabolic improvements following bariatric surgery or whether they simply reflect the adipose remodeling remain to be elucidated. However, it is worth noting that one study has shown that adiponectin levels are elevated only 2 weeks following bariatric surgery, before significant weight loss has occurred, suggesting that adipokine responses may be independent from weight loss (619).
Following bariatric surgery, obesity-associated systemic inflammation persists for as much as 1 month, as indicated by IL-6 and CRP levels (549, 550, 620). Some of this inflammation has been attributed to the surgery itself (545). However, by 6 to 12 months post-surgery, circulating IL-6, CRP, and MCP-1 are typically reduced below pre-surgery levels (548, 549, 620–627), an effect that may be due to fat loss. Importantly, it is not yet clear what effect weight loss due to bariatric surgery has specifically on adipose tissue inflammation. Some studies have reported reduced levels of adipose tissue inflammation following 15–17% weight loss mediated by bariatric surgery (547, 548, 628, 629), while others have shown no changes in adipose tissue inflammation following 7–37% weight loss (549, 550, 630). With insulin sensitivity being substantially improved in all of these studies, these latter studies present a potential disconnect between adipose tissue inflammation and insulin sensitivity that requires further study. However, it must be noted that the adipose tissue sampled in these studies was from subcutaneous depots, due to ease of sampling. Given that visceral WAT is more prone to inflammatory changes, it is possible that visceral WAT inflammation is more impacted by bariatric surgery than subcutaneous WAT.
Bariatric surgery has been shown to upregulate FGF21 in humans, an effect that appears to be specific to RYGB-induced weight loss, as this effect is not observed following weight loss due to caloric restriction or sleeve gastrectomy (631–635). Importantly, it is not known if such FGF21 derives from the liver or adipose tissue. One study has shown that increased FGF21 is associated with improved HOMA-IR in RYGB subjects, an effect that remains when adjusted for adiposity (634), introducing the possibility that elevated FGF21 levels serve to impact glucose homeostasis. Given that FGF21 has been shown to be elevated in obesity, and in particular in subjects with insulin resistance (634), the notion that FGF21 levels would become even further elevated following RYGB surgery, a procedure which rapidly improves insulin sensitivity, represents a paradox. It has been proposed that obesity-associated increased FGF21 levels reflect a “spill-over” from cells that are experiencing metabolic stress (636); however, this hypothesis does not explain the further increased FGF21 levels that accompany RYGB.
Various forms of bariatric surgery have been shown to evoke long-term benefits including sustained and considerable weight loss as well as rapid and sustained remission of T2DM and reduced risk of CVD-related mortality (489). A recent meta-analysis has estimated that on average, patients exhibit a 48% reduction in macrovascular events with a 79% reduction in mortality more than 5 years following bariatric surgery (637). Similarly, long-term follow-up (>17 years) post-surgery in the Swedish Obesity Study showed a 32% reduction in macrovascular complications in T2DM subjects, with 29% fewer myocardial infarctions and a 29% decrease in stroke incidence (638, 639). Bariatric surgery also is associated with improved hypertension, but not a reduced risk of incident hypertension (640). Interestingly, the CRP reduction observed following bariatric surgery was most pronounced in subjects that regained the most insulin sensitivity (624), suggesting an important link between improved glucose metabolism and CVD.
Thiazolidinediones (TZDs)
TZDs are synthetic peroxisome proliferator-activated receptor gamma (PPARγ) activators that have been used to treat T2DM for decades (641–643). The ability of TZDs to improve insulin resistance is clear; however, TZDs also promote adipogenesis and subsequent weight gain (644–647), and are thus not popular choices among patients who don't want to gain weight, even if it is “metabolically healthy” weight gain. The mechanism for such improvements in insulin sensitivity in the face of weight gain appears to be through the induction of adiponectin by TZDs (648), which has known insulin-sensitizing properties as described above.
Activation of PPARγ by TZDs not only enhances adipogenesis, it also alleviates inflammatory cytokine secretion associated with obesity (649) and reduces ectopic fat deposition in tissues such as the liver and skeletal muscle (650). There appears to be a reciprocal relationship between inflammatory cytokines and adiponectin. For example, in vitro experiments in cultured adipocytes revealed that treatment with adiponectin reduces cytokine secretion (651, 652), while treatment with cytokines drastically reduces adiponectin expression and secretion (648, 653, 654). Due to greater adipose lipid storage potential, TZDs should therefore reduce plasma triglyceride levels, which appears to be the case for pioglitazone but not rosiglitazone (655–657). This may in part account for the beneficial cardiovascular effects of pioglitazone in a clinical trial (658).
Anti-Inflammatory Approaches
Characteristic features of MUHO and the metabolic syndrome include adipose tissue and systemic inflammation, which may play a role in the pathogenesis of atherosclerotic CVD. Therefore, an approach that inhibits inflammation would seem logical.
The CANTOS trial, in which CVD events were reduced using an IL-1β antagonist, canakinumab (659), was the first successful proof of concept study using an anti-inflammatory approach for the prevention of recurrent CVD events. A more recent study showed that colchicine, an old drug that has powerful anti-inflammatory properties, reduced recurrent ischemic events when administered after a myocardial infarction (660). Statins, which inhibit 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG-CoA reductase) to reduce LDL cholesterol levels, also have anti-inflammatory properties (661–664). Whether this anti-inflammatory effect of statins plays a role in the well-documented effect of statins in inhibiting clinical CVD events and CVD mortality (665, 666) is unknown. Even less is known about the effect of statins on inhibiting inflammation in adipose tissue, although statins have been shown to reduce epicardial fat accumulation (667). A clue to the potential role of statins in adipose tissue inflammation is provided by the recent demonstration that myeloid-specific deletion of HMG-CoA reductase improved glucose tolerance in obesity induced by a high fat diet, as a result of decreased macrophage recruitment into adipose tissue (668). These changes occurred independently of weight loss and provide impetus for further studies on the effect of statins on adipose tissue inflammation. Regardless, the effect of statins on adipose tissue inflammation is an area that warrants further investigation.
Modulating the Gut Microbiota
The trillions of bacteria that reside within our digestive tract, termed gut microbiota, play an important symbiotic role in shaping our metabolic health. The specific bacterial populations that inhabit our gut can have substantial metabolic impact in relation to obesity, as it is becoming increasingly recognized that that the gut microbiota may contribute to the pathology of obesity (669–671). Dysbiosis, or microbial imbalance in the body, has been associated with obesity in both humans and mice, and can be reversed with weight loss (672–675). Germ-free mice that do not possess gut microbiota are protected from diet-induced obesity and insulin resistance (676, 677), and the obesity phenotype can be conferred by transplantation of cecal contents from obese mice into lean germ-free mice (678), suggesting that the “obese microbiome” is sufficient to cause obesity. It is known that gut bacteria can influence distinct host organ systems indirectly and specifically through the release of particular microbial metabolites such as bile acids, short-chain fatty acids (SCFA), and others. Adipose tissue is a notable target of these microbial metabolites (679). As such, treatments that target the microbiome and modulate microbial metabolism could improve metabolic health.
There is growing evidence that gut dysbiosis can contribute directly to atherosclerotic CVD (669–671, 680). Gut microbial imbalance could modulate atherosclerosis by several mechanisms, including but not limited to: (1) promotion of metabolic endotoxemia due to decreased intestinal barrier integrity, leading to systemic inflammation; (2) altered cholesterol metabolism through the modification of bile acid metabolism, or (3) microbial production of specific beneficial or harmful metabolites with local and/or systemic activity, such as short-chain fatty acids (SCFA). These processes are described below.
Metabolic Endotoxemia
Gut dysbiosis has been associated with elevated intestinal permeability, or a “leaky gut” (681–683). Increased intestinal permeability allows inflammatory bacterial components to enter the systemic circulation to trigger an inflammatory response in diverse tissues such as the liver and adipose tissue. Obese mice and humans have been shown to exhibit gut dysbiosis (670), with increased proportions of endotoxin-producing gut bacteria and elevated circulating levels of lipopolysacharide that correlate with metabolic disease state such as obesity or T2DM (684, 685). Such metabolic endotoxemia is reduced following antibiotic treatment (681) or RYGB surgery-induced weight loss (627). Thus, a compromised intestinal barrier may contribute to systemic inflammation that is characteristic of obesity and CVD (686).
Bile Acid Metabolism
Gut dysbiosis contributes to dysregulated bile acid metabolism (687), leading to hyperlipidemia and hyperglycemia (688, 689). Bile acids produced by the liver facilitate the absorption of dietary fat in the small intestine, and are known to regulate lipid and glucose metabolism through the FXR (690, 691). FXR activation by bile acids initiates a negative feedback pathway, such that bile acid synthesis is inhibited when FXR is activated. Intestinal microbiota are capable of producing secondary bile acids from ~5 to 10% of hepatic-derived bile acids that affect host physiology by serving as FXR agonists, thus resulting in a smaller bile acid pool due to the inhibition of primary bile acid synthesis (692, 693). Subjects with obesity and/or T2DM have been shown to have fewer plasma secondary bile acids in comparison to healthy control subjects (694), an effect that may reflect the altered gut microbial composition observed in obesity. Adipocytes express the major G-protein-coupled bile acid receptor, TGR5, and FXR (695–697), and obesity is accompanied by reduced FXR expression in WAT (697); thus, bile acid signaling to adipose tissue could play a role in modulating adipose tissue inflammation and/or lipid metabolism (698). Secondary bile acids have been shown to exert an anti-inflammatory phenotype in macrophages and hepatocytes (699–701). Bariatric surgery increases plasma bile acid concentrations before any significant weight loss has been achieved (702–704). Metabolic benefits from bariatric surgery, including weight loss and improved glucose metabolism, were absent in mice lacking the TGR5 receptor (705), suggesting an important role for bile acids in the metabolic improvements associated with bariatric surgery. Indeed, adipocyte TGR5 is required for adipogenesis and a metabolically healthy adipokine profile, including secretion of adiponectin and repression of inflammatory cytokines (706, 707). Similarly, deficiency of FXR promotes adipocyte dysfunction, exemplified by impaired adipogenesis, defective insulin signaling, and reduced lipid storage capacity (697). Collectively, these previous studies suggest that intact bile acid signaling is required for adipocyte homeostasis. Thus, equilibrium between dietary-intestinal- and microbiome-intestinal-derived bile acids is important for metabolic health associated with lipid metabolism. The gut microbiota composition and metabolism are therefore important contributors to metabolic health.
SCFA Metabolism
SCFA, including predominantly acetate, propionate, and butyrate, are produced in the gut to varying degrees, depending on the fermentable carbohydrate-based substrates available (i.e., dietary fiber quantity and type) and the particular bacterial populations that are present (i.e., microbiota composition). SCFA serve as signaling molecules to remote organ systems, with impacts on autonomic regulation of systemic blood pressure, systemic inflammation, and other cellular functions. Dysbiotic gut bacteria that is observed in metabolic pathologies such as obesity and T2DM has been characterized by taxonomic shifts that produce fewer SCFA, with notably less butrate produced in the gut (708–710). Evidence from pre-clinical models suggests that SCFA administration could improve metabolic disease states such as obesity, T2DM, and atherosclerosis (711–714). Adipocytes express high levels of key receptors for SCFA, including GPR43 (715). Genetic deletion of GPR43 from adipocytes results in spontaneous obesity, while overexpression of adipocyte GPR43 protects mice from obesity (713). As such, adipose homeostasis can be directly modulated by the gut microbial composition and subsequent SCFA profile. Health benefits of giving SCFA to obese rodents include weight loss (712), improved glucose metabolism, reduced inflammation (716–719), and reduced LDL-cholesterol (720, 721), among others.
Gut Inflammation/Adipose Tissue Cross Talk
The gut microbiota are now considered to be a distinct organ system with endocrine properties that can directly and profoundly modulate the host immune system (722, 723). Under healthy conditions, the commensal (“normal”) gut microbiota play a prominent role in host homeostatic immunity, an essential function to limit the pathogenic potential of gut microbes, via innate and adaptive mechanisms (724, 725). When gut bacteria become dysbiotic, resulting immune deficiencies may contribute to the pathogenesis of obesity, T2DM, and CVD (726). The precise mechanisms by which gut microbiota modulate host immunity [reviewed in (709)] are beyond the scope of this review. However, some mechanisms by which microbial-derived metabolites can modulate adipose tissue function will be described herein. SCFA such as butyrate have been shown to dampen subcutaneous and visceral WAT inflammation by inhibiting NFκB activation (727, 728). Similarly, secondary bile acids negatively correlate with inflammatory pathways in WAT, suggesting an anti-inflammatory effect (729). Bacterial endotoxin, circulating levels of which increase during metabolic diseases that exhibit metabolic endotoxemia, readily promotes adipose tissue inflammation by activating toll-like receptor 4 (TLR4), which is highly expressed in adipocytes as well as macrophages (730). Thus, various metabolites produced by the gut microbiota are known to modulate adipose tissue inflammation directly through the circulation.
Probiotics, Prebiotics, and Synbiotics
Probiotics, prebiotics, and the combined synbiotics could provide an avenue for increased endogenous production of secondary bile acids and/or particular SCFA by modulating the composition of the gut microbiota. Probiotics are commercial preparations of live bacteria designed to be ingested, with the intention of colonizing the gut with the ingested bacteria, or at a minimum to confer a health benefit to the host. Prebiotics, on the other hand, are non-digestible dietary substrates designed to promote an abundance of gut-healthy bacteria, with inferred benefit to the host (731). Synbiotics are preparations that combine particular pre- and pro-biotics, as it is becoming clear that defined fiber substrates increase probiotic colonization efficiency. Pre- and pro-biotics and synbiotics are relatively inexpensive alternatives to conventional CVD medications, with fewer side effects (732). Mechanisms by which pre- and pro-biotic-mediated changes in the gut microbiota may improve adipose function are still emerging, but may include the promotion of an anti-inflammatory milieu (including reducing intestinal permeability to decrease circulating endotoxins), enhancing fat oxidation, recruitment of beige adipocytes, increased energy expenditure, and improved lipoprotein profile, which collectively could improve insulin sensitivity and reduce ectopic fat to combat T2DM and CVD (733–739). While it is generally accepted that particular pre- and pro-biotics reduce diet-induced weight and adiposity gain in animal models (736, 740–743), human intervention studies to date showing efficacy of probiotic treatment are still emerging (744, 745), warranting further study (746).
Concluding Remarks
Obesity results in many changes to adipose tissue, including adipocyte hypertrophy and hyperplasia, infiltration of inflammatory cells, changes in the ECM, and altered adipokine secretion patterns. A critical determinant of whether obesity is likely to lead to metabolic complications such as insulin resistance, the metabolic syndrome, T2DM and CVD is the site where adiposity increases, particularly intra-abdominal, epicardial and perivascular depots, as well as other ectopic sites such as liver, skeletal muscle and pancreas. Ectopic fat accumulation at these sites demonstrate different metabolic, adipokine, and inflammatory profiles from excess white adipose tissue that accumulates subcutaneously, which is predominantly in a lower body distribution and contributes to a less unhealthy form of obesity. Several mechanisms by which these metabolic and inflammatory changes to different adipose tissue depots could influence the metabolic syndrome and its downstream consequences are potential targets for intervention. Various strategies for the treatment of T2DM and/or CVD, including lifestyle- and surgically-mediated weight loss as well as pharmacological or naturopathic methods, also have notable impacts on adipose tissue, which are important to consider.
Author Contributions
LH and AC reviewed the literature and contributed to the preparation of this manuscript.
Funding
This work was supported by the NIH grant # HL092969.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALT, Alanine transaminase; M2, Alternatively activated macrophages; AMPK, AMP-activated protein kinase; ARG1, arginase-1; AST, aspartate transaminase; β-AR, beta-adrenergic receptor; BMI, body mass index; BAT, brown adipose tissue; CRP, C-reactive protein; CVD, cardiovascular disease; CT, computed tomography; DAGs, diacylglycerols; DEXA, dual-energy X-ray absorptiometry; eNOS, endothelial nitric oxide synthase; epiWAT, epicardial white adipose tissue; ECM, extracellular matrix; FXR, farnesoid X receptor; FGFs, fibroblast growth factors; FGF21, fibroblast growth factor 21; FDG, 2-[18F]fluoro-2-deoxyglucose; FFA, free fatty acid; TGR5, G-protein-coupled bile acid receptor; GLP-1, glucagon-like peptide-1; HDL, high-density lipoprotein; HMG-CoA reductase, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; HOMA-IR, homeostatic model assessment of insulin resistance; HSL, hormone sensitive lipase; IKK, IkB kinase; IRS-1, insulin receptor substrate-1; ICAM-1, intercellular adhesion molecule-1; IFNγ, interferon γ; IRF3, interferon regulatory factor 3; IL-1Ra, interleukin-1 receptor antagonist; IL-6, interleukin-6; JNK, c-Jun N-terminal kinase; LEPR, leptin receptor; LXR, liver X receptor; LDL, low-density lipoprotein; MGL1, macrophage galactose type C-type Lectin/CD301a/CLEC10A; MHC, major histocompatibility complex; CD206, mannose receptor; MWAT, mesenteric white adipose tissue; MMe, metabolically activated macrophage; MHO, metabolically healthy obesity; MUHO, metabolically unhealthy obesity; MCP-1, monocyte chemotactic protein-1; NKT cells, natural killer T cells; NAFLD, non-alcoholic fatty liver disease; NE, norepinephrine; NFκB, nuclear factor kappa-B; OWAT, omental white adipose tissue; PVAT, perivascular adipose tissue; PPARα, peroxisome proliferator-activated receptor alpha; PPARγ, peroxisome proliferator-activated receptor gamma; RWAT, retroperitoneal white adipose tissue; SAA, serum amyloid A; SCFA, short-chain fatty acids; SGLT-2, sodium glucose co-transporter-2; SCD1, steroyl-CoA desaturase-1; SNS, sympathetic nervous system; TZDs, thiazolidinediones; TLR4, toll-like receptor 4; TNFα, tumor necrosis factor alpha; T2DM, type 2 diabetes mellitus; UCP-1, uncoupling protein-1; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VLDL, very low-density lipoprotein; WAT, white adipose tissue.
References
1. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among us adults, 1999-2010. JAMA. (2012) 307:491–7. doi: 10.1001/jama.2012.39
2. Abdullah A, Peeters A, de Courten M, Stoelwinder J. The magnitude of association between overweight and obesity and the risk of diabetes: a meta-analysis of prospective cohort studies. Diabetes Res Clin Pract. (2010) 89:309–19. doi: 10.1016/j.diabres.2010.04.012
3. Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960-1994. Int J Obes Relat Metab Disord. (1998) 22:39–47. doi: 10.1038/sj.ijo.0800541
4. Centers for Disease Control and Prevention (CDC). Prevalence of overweight and obesity among adults with diagnosed diabetes–United States, 1988-1994 and 1999-2002. MMWR Morb Mortal Wkly Rep. (2004) 53:1066–8.
5. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet. (2016) 387:1513–30. doi: 10.1016/S0140-6736(16)00618-8
6. Defronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. (2009) 58:773–95. doi: 10.2337/db09-9028
7. Rao Kondapally Seshasai S, Kaptoge S, Thompson A, Di Angelantonio E, Gao P, Sarwar N, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. (2011) 364:829–41. doi: 10.1056/NEJMoa1008862
8. Lloyd-Jones DM, Hong Y, Labarthe D, Mozaffarian D, Appel LJ, Van Horn L, et al. Defining and setting national goals for cardiovascular health promotion and disease reduction: the American heart association's strategic impact goal through 2020 and beyond. Circulation. (2010) 121:586–613. doi: 10.1161/CIRCULATIONAHA.109.192703
9. Cornier MA, Després JP, Davis N, Grossniklaus DA, Klein S, Lamarche B, et al. Assessing adiposity: a scientific statement from the American heart association. Circulation. (2011) 124:1996–2019. doi: 10.1161/CIR.0b013e318233bc6a
10. Flint AJ, Hu FB, Glynn RJ, Caspard H, Manson JE, Willett WC, et al. Excess weight and the risk of incident coronary heart disease among men and women. Obesity. (2010) 18:377–83. doi: 10.1038/oby.2009.223
11. Lee CM, Huxley RR, Wildman RP, Woodward M. Indices of abdominal obesity are better discriminators of cardiovascular risk factors than bmi: a meta-analysis. J Clin Epidemiol. (2008) 61:646–53. doi: 10.1016/j.jclinepi.2007.08.012
12. Chusyd DE, Wang D, Huffman DM, Nagy TR. Relationships between rodent white adipose fat pads and human white adipose fat depots. Front Nutr. (2016) 3:10. doi: 10.3389/fnut.2016.00010
13. Reddy P, Lent-Schochet D, Ramakrishnan N, McLaughlin M, Jialal I. Metabolic syndrome is an inflammatory disorder: a conspiracy between adipose tissue and phagocytes. Clin Chim Acta. (2019) 496:35–44. doi: 10.1016/j.cca.2019.06.019
14. Freedl. Role of a critical visceral adipose tissue threshold (cvatt) in metabolic syndrome: implications for controlling dietary carbohydrates: a review. Nutr Metab. (2004) 1:12. doi: 10.1186/1743-7075-1-12
15. Kwok KH, Lam KS, Xu A. Heterogeneity of white adipose tissue: molecular basis and clinical implications. Exp Mol Med. (2016) 48:e215. doi: 10.1038/emm.2016.5
16. Gesta S, Blüher M, Yamamoto Y, Norris AW, Berndt J, Kralisch S, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci USA. (2006) 103:6676–81. doi: 10.1073/pnas.0601752103
17. Baker M, Gaukrodger N, Mayosi BM, Imrie H, Farrall M, Watkins H, et al. Association between common polymorphisms of the proopiomelanocortin gene and body fat distribution: a family study. Diabetes. (2005) 54:2492–6. doi: 10.2337/diabetes.54.8.2492
18. Ho E, Shimada Y. Formation of the epicardium studied with the scanning electron microscope. Dev Biol. (1978) 66:579–85. doi: 10.1016/0012-1606(78)90263-4
19. Iacobellis G, Corradi D, Sharma AM. Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat Clin Pract Cardiovasc Med. (2005) 2:536–43. doi: 10.1038/ncpcardio0319
20. Marchington JM, Mattacks CA, Pond CM. Adipose tissue in the mammalian heart and pericardium: structure, foetal development and biochemical properties. Comp Biochem Physiol B. (1989) 94:225–32. doi: 10.1016/0305-0491(89)90337-4
21. Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. (2003) 108:2460–6. doi: 10.1161/01.CIR.0000099542.57313.C5
22. Iacobellis G, Malavazos AE, Corsi MM. Epicardial fat: from the biomolecular aspects to the clinical practice. Int J Biochem Cell Biol. (2011) 43:1651–4. doi: 10.1016/j.biocel.2011.09.006
23. Szasz T, Webb RC. Perivascular adipose tissue: more than just structural support. Clin Sci. (2012) 122:1–12. doi: 10.1042/CS20110151
24. Qi XY, Qu SL, Xiong WH, Rom O, Chang L, Jiang ZS. Perivascular adipose tissue (pvat) in atherosclerosis: a double-edged sword. Cardiovasc Diabetol. (2018) 17:134. doi: 10.1186/s12933-018-0777-x
25. Björntorp P. “Portal” Adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis. (1990) 10:493–496. doi: 10.1161/01.ATV.10.4.493
26. Zhang F, Hao G, Shao M, Nham K, An Y, Wang Q, et al. An adipose tissue atlas: an image-guided identification of human-like bat and beige depots in rodents. Cell Metab. (2018) 27:252–62.e253. doi: 10.1016/j.cmet.2017.12.004
27. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
28. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. (2009) 360:1509–17. doi: 10.1056/NEJMoa0810780
29. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elía E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. (2015) 21:33–8. doi: 10.1016/j.cmet.2014.12.009
30. Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. (2013) 19:635–9. doi: 10.1038/nm.3112
31. Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. (2019) 129:3990–4000. doi: 10.1172/JCI129187
32. Hany TF, Gharehpapagh E, Kamel EM, Buck A, Himms-Hagen J, von Schulthess GK. Brown adipose tissue: a factor to consider in symmetrical tracer uptake in the neck and upper chest region. Eur J Nucl Med Mol Imaging. (2002) 29:1393–8. doi: 10.1007/s00259-002-0902-6
33. Lee MW, Lee M, Oh KJ. Adipose tissue-derived signatures for obesity and type 2 diabetes: adipokines, batokines and micrornas. J Clin Med. (2019) 8:E854. doi: 10.3390/jcm8060854
34. Srivastava S, Veech RL. Brown and brite: the fat soldiers in the anti-obesity fight. Front Physiol. (2019) 10:38. doi: 10.3389/fphys.2019.00038
35. Waldén TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J. Recruited vs. Nonrecruited molecular signatures of brown, “Brite,” and white adipose tissues. Am J Physiol Endocrinol Metab. (2012) 302:E19–31. doi: 10.1152/ajpendo.00249.2011
36. Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. (2012) 150:366–76. doi: 10.1016/j.cell.2012.05.016
37. Jespersen NZ, Larsen TJ, Peijs L, Daugaard S, Homøe P, Loft A, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab. (2013) 17:798–805. doi: 10.1016/j.cmet.2013.04.011
38. Sharp LZ, Shinoda K, Ohno H, Scheel DW, Tomoda E, Ruiz L, et al. Human bat possesses molecular signatures that resemble beige/brite cells. PLoS ONE. (2012) 7:e49452. doi: 10.1371/journal.pone.0049452
39. Shinoda K, Luijten IH, Hasegawa Y, Hong H, Sonne SB, Kim M, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat Med. (2015) 21:389–94. doi: 10.1038/nm.3819
40. Zuriaga MA, Fuster JJ, Gokce N, Walsh K. Humans and mice display opposing patterns of “Browning” Gene expression in visceral and subcutaneous white adipose tissue depots. Front Cardiovasc Med. (2017) 4:27. doi: 10.3389/fcvm.2017.00027
41. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour-derived pth-related protein triggers adipose tissue browning and cancer cachexia. Nature. (2014) 513:100–4. doi: 10.1038/nature13528
42. Neinast MD, Frank AP, Zechner JF, Li Q, Vishvanath L, Palmer BF, et al. Activation of natriuretic peptides and the sympathetic nervous system following roux-en-y gastric bypass is associated with gonadal adipose tissues browning. Mol Metab. (2015) 4:427–36. doi: 10.1016/j.molmet.2015.02.006
43. Sidossis LS, Porter C, Saraf MK, Børsheim E, Radhakrishnan RS, Chao T, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. (2015) 22:219–27. doi: 10.1016/j.cmet.2015.06.022
44. den Hartigh LJ, Wang S, Goodspeed L, Wietecha T, Houston B, Omer M, et al. Metabolically distinct weight loss by 10,12 cla and caloric restriction highlight the importance of subcutaneous white adipose tissue for glucose homeostasis in mice. PLoS ONE. (2017) 12:e0172912. doi: 10.1371/journal.pone.0172912
45. Sahuri-Arisoylu M, Brody LP, Parkinson JR, Parkes H, Navaratnam N, Miller AD, et al. Reprogramming of hepatic fat accumulation and 'browning' of adipose tissue by the short-chain fatty acid acetate. Int J Obes. (2016) 40:955–63. doi: 10.1038/ijo.2016.23
46. Ghorbani M, Himms-Hagen J. Appearance of brown adipocytes in white adipose tissue during cl 316,243-induced reversal of obesity and diabetes in zucker fa/fa rats. Int J Obes Relat Metab Disord. (1997) 21:465–75. doi: 10.1038/sj.ijo.0800432
47. Baskaran P, Krishnan V, Ren J, Thyagarajan B. Capsaicin induces browning of white adipose tissue and counters obesity by activating trpv1 channel-dependent mechanisms. Br J Pharmacol. (2016) 173:2369–89. doi: 10.1111/bph.13514
48. Chen LH, Chien YW, Liang CT, Chan CH, Fan MH, Huang HY. Green tea extract induces genes related to browning of white adipose tissue and limits weight-gain in high energy diet-fed rat. Food Nutr Res. (2017) 61:1347480. doi: 10.1080/16546628.2017.1347480
49. Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator-activated receptor gamma (ppargamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, ucp1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. (2010) 285:7153–64. doi: 10.1074/jbc.M109.053942
50. Lin HV, Frassetto A, Kowalik EJ, Nawrocki AR, Lu MM, Kosinski JR, et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE. (2012) 7:e35240. doi: 10.1371/journal.pone.0035240
51. Fukui Y, Masui S, Osada S, Umesono K, Motojima K. A new thiazolidinedione, nc-2100, which is a weak ppar-gamma activator, exhibits potent antidiabetic effects and induces uncoupling protein 1 in white adipose tissue of kkay obese mice. Diabetes. (2000) 49:759–67. doi: 10.2337/diabetes.49.5.759
52. Stanford KI, Middelbeek RJ, Goodyear LJ. Exercise effects on white adipose tissue: beiging and metabolic adaptations. Diabetes. (2015) 64:2361–8. doi: 10.2337/db15-0227
53. Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. Prdm16 controls a brown fat/skeletal muscle switch. Nature. (2008) 454:961–7. doi: 10.1038/nature07182
54. Shao M, Wang QA, Song A, Vishvanath L, Busbuso NC, Scherer PE, et al. Cellular origins of beige fat cells revisited. Diabetes. (2019) 68:1874–85. doi: 10.2337/db19-0308
55. Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristiansen K, et al. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab. (2010) 298:E1244–53. doi: 10.1152/ajpendo.00600.2009
56. Cinti S. Transdifferentiation properties of adipocytes in the adipose organ. Am J Physiol Endocrinol Metab. (2009) 297:E977–86. doi: 10.1152/ajpendo.00183.2009
57. Altshuler-Keylin S, Shinoda K, Hasegawa Y, Ikeda K, Hong H, Kang Q, et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metab. (2016) 24:402–19. doi: 10.1016/j.cmet.2016.08.002
58. Xue R, Lynes MD, Dreyfuss JM, Shamsi F, Schulz TJ, Zhang H, et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat Med. (2015) 21:760–8. doi: 10.1038/nm.3881
59. Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. (1998) 102:412–20. doi: 10.1172/JCI3155
60. Almind K, Kahn CR. Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes. (2004) 53:3274–85. doi: 10.2337/diabetes.53.12.3274
61. Collins S, Daniel KW, Petro AE, Surwit RS. Strain-specific response to beta 3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. (1997) 138:405–13. doi: 10.1210/endo.138.1.4829
62. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. (2011) 121:96–105. doi: 10.1172/JCI44271
63. Shao M, Ishibashi J, Kusminski CM, Wang QA, Hepler C, Vishvanath L, et al. Zfp423 maintains white adipocyte identity through suppression of the beige cell thermogenic gene program. Cell Metab. (2016) 23:1167–84. doi: 10.1016/j.cmet.2016.04.023
64. Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. (2007) 21:1443–55. doi: 10.1101/gad.1550907
65. Schoettl T, Fischer IP, Ussar S. Heterogeneity of adipose tissue in development and metabolic function. J Exp Biol. (2018) 221:jeb162958. doi: 10.1242/jeb.162958
66. Chondronikola M, Volpi E, Børsheim E, Chao T, Porter C, Annamalai P, et al. Brown adipose tissue is linked to a distinct thermoregulatory response to mild cold in people. Front Physiol. (2016) 7:129. doi: 10.3389/fphys.2016.00129
67. Frontini A, Cinti S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. (2010) 11:253–6. doi: 10.1016/j.cmet.2010.03.004
68. Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. (2013) 123:3404–8. doi: 10.1172/JCI67803
69. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. (2013) 19:1252–63. doi: 10.1038/nm.3361
70. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. (2011) 480:104–8. doi: 10.1038/nature10653
71. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. (2014) 157:1292–308. doi: 10.1016/j.cell.2014.03.066
72. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic er stress sensor ire1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol. (2017) 18:519–29. doi: 10.1038/ni.3709
73. Vargovic P, Manz G, Kvetnansky R. Continuous cold exposure induces an anti-inflammatory response in mesenteric adipose tissue associated with catecholamine production and thermogenin expression in rats. Endocr Regul. (2016) 50:137–44. doi: 10.1515/enr-2016-0015
74. Liu PS, Lin YW, Burton FH, Wei LN. Injecting engineered anti-inflammatory macrophages therapeutically induces white adipose tissue browning and improves diet-induced insulin resistance. Adipocyte. (2015) 4:123–8. doi: 10.4161/21623945.2014.981438
75. Liu PS, Lin YW, Lee B, McCrady-Spitzer SK, Levine JA, Wei LN. Reducing rip140 expression in macrophage alters atm infiltration, facilitates white adipose tissue browning, and prevents high-fat diet-induced insulin resistance. Diabetes. (2014) 63:4021–31. doi: 10.2337/db14-0619
76. Kusminski CM, Holl WL, Sun K, Park J, Spurgin SB, Lin Y, et al. Mitoneet-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. (2012) 18:1539–49. doi: 10.1038/nm.2899
77. Hallowell RW, Collins SL, Craig JM, Zhang Y, Oh M, Illei PB, et al. Mtorc2 signalling regulates m2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat Commun. (2017) 8:14208. doi: 10.1038/ncomms14208
78. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. (2015) 519:242–6. doi: 10.1038/nature14115
79. Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. (2015) 160:74–87. doi: 10.1016/j.cell.2014.12.011
80. Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med. (2017) 23:623–30. doi: 10.1038/nm.4316
81. Ahima RS, Lazar MA. Adipokines and the peripheral and neural control of energy balance. Mol Endocrinol. (2008) 22:1023–31. doi: 10.1210/me.2007-0529
83. Farooqi IS, O'Rahilly S. 20 years of leptin: human disorders of leptin action. J Endocrinol. (2014) 223:T63–70. doi: 10.1530/JOE-14-0480
84. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. (1996) 334:292–5. doi: 10.1056/NEJM199602013340503
85. Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. (1996) 81:3419–23. doi: 10.1210/jcem.81.9.8784108
86. Hube F, Lietz U, Igel M, Jensen PB, Tornqvist H, Joost HG, et al. Difference in leptin mRNA levels between omental and subcutaneous abdominal adipose tissue from obese humans. Horm Metab Res. (1996) 28:690–3. doi: 10.1055/s-2007-979879
87. Hu E, Liang P, Spiegelman BM. Adipoq is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. (1996) 271:10697–703. doi: 10.1074/jbc.271.18.10697
88. Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. Cdna cloning and expression of a novel adipose specific collagen-like factor, apm1 (adipose most abundant gene transcript 1). Biochem Biophys Res Commun. (1996) 221:286–9. doi: 10.1006/bbrc.1996.0587
89. Nakano Y, Tobe T, Choi-Miura NH, Mazda T, Tomita M. Isolation and characterization of gbp28, a novel gelatin-binding protein purified from human plasma. J Biochem. (1996) 120:803–12. doi: 10.1093/oxfordjournals.jbchem.a021483
90. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to c1q, produced exclusively in adipocytes. J Biol Chem. (1995) 270:26746–9. doi: 10.1074/jbc.270.45.26746
91. Cnop M, Havel PJ, Utzschneider KM, Carr DB, Sinha MK, Boyko EJ, et al. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia. (2003) 46:459–69. doi: 10.1007/s00125-003-1074-z
92. Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes. (2003) 52:1779–85. doi: 10.2337/diabetes.52.7.1779
93. Kishida K, Kim KK, Funahashi T, Matsuzawa Y, Kang HC, Shimomura I. Relationships between circulating adiponectin levels and fat distribution in obese subjects. J Atheroscler Thromb. (2011) 18:592–5. doi: 10.5551/jat.7625
94. Samaras K, Botelho NK, Chisholm DJ, Lord RV. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity. (2010) 18:884–9. doi: 10.1038/oby.2009.443
95. Lihn AS, Bruun JM, He G, Pedersen SB, Jensen PF, Richelsen B. Lower expression of adiponectin mRNA in visceral adipose tissue in lean and obese subjects. Mol Cell Endocrinol. (2004) 219:9–15. doi: 10.1016/j.mce.2004.03.002
96. Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/acrp30. Nat Med. (2002) 8:731–7. doi: 10.1038/nm724
97. Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. (2007) 117:2621–37. doi: 10.1172/JCI31021
98. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. (2003) 423:762–9. doi: 10.1038/nature01705
99. Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol. (2018) 8:1031–63. doi: 10.1002/cphy.c170046
100. Ye R, Wang M, Wang QA, Scherer PE. Adiponectin-mediated antilipotoxic effects in regenerating pancreatic islets. Endocrinology. (2015) 156:2019–28. doi: 10.1210/en.2015-1066
101. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent m2 macrophage polarization: link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. (2011) 286:13460–9. doi: 10.1074/jbc.M110.204644
102. Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology. (2008) 47:668–76. doi: 10.1002/hep.21995
103. Jamaluddin MS, Weakley SM, Yao Q, Chen C. Resistin: functional roles and therapeutic considerations for cardiovascular disease. Br J Pharmacol. (2012) 165:622–32. doi: 10.1111/j.1476-5381.2011.01369.x
104. Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, et al. Resistin / fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. (2001) 50:2199–202. doi: 10.2337/diabetes.50.10.2199
105. Tsiotra PC, Tsigos C, Anastasiou E, Yfanti E, Boutati E, Souvatzoglou E, et al. Peripheral mononuclear cell resistin mRNA expression is increased in type 2 diabetic women. Mediators Inflamm. (2008) 2008:892864. doi: 10.1155/2008/892864
106. Ghosh S, Singh AK, Aruna B, Mukhopadhyay S, Ehtesham NZ. The genomic organization of mouse resistin reveals major differences from the human resistin: functional implications. Gene. (2003) 305:27–34. doi: 10.1016/S0378-1119(02)01213-1
107. Park HK, Ahima RS. Resistin in rodents and humans. Diabetes Metab J. (2013) 37:404–14. doi: 10.4093/dmj.2013.37.6.404
108. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. (2001) 409:307–12. doi: 10.1038/35053000
109. Sheng CH, Di J, Jin Y, Zhang YC, Wu M, Sun Y, et al. Resistin is expressed in human hepatocytes and induces insulin resistance. Endocrine. (2008) 33:135–43. doi: 10.1007/s12020-008-9065-y
110. Yannakoulia M, Yiannakouris N, Blüher S, Matalas AL, Klimis-Zacas D, Mantzoros CS. Body fat mass and macronutrient intake in relation to circulating soluble leptin receptor, free leptin index, adiponectin, and resistin concentrations in healthy humans. J Clin Endocrinol Metab. (2003) 88:1730–6. doi: 10.1210/jc.2002-021604
111. Degawa-Yamauchi M, Bovenkerk JE, Juliar BE, Watson W, Kerr K, Jones R, et al. Serum resistin (fizz3) protein is increased in obese humans. J Clin Endocrinol Metab. (2003) 88:5452–5. doi: 10.1210/jc.2002-021808
112. McTernan CL, McTernan PG, Harte AL, Levick PL, Barnett AH, Kumar S. Resistin, central obesity, and type 2 diabetes. Lancet. (2002) 359:46–7. doi: 10.1016/S0140-6736(02)07281-1
113. McTernan PG, McTernan CL, Chetty R, Jenner K, Fisher FM, Lauer MN, et al. Increased resistin gene and protein expression in human abdominal adipose tissue. J Clin Endocrinol Metab. (2002) 87:2407. doi: 10.1210/jcem.87.5.8627
114. Valsamakis G, McTernan PG, Chetty R, Al Daghri N, Field A, Hanif W, et al. Modest weight loss and reduction in waist circumference after medical treatment are associated with favorable changes in serum adipocytokines. Metabolism. (2004) 53:430–4. doi: 10.1016/j.metabol.2003.11.022
115. Way JM, Görgün CZ, Tong Q, Uysal KT, Brown KK, Harrington WW, et al. Adipose tissue resistin expression is severely suppressed in obesity and stimulated by peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. (2001) 276:25651–53. doi: 10.1074/jbc.C100189200
116. Janke J, Engeli S, Gorzelniak K, Luft FC, Sharma AM. Resistin gene expression in human adipocytes is not related to insulin resistance. Obes Res. (2002) 10:1–5. doi: 10.1038/oby.2002.1
117. Vidal-Puig A, O'Rahilly S. Resistin: a new link between obesity and insulin resistance? Clin Endocrinol. (2001) 55:437–8. doi: 10.1046/j.1365-2265.2001.01377.x
118. Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J Immunol. (2005) 174:5789–95. doi: 10.4049/jimmunol.174.9.5789
119. Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. (2006) 290:E1253–61. doi: 10.1152/ajpendo.00572.2004
120. Landecho MF, Tuero C, Valentí V, Bilbao I, de la Higuera M, Frühbeck G. Relevance of leptin and other adipokines in obesity-associated cardiovascular risk. Nutrients. (2019) 11:2664. doi: 10.3390/nu11112664
121. Schäffler A, Neumeier M, Herfarth H, Fürst A, Schölmerich J, Büchler C. Genomic structure of human omentin, a new adipocytokine expressed in omental adipose tissue. Biochim Biophys Acta. (2005) 1732:96–102. doi: 10.1016/j.bbaexp.2005.11.005
122. Auguet T, Quintero Y, Riesco D, Morancho B, Terra X, Crescenti A, et al. New adipokines vaspin and omentin. Circulating levels and gene expression in adipose tissue from morbidly obese women. BMC Med Genet. (2011) 12:60. doi: 10.1186/1471-2350-12-60
123. de Souza Batista CM, Yang RZ, Lee MJ, Glynn NM, Yu DZ, Pray J, et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes. (2007) 56:1655–61. doi: 10.2337/db06-1506
124. Pan HY, Guo L, Li Q. Changes of serum omentin-1 levels in normal subjects and in patients with impaired glucose regulation and with newly diagnosed and untreated type 2 diabetes. Diabetes Res Clin Pract. (2010) 88:29–33. doi: 10.1016/j.diabres.2010.01.013
125. Tan BK, Adya R, Farhatullah S, Lewandowski KC, O'Hare P, Lehnert H, et al. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes. (2008) 57:801–8. doi: 10.2337/db07-0990
126. Cai RC, Wei L, DI JZ, Yu HY, Bao YQ, Jia WP. [Expression of omentin in adipose tissues in obese and type 2 diabetic patients]. Zhonghua Yi Xue Za Zhi. (2009) 89:381–4.
127. Elsaid NH, Sadik NA, Ahmed NR, Fayez SE, Mohammed NAE. Serum omentin-1 levels in type 2 diabetic obese women in relation to glycemic control, insulin resistance and metabolic parameters. J Clin Transl Endocrinol. (2018) 13:14–9. doi: 10.1016/j.jcte.2018.05.003
128. Zhang M, Tan X, Yin C, Wang L, Tie Y, Xiao Y. Serum levels of omentin-1 are increased after weight loss and are particularly associated with increases in obese children with metabolic syndrome. Acta Paediatr. (2017) 106:1851–6. doi: 10.1111/apa.14026
129. Moreno-Navarrete JM, Catalán V, Ortega F, Gómez-Ambrosi J, Ricart W, Frühbeck G, et al. Circulating omentin concentration increases after weight loss. Nutr Metab. (2010) 7:27. doi: 10.1186/1743-7075-7-27
130. Yamawaki H, Kuramoto J, Kameshima S, Usui T, Okada M, Hara Y. Omentin, a novel adipocytokine inhibits tnf-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. (2011) 408:339–43. doi: 10.1016/j.bbrc.2011.04.039
131. Kazama K, Usui T, Okada M, Hara Y, Yamawaki H. Omentin plays an anti-inflammatory role through inhibition of TNF-α-induced superoxide production in vascular smooth muscle cells. Eur J Pharmacol. (2012) 686:116–23. doi: 10.1016/j.ejphar.2012.04.033
132. Kazama K, Okada M, Yamawaki H. A novel adipocytokine, omentin, inhibits platelet-derived growth factor-bb-induced vascular smooth muscle cell migration through antioxidative mechanism. Am J Physiol Heart Circ Physiol. (2014) 306:H1714–9. doi: 10.1152/ajpheart.00048.2014
133. Wang J, Gao Y, Lin F, Han K, Wang X. Omentin-1 attenuates lipopolysaccharide (LPS)-induced U937 macrophages activation by inhibiting the TLR4/MyD88/NF-κB signaling. Arch Biochem Biophys. (2019) 679:108187. doi: 10.1016/j.abb.2019.108187
134. Kazama K, Okada M, Yamawaki H. Adipocytokine, omentin inhibits doxorubicin-induced h9c2 cardiomyoblasts apoptosis through the inhibition of mitochondrial reactive oxygen species. Biochem Biophys Res Commun. (2015) 457:602–7. doi: 10.1016/j.bbrc.2015.01.032
135. Fernández-Trasancos Á, Agra RM, García-Acuña JM, Fernández Á, González-Juanatey JR, Eiras S. Omentin treatment of epicardial fat improves its anti-inflammatory activity and paracrine benefit on smooth muscle cells. Obesity. (2017) 25:1042–9. doi: 10.1002/oby.21832
136. Zabetian-Targhi F, Mirzaei K, Keshavarz SA, Hossein-Nezhad A. Modulatory role of omentin-1 in inflammation: cytokines and dietary intake. J Am Coll Nutr. (2016) 35:670–8. doi: 10.1080/07315724.2015.1126207
137. Shibata R, Ouchi N, Takahashi R, Terakura Y, Ohashi K, Ikeda N, et al. Omentin as a novel biomarker of metabolic risk factors. Diabetol Metab Syndr. (2012) 4:37. doi: 10.1186/1758-5996-4-37
138. Cuevas-Ramos D, Almeda-Valdes P, Aguilar-Salinas CA, Cuevas-Ramos G, Cuevas-Sosa AA, Gomez-Perez FJ. The role of fibroblast growth factor 21 (fgf21) on energy balance, glucose and lipid metabolism. Curr Diabetes Rev. (2009) 5:216–20. doi: 10.2174/157339909789804396
139. Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. (2008) 149:6018–27. doi: 10.1210/en.2008-0816
140. Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, et al. Circulating fgf21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. (2014) 63:4057–63. doi: 10.2337/db14-0595
141. BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR, et al. Fgf21 regulates metabolism through adipose-dependent and -independent mechanisms. Cell Metab. (2017) 25:935–44.e934. doi: 10.1016/j.cmet.2017.03.005
142. Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, et al. A long-acting fgf21 molecule, pf-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metab. (2016) 23:427–40. doi: 10.1016/j.cmet.2016.02.001
143. Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, Walsh K. fgf21 is an akt-regulated myokine. FEBS Lett. (2008) 582:3805–10. doi: 10.1016/j.febslet.2008.10.021
144. Lin X, Liu YB, Hu H. metabolic role of fibroblast growth factor 21 in liver, adipose and nervous system tissues. Biomed Rep. (2017) 6:495–502. doi: 10.3892/br.2017.890
145. Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, Liu F, et al. Serum fgf21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. (2008) 57:1246–53. doi: 10.2337/db07-1476
146. Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, et al. Obesity is a fibroblast growth factor 21 (fgf21)-resistant state. Diabetes. (2010) 59:2781–9. doi: 10.2337/db10-0193
147. Crujeiras AB, Gomez-Arbelaez D, Zulet MA, Carreira MC, Sajoux I, de Luis D, et al. Plasma fgf21 levels in obese patients undergoing energy-restricted diets or bariatric surgery: a marker of metabolic stress? Int J Obes. (2017) 41:1570–8. doi: 10.1038/ijo.2017.138
148. Berti L, Irmler M, Zdichavsky M, Meile T, Böhm A, Stefan N, et al. Fibroblast growth factor 21 is elevated in metabolically unhealthy obesity and affects lipid deposition, adipogenesis, and adipokine secretion of human abdominal subcutaneous adipocytes. Mol Metab. (2015) 4:519–27. doi: 10.1016/j.molmet.2015.04.002
149. Mashili FL, Ramaiya K, Lutale J, Njelekela M, Francis F, Zierath J, et al. Adiposity is a key correlate of circulating fibroblast growth factor-21 levels in African males with or without type 2 diabetes mellitus. J Obes. (2018) 2018:7461903. doi: 10.1155/2018/7461903
150. Sandoval V, Rodríguez-Rodríguez R, Martínez-Garza Ú, Rosell-Cardona C, Lamuela-Raventós RM, Marrero PF, et al. Mediterranean tomato-based sofrito sauce improves fibroblast growth factor 21 (fgf21) signaling in white adipose tissue of obese zucker rats. Mol Nutr Food Res. (2018) 62:1–6. doi: 10.1002/mnfr.201700606
151. Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. (2010) 24:2050–64. doi: 10.1210/me.2010-0142
152. Muise ES, Azzolina B, Kuo DW, El-Sherbeini M, Tan Y, Yuan X, et al. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Mol Pharmacol. (2008) 74:403–12. doi: 10.1124/mol.108.044826
153. Miehle K, Ebert T, Kralisch S, Hoffmann A, Kratzsch J, Schlögl H, et al. Serum concentrations of fibroblast growth factor 21 are elevated in patients with congenital or acquired lipodystrophy. Cytokine. (2016) 83:239–44. doi: 10.1016/j.cyto.2016.04.015
154. Keipert S, Kutschke M, Lamp D, Brachthäuser L, Neff F, Meyer CW, et al. Genetic disruption of uncoupling protein 1 in mice renders brown adipose tissue a significant source of fgf21 secretion. Mol Metab. (2015) 4:537–42. doi: 10.1016/j.molmet.2015.04.006
155. Gerhard GS, Styer AM, Strodel WE, Roesch SL, Yavorek A, Carey DJ, et al. Gene expression profiling in subcutaneous, visceral and epigastric adipose tissues of patients with extreme obesity. Int J Obes. (2014) 38:371–8. doi: 10.1038/ijo.2013.152
156. van Baak MA, Vink RG, Roumans NJT, Cheng CC, Adams AC, Mariman ECM. Adipose tissue contribution to plasma fibroblast growth factor 21 and fibroblast activation protein in obesity. Int J Obes. (2019) 44:544–547. doi: 10.1038/s41366-019-0433-x
157. Hong ES, Lim C, Choi HY, Lee YK, Ku EJ, Moon JH, et al. Plasma fibroblast growth factor 21 levels increase with ectopic fat accumulation and its receptor levels are decreased in the visceral fat of patients with type 2 diabetes. BMJ Open Diabetes Res Care. (2019) 7:e000776. doi: 10.1136/bmjdrc-2019-000776
158. Mraz M, Bartlova M, Lacinova Z, Michalsky D, Kasalicky M, Haluzikova D, et al. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin Endocrinol. (2009) 71:369–75. doi: 10.1111/j.1365-2265.2008.03502.x
159. Kotulák T, Drápalová J, Kopecký P, Lacinová Z, Kramár P, Ríha H, Netuka I, et al. Increased circulating and epicardial adipose tissue mRNA expression of fibroblast growth factor-21 after cardiac surgery: possible role in postoperative inflammatory response and insulin resistance. Physiol Res. (2011) 60:757–67.
160. Lee JH, Kim JM, Choi MJ, Kang YE, Joung KH, Yi HS, et al. Clinical implications of ucp1 mRNA expression in human cervical adipose tissue under physiological conditions. Obesity. (2018) 26:1008–16. doi: 10.1002/oby.22188
161. Lee P, Werner CD, Kebebew E, Celi FS. Functional thermogenic beige adipogenesis is inducible in human neck fat. Int J Obes. (2014) 38:170–6. doi: 10.1038/ijo.2013.82
162. Vargas D, López C, Acero E, Benitez E, Wintaco A, Camacho J, et al. Thermogenic capacity of human periaortic adipose tissue is transformed by body weight. PLoS ONE. (2018) 13:e0194269. doi: 10.1371/journal.pone.0194269
163. Vargas D, Camacho J, Duque J, Carreño M, Acero E, Pérez M, et al. Functional characterization of preadipocytes derived from human periaortic adipose tissue. Int J Endocrinol. (2017) 2017:2945012. doi: 10.1155/2017/2945012
164. Cuevas-Ramos D, Almeda-Valdes P, Gómez-Pérez FJ, Meza-Arana CE, Cruz-Bautista I, Arellano-Campos O, et al. Daily physical activity, fasting glucose, uric acid, and body mass index are independent factors associated with serum fibroblast growth factor 21 levels. Eur J Endocrinol. (2010) 163:469–77. doi: 10.1530/EJE-10-0454
165. Gómez-Sámano M, Grajales-Gómez M, Zuarth-Vázquez JM, Navarro-Flores MF, Martínez-Saavedra M, Juárez-León Ó, et al. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. (2017) 11:335–41. doi: 10.1016/j.redox.2016.12.024
166. Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, et al. Thermogenic activation induces fgf21 expression and release in brown adipose tissue. J Biol Chem. (2011) 286:12983–90. doi: 10.1074/jbc.M110.215889
167. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature. (2008) 453:783–7. doi: 10.1038/nature06902
168. Goossens GH, Blaak EE. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front Endocrinol. (2015) 6:55. doi: 10.3389/fendo.2015.00055
169. Wang QA, Scherer PE. The adipochaser mouse: a model tracking adipogenesis in vivo. Adipocyte. (2014) 3:146–150. doi: 10.4161/adip.27656
170. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. (2013). 19:1338–44 doi: 10.1038/nm.3324
171. Kim SM, Lun M, Wang M, Senyo SE, Guillermier C, Patwari P, et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. (2014) 20:1049–58. doi: 10.1016/j.cmet.2014.10.010
172. Guillermier C, Fazeli PK, Kim S, Lun M, Zuflacht JP, Milian J, et al. Imaging mass spectrometry demonstrates age-related decline in human adipose plasticity. JCI Insight. (2017) 2:e90349. doi: 10.1172/jci.insight.90349
173. Denis GV, Obin MS. ‘Metabolically healthy obesity': origins and implications. Mol Aspects Med. (2013) 34:59–70. doi: 10.1016/j.mam.2012.10.004
174. Stefan N, Häring HU, Hu FB, Schulze MB. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. (2013) 1:152–62. doi: 10.1016/S2213-8587(13)70062-7
175. Samocha-Bonet D, Dixit VD, Kahn CR, Leibel RL, Lin X, Nieuwdorp M, et al. Metabolically healthy and unhealthy obese–the 2013 stock conference report. Obes Rev. (2014) 15:697–708. doi: 10.1111/obr.12199
176. Smith GI, Mittendorfer B, Klein S. Metabolically healthy obesity: facts and fantasies. J Clin Invest. (2019) 129:3978–89. doi: 10.1172/JCI129186
177. Naukkarinen J, Heinonen S, Hakkarainen A, Lundbom J, Vuolteenaho K, Saarinen L, et al. Characterising metabolically healthy obesity in weight-discordant monozygotic twins. Diabetologia. (2014) 57:167–76. doi: 10.1007/s00125-013-3066-y
178. Rey-López JP, de Rezende LF, Pastor-Valero M, Tess BH. The prevalence of metabolically healthy obesity: a systematic review and critical evaluation of the definitions used. Obes Rev. (2014) 15:781–90. doi: 10.1111/obr.12198
179. Karelis AD, Faraj M, Bastard JP, St-Pierre DH, Brochu M, Prud'homme D, et al. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. (2005) 90:4145–50. doi: 10.1210/jc.2005-0482
180. Blüher M. The distinction of metabolically ‘healthy' from ‘unhealthy' obese individuals. Curr Opin Lipidol. (2010) 21:38–43. doi: 10.1097/MOL.0b013e3283346ccc
181. Echouffo-Tcheugui JB, Short MI, Xanthakis V, Field P, Sponholtz TR, Larson MG, et al. Natural history of obesity subphenotypes: dynamic changes over two decades and prognosis in the framingham heart study. J Clin Endocrinol Metab. (2019) 104:738–52. doi: 10.1210/jc.2018-01321
182. Mongraw-Chaffin M, Foster MC, Anderson CAM, Burke GL, Haq N, Kalyani RR, et al. Metabolically healthy obesity, transition to metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol. (2018) 71:1857–65. doi: 10.1016/j.jacc.2018.02.055
183. Soriguer F, Gutiérrez-Repiso C, Rubio-Martín E, García-Fuentes E, Almaraz MC, Colomo N, et al. Metabolically healthy but obese, a matter of time? Findings from the prospective pizarra study. J Clin Endocrinol Metab. (2013) 98:2318–25. doi: 10.1210/jc.2012-4253
184. Kouvari M, Panagiotakos DB, Yannakoulia M, Georgousopoulou E, Critselis E, Chrysohoou C, et al. Transition from metabolically benign to metabolically unhealthy obesity and 10-year cardiovascular disease incidence: the attica cohort study. Metabolism. (2019) 93:18–24. doi: 10.1016/j.metabol.2019.01.003
185. Caleyachetty R, Thomas GN, Toulis KA, Mohammed N, Gokhale KM, Balachandran K, et al. Metabolically healthy obese and incident cardiovascular disease events among 3.5 million men and women. J Am Coll Cardiol. (2017) 70:1429–37. doi: 10.1016/j.jacc.2017.07.763
186. Twig G, Afek A, Derazne E, Tzur D, Cukierman-Yaffe T, Gerstein HC, et al. Diabetes risk among overweight and obese metabolically healthy young adults. Diabetes Care. (2014) 37:2989–95. doi: 10.2337/dc14-0869
187. Guo F, Moellering DR, Garvey WT. The progression of cardiometabolic disease: validation of a new cardiometabolic disease staging system applicable to obesity. Obesity. (2014) 22:110–8. doi: 10.1002/oby.20585
188. Johnson W, Bell JA, Robson E, Norris T, Kivimäki M, Hamer M. Do worse baseline risk factors explain the association of healthy obesity with increased mortality risk? Whitehall II study. Int J Obes. (2019) 43:1578–89. doi: 10.1038/s41366-018-0192-0
189. Shin MJ, Hyun YJ, Kim OY, Kim JY, Jang Y, Lee JH. Weight loss effect on inflammation and ldl oxidation in metabolically healthy but obese (mho) individuals: low inflammation and ldl oxidation in mho women. Int J Obes. (2006) 30:1529–34. doi: 10.1038/sj.ijo.0803304
190. Karelis AD, Messier V, Brochu M, Rabasa-Lhoret R. Metabolically healthy but obese women: effect of an energy-restricted diet. Diabetologia. (2008) 51:1752–4. doi: 10.1007/s00125-008-1038-4
191. Kantartzis K, Machann J, Schick F, Rittig K, Machicao F, Fritsche A, et al. Effects of a lifestyle intervention in metabolically benign and malign obesity. Diabetologia. (2011) 54:864–8. doi: 10.1007/s00125-010-2006-3
192. Blüher M. Are metabolically healthy obese individuals really healthy? Eur J Endocrinol. (2014) 171:R209–19. doi: 10.1530/EJE-14-0540
193. Yang X, Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia. (2007) 50:1127–39. doi: 10.1007/s00125-007-0640-1
194. Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. (2014) 2:901–10. doi: 10.1016/S2213-8587(14)70032-4
195. Ruderman NB, Schneider SH, Berchtold P. The “Metabolically-Obese,” Normal-weight individual. Am J Clin Nutr. (1981) 34:1617–21. doi: 10.1093/ajcn/34.8.1617
196. Newell-Morris LL, Treder RP, Shuman WP, Fujimoto WY. Fatness, fat distribution, and glucose tolerance in second-generation Japanese-American (nisei) men. Am J Clin Nutr. (1989) 50:9–18. doi: 10.1093/ajcn/50.1.9
197. Jensen MD. Is visceral fat involved in the pathogenesis of the metabolic syndrome? Human model. Obesity. (2006) 14(Suppl. 1):20S−4S. doi: 10.1038/oby.2006.278
198. Karpe F, Pinnick KE. Biology of upper-body and lower-body adipose tissue–link to whole-body phenotypes. Nat Rev Endocrinol. (2015) 11:90–100. doi: 10.1038/nrendo.2014.185
199. Vega GL, Adams-Huet B, Peshock R, Willett D, Shah B, Grundy SM. Influence of body fat content and distribution on variation in metabolic risk. J Clin Endocrinol Metab. (2006) 91:4459–66. doi: 10.1210/jc.2006-0814
200. Søndergaard E, Gormsen LC, Nellemann B, Jensen MD, Nielsen S. Body composition determines direct ffa storage pattern in overweight women. Am J Physiol Endocrinol Metab. (2012) 302:E1599–604. doi: 10.1152/ajpendo.00015.2012
201. Jensen MD. Role of body fat distribution and the metabolic complications of obesity. J Clin Endocrinol Metab. (2008) 93:S57–63. doi: 10.1210/jc.2008-1585
202. Fu J, Li Y, Esangbedo IC, Li G, Feng D, Li L, et al. Circulating osteonectin and adipokine profiles in relation to metabolically healthy obesity in Chinese children: findings from bcams. J Am Heart Assoc. (2018) 7:e009169. doi: 10.1161/JAHA.118.009169
203. Klöting N, Fasshauer M, Dietrich A, Kovacs P, Schön MR, Kern M, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. (2010) 299:E506–15. doi: 10.1152/ajpendo.00586.2009
204. Straznicky NE, Lambert GW, Masuo K, Dawood T, Eikelis N, Nestel PJ, et al. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am J Clin Nutr. (2009) 89:27–36. doi: 10.3945/ajcn.2008.26299
205. Genelhu VA, Celoria BM, Duarte SF, Cabello PH, Francischetti EA. Not all obese subjects of multiethnic origin are at similar risk for developing hypertension and type 2 diabetes. Eur J Intern Med. (2009) 20:289–95. doi: 10.1016/j.ejim.2008.09.009
206. Ahl S, Guenther M, Zhao S, James R, Marks J, Szabo A, et al. Adiponectin levels differentiate metabolically healthy vs unhealthy among obese and nonobese white individuals. J Clin Endocrinol Metab. (2015) 100:4172–80. doi: 10.1210/jc.2015-2765
207. Weiss R, Taksali SE, Dufour S, Yeckel CW, Papademetris X, Cline G, et al. The “Obese insulin-sensitive” adolescent: importance of adiponectin and lipid partitioning. J Clin Endocrinol Metab. (2005) 90:3731–7. doi: 10.1210/jc.2004-2305
208. Indulekha K, Surendar J, Anjana RM, Geetha L, Gokulakrishnan K, Pradeepa R, et al. Metabolic obesity, adipocytokines, and inflammatory markers in asian indians–cures-124. Diabetes Technol Ther. (2015) 17:134–41. doi: 10.1089/dia.2014.0202
209. Doumatey AP, Bentley AR, Zhou J, Huang H, Adeyemo A, Rotimi CN. Paradoxical hyperadiponectinemia is associated with the metabolically healthy obese (mho) phenotype in African Americans. J Endocrinol Metab. (2012) 2:51–65. doi: 10.4021/jem95w
210. Alizadeh S, Mirzaei K, Mohammadi C, Keshavarz SA, Maghbooli Z. Circulating omentin-1 might be associated with metabolic health status in different phenotypes of body size. Arch Endocrinol Metab. (2017) 61:567–74. doi: 10.1590/2359-3997000000269
211. Bremer AA, Jialal I. Adipose tissue dysfunction in nascent metabolic syndrome. J Obes. (2013) 2013:393192. doi: 10.1155/2013/393192
212. Xu H, Barnes G, Yang Q, Tan G, Yang D, Chou C, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI200319451
213. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI200319246
214. Han CY, Subramanian S, Chan CK, Omer M, Chiba T, Wight TN, et al. Adipocyte-derived serum amyloid a3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes. (2007) 56:2260–73. doi: 10.2337/db07-0218
215. Haase J, Weyer U, Immig K, Klöting N, Blüher M, Eilers J, et al. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia. (2014) 57:562–71. doi: 10.1007/s00125-013-3139-y
216. Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation. (2008) 117:806–15. doi: 10.1161/CIRCULATIONAHA.107.724096
217. Cancello R, Tordjman J, Poitou C, Guilhem G, Bouillot JL, Hugol D, et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes. (2006) 55:1554–61. doi: 10.2337/db06-0133
218. Subramanian S, Han C, Chiba T, McMillen T, Wang S, Haw Ar, et al. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese ldl receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:685–91. doi: 10.1161/ATVBAHA.107.157685
219. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. (2008) 49:1562–8. doi: 10.1194/jlr.M800019-JLR200
220. Harman-Boehm I, Blüher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab. (2007) 92:2240–7. doi: 10.1210/jc.2006-1811
221. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
222. Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, et al. Pro-inflammatory cd11c+cd206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. (2010) 59:1648–56. doi: 10.2337/db09-0287
223. Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. (2007) 56:16–23. doi: 10.2337/db06-1076
224. Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care. (2011) 14:341–6. doi: 10.1097/MCO.0b013e328347970b
225. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. (2014) 20:614–25. doi: 10.1016/j.cmet.2014.08.010
226. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. (2009) 15:921–9. doi: 10.1038/nm.2001
227. O'Rourke RW, Metcalf MD, White AE, Madala A, Winters BR, Maizlin II, et al. Depot-specific differences in inflammatory mediators and a role for nk cells and ifn-gamma in inflammation in human adipose tissue. Int J Obes. (2009) 33:978–90. doi: 10.1038/ijo.2009.133
228. Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, et al. Interferon-gamma, a th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. (2008) 103:467–76. doi: 10.1161/CIRCRESAHA.108.177105
229. Bendelac A, Savage PB, Teyton L. The biology of nkt cells. Annu Rev Immunol. (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
230. Godfrey DI, Stankovic S, Baxter AG. Raising the nkt cell family. Nat Immunol. (2010) 11:197–206. doi: 10.1038/ni.1841
231. Kronenberg M. Toward an understanding of nkt cell biology: progress and paradoxes. Annu Rev Immunol. (2005) 23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742
232. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, et al. Activation of natural killer t cells promotes m2 macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (il-4)/stat6 protein signaling axis in obesity. J Biol Chem. (2012) 287:13561–71. doi: 10.1074/jbc.M112.350066
233. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant nkt cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. (2012) 37:574–87. doi: 10.1016/j.immuni.2012.06.016
234. Lynch L, O'Shea D, Winter DC, Geoghegan J, Doherty DG, O'Farrelly C. Invariant nkt cells and cd1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol. (2009) 39:1893–901. doi: 10.1002/eji.200939349
235. Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer t cells in adipose tissue prevent insulin resistance. J Clin Invest. (2012) 122:3343–54. doi: 10.1172/JCI62739
236. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of t cells and production of pathogenic igg antibodies. Nat Med. (2011) 17:610–7. doi: 10.1038/nm.2353
237. Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. (2009) 15:940–5. doi: 10.1038/nm.1994
238. Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, et al. Adipose tissue dendritic cells are independent contributors to obesity-induced inflammation and insulin resistance. J Immunol. (2016) 197:3650–61. doi: 10.4049/jimmunol.1600820
239. Sundara Rajan S, Longhi MP. Dendritic cells and adipose tissue. Immunology. (2016) 149:353–61. doi: 10.1111/imm.12653
240. Mortensen RF. C-reactive protein, inflammation, and innate immunity. Immunol Res. (2001) 24:163–76. doi: 10.1385/IR:24:2:163
241. Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis. (2000) 148:209–14. doi: 10.1016/S0021-9150(99)00463-3
242. Schmidt-Arras D, Rose-John S. Il-6 pathway in the liver: from physiopathology to therapy. J Hepatol. (2016) 64:1403–15. doi: 10.1016/j.jhep.2016.02.004
243. Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated c-reactive protein levels in overweight and obese adults. JAMA. (1999) 282:2131–5. doi: 10.1001/jama.282.22.2131
244. Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, et al. Acute-phase serum amyloid a: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. (2006) 3:e287. doi: 10.1371/journal.pmed.0030287
245. Uemura H, Katsuura-Kamano S, Yamaguchi M, Bahari T, Ishizu M, Fujioka M, et al. Relationships of serum high-sensitivity c-reactive protein and body size with insulin resistance in a japanese cohort. PLoS ONE. (2017) 12:e0178672. doi: 10.1371/journal.pone.0178672
246. González AS, Guerrero DB, Soto MB, Díaz SP, Martinez-Olmos M, Vidal O. Metabolic syndrome, insulin resistance and the inflammation markers c-reactive protein and ferritin. Eur J Clin Nutr. (2006) 60:802–9. doi: 10.1038/sj.ejcn.1602384
247. Cozlea DL, Farcas DM, Nagy A, Keresztesi AA, Tifrea R, Cozlea L, et al. The impact of c reactive protein on global cardiovascular risk on patients with coronary artery disease. Curr Health Sci J. (2013) 39:225–31.
248. Filippin-Monteiro FB, de Oliveira EM, Sandri S, Knebel FH, Albuquerque RC, Campa A. Serum amyloid a is a growth factor for 3t3-l1 adipocytes, inhibits differentiation and promotes insulin resistance. Int J Obes. (2012) 36:1032–9. doi: 10.1038/ijo.2011.193
249. Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, et al. Serum amyloid a as a predictor of coronary artery disease and cardiovascular outcome in women: The national heart, lung, and blood institute-sponsored women's ischemia syndrome evaluation (wise). Circulation. (2004) 109:726–32. doi: 10.1161/01.CIR.0000115516.54550.B1
250. Targonska-Stepniak B, Majdan M. Serum amyloid a as a marker of persistent inflammation and an indicator of cardiovascular and renal involvement in patients with rheumatoid arthritis. Mediators Inflamm. (2014) 2014:793628. doi: 10.1155/2014/793628
251. Getz GS, Krishack PA, Reardon CA. Serum amyloid a and atherosclerosis. Curr Opin Lipidol. (2016) 27:531–5. doi: 10.1097/MOL.0000000000000331
252. Lewis KE, Kirk EA, McDonald TO, Wang S, Wight TN, O'Brien KD, et al. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. (2004) 110:540–5. doi: 10.1161/01.CIR.0000136819.93989.E1
253. Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid a mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid a function. Proc Natl Acad Sci USA. (1994) 91:3186–90. doi: 10.1073/pnas.91.8.3186
254. Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, et al. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. (2000) 85:3338–42. doi: 10.1210/jcem.85.9.6839
255. Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. (2000) 67:291–300. doi: 10.1016/S0024-3205(00)00622-6
256. Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab. (1997) 82:4196–200. doi: 10.1210/jc.82.12.4196
257. Yeop Han C, Kargi A, Omer M, Chan C, Wabitsch M, O'Brien K, et al. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: dissociation of adipocyte hypertrophy from inflammation. Diabetes. (2010) 59:386–96. doi: 10.2337/db09-0925
258. Zhang W, Mottillo EP, Zhao J, Gartung A, VanHecke GC, Lee JF, et al. Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J Biol Chem. (2014) 289:32178–85. doi: 10.1074/jbc.M114.601096
259. Matsubara T, Mita A, Minami K, Hosooka T, Kitazawa S, Takahashi K, et al. Pgrn is a key adipokine mediating high fat diet-induced insulin resistance and obesity through il-6 in adipose tissue. Cell Metab. (2012) 15:38–50. doi: 10.1016/j.cmet.2011.12.002
260. Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. (1998) 83:847–50. doi: 10.1210/jc.83.3.847
261. Carey DG, Jenkins AB, Campbell LV, Freund J, Chisholm DJ. Abdominal fat and insulin resistance in normal and overweight women: direct measurements reveal a strong relationship in subjects at both low and high risk of niddm. Diabetes. (1996) 45:633–8. doi: 10.2337/diabetes.45.5.633
262. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. (2008) 322:1539–43. doi: 10.1126/science.1160794
263. Rotter V, Nagaev I, Smith U. Interleukin-6 (Il-6) induces insulin resistance in 3t3-l1 adipocytes and is, like il-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. (2003) 278:45777–84. doi: 10.1074/jbc.M301977200
264. Wunderlich FT, Ströhle P, Könner AC, Gruber S, Tovar S, Brönneke HS, et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. (2010) 12:237–49. doi: 10.1016/j.cmet.2010.06.011
265. Braune J, Weyer U, Hobusch C, Mauer J, Brüning JC, Bechmann I, et al. Il-6 regulates m2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol. (2017) 198:2927–34. doi: 10.4049/jimmunol.1600476
266. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
267. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. (2001) 280:E745–51. doi: 10.1152/ajpendo.2001.280.5.E745
268. Zahorska-Markiewicz B, Janowska J, Olszanecka-Glinianowicz M, Zurakowski A. Serum concentrations of tnf-alpha and soluble tnf-alpha receptors in obesity. Int J Obes Relat Metab Disord. (2000) 24:1392–5. doi: 10.1038/sj.ijo.0801398
269. Zahorska-Markiewicz B. Metabolic effects associated with adipose tissue distribution. Adv Med Sci. (2006) 51:111–4.
270. Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, et al. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of irs-1 and irs-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. (1997) 272:29911–8. doi: 10.1074/jbc.272.47.29911
271. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. (2003) 112:1785–8. doi: 10.1172/JCI20514
272. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. Ccr2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. (2006) 116:115–24. doi: 10.1172/JCI24335
273. Christiansen T, Richelsen B, Bruun JM. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes. (2005) 29:146–50. doi: 10.1038/sj.ijo.0802839
274. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. (2006) 116:1494–505. doi: 10.1172/JCI26498
275. Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA. (2003) 100:7265–70. doi: 10.1073/pnas.1133870100
276. Bruun JM, Lihn AS, Pedersen SB, Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (at): implication of macrophages resident in the at. J Clin Endocrinol Metab. (2005) 90:2282–9. doi: 10.1210/jc.2004-1696
277. Poitou C, Viguerie N, Cancello R, De Matteis R, Cinti S, Stich V, et al. Serum amyloid a: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia. (2005) 48:519–28. doi: 10.1007/s00125-004-1654-6
278. Poitou C, Coussieu C, Rouault C, Coupaye M, Cancello R, Bedel JF, et al. Serum amyloid a: a marker of adiposity-induced low-grade inflammation but not of metabolic status. Obesity. (2006) 14:309–18. doi: 10.1038/oby.2006.40
279. Lappalainen T, Kolehmainen M, Schwab U, Pulkkinen L, Laaksonen DE, Rauramaa R, et al. Serum concentrations and expressions of serum amyloid a and leptin in adipose tissue are interrelated: the genobin study. Eur J Endocrinol. (2008) 158:333–41. doi: 10.1530/EJE-07-0598
280. Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. (2004) 92:347–55. doi: 10.1079/BJN20041213
281. Gómez-Ambrosi J, Salvador J, Rotellar F, Silva C, Catalán V, Rodríguez A, et al. Increased serum amyloid a concentrations in morbid obesity decrease after gastric bypass. Obes Surg. (2006) 16:262–9. doi: 10.1381/096089206776116525
282. den Hartigh LJ, Wang S, Goodspeed L, Ding Y, Averill M, Subramanian S, et al. Deletion of serum amyloid a3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS ONE. (2014) 9:e108564. doi: 10.1371/journal.pone.0108564
283. Sjöholm K, Palming J, Olofsson LE, Gummesson A, Svensson PA, Lystig TC, et al. A microarray search for genes predominantly expressed in human omental adipocytes: adipose tissue as a major production site of serum amyloid a. J Clin Endocrinol Metab. (2005) 90:2233–9. doi: 10.1210/jc.2004-1830
284. Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol. (2015) 208:501–12. doi: 10.1083/jcb.201409063
285. Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1h nmr spectroscopy study. Diabetologia. (1999) 42:113–6. doi: 10.1007/s001250051123
286. Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. (2000) 21:697–738. doi: 10.1210/edrv.21.6.0415
287. Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (irs-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. (2002) 277:50230–6. doi: 10.1074/jbc.M200958200
288. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase c, and ikappab-alpha. Diabetes. (2002) 51:2005–11. doi: 10.2337/diabetes.51.7.2005
289. Turinsky J, O'Sullivan DM, Bayly BP. 1,2-diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. (1990) 265:16880–5.
290. Holl WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. (2007) 5:167–79. doi: 10.1016/j.cmet.2007.01.002
291. Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci USA. (2009) 106:15430–5. doi: 10.1073/pnas.0904944106
292. Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. (2008) 134:1369–75. doi: 10.1053/j.gastro.2008.01.075
293. Magkos F, Fabbrini E, Mohammed BS, Patterson BW, Klein S. Increased whole-body adiposity without a concomitant increase in liver fat is not associated with augmented metabolic dysfunction. Obesity. (2010) 18:1510–5. doi: 10.1038/oby.2010.90
294. Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, et al. Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance. Am J Physiol Endocrinol Metab. (2005) 288:E454–61. doi: 10.1152/ajpendo.00203.2004
295. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. (2005) 115:1343–51. doi: 10.1172/JCI23621
296. Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. (2014) 510:84–91. doi: 10.1038/nature13478
297. Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of akt/protein kinase b. J Biol Chem. (2004) 279:36608–15. doi: 10.1074/jbc.M406499200
298. Blouin CM, Prado C, Takane KK, Lasnier F, Garcia-Ocana A, Ferré P, et al. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes. (2010) 59:600–10. doi: 10.2337/db09-0897
299. Xia JY, Holl WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab. (2015) 22:266–78. doi: 10.1016/j.cmet.2015.06.007
300. Liu Q, Yuan B, Lo KA, Patterson HC, Sun Y, Lodish HF. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc Natl Acad Sci USA. (2012) 109:14568–73. doi: 10.1073/pnas.1211611109
301. Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol. (2016) 8:101–9. doi: 10.1093/jmcb/mjw014
302. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating amp-activated protein kinase. Nat Med. (2002) 8:1288–95. doi: 10.1038/nm788
303. Bézaire V, Mairal A, Anesia R, Lefort C, Langin D. Chronic tnfalpha and camp pre-treatment of human adipocytes alter hsl, atgl and perilipin to regulate basal and stimulated lipolysis. FEBS Lett. (2009) 583:3045–9. doi: 10.1016/j.febslet.2009.08.019
304. Laurencikiene J, van Harmelen V, Arvidsson Nordström E, Dicker A, Blomqvist L, Näslund E, et al. Nf-kappab is important for tnf-alpha-induced lipolysis in human adipocytes. J Lipid Res. (2007) 48:1069–77. doi: 10.1194/jlr.M600471-JLR200
305. Gregory SH, Wing EJ. Neutrophil-kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J Leukoc Biol. (2002) 72:239–48.
306. Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol. (2009) 51:212–23. doi: 10.1016/j.jhep.2009.03.008
307. Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. (2014) 5:683. doi: 10.3389/fimmu.2014.00683
308. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 116:1793–801. doi: 10.1172/JCI29069
309. Jager J, Aparicio-Vergara M, Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of kupffer cells. J Intern Med. (2016) 280:209–20. doi: 10.1111/joim.12483
310. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem. (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
311. Gallagher D, Kuznia P, Heshka S, Albu J, Heymsfield SB, Goodpaster B, et al. Adipose tissue in muscle: a novel depot similar in size to visceral adipose tissue. Am J Clin Nutr. (2005) 81:903–10. doi: 10.1093/ajcn/81.4.903
312. Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, et al. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes. (2001) 50:2579–84. doi: 10.2337/diabetes.50.11.2579
313. Sciorati C, Clementi E, Manfredi AA, Rovere-Querini P. Fat deposition and accumulation in the damaged and inflamed skeletal muscle: cellular and molecular players. Cell Mol Life Sci. (2015) 72:2135–56. doi: 10.1007/s00018-015-1857-7
314. Addison O, Marcus RL, Lastayo PC, Ryan AS. Intermuscular fat: a review of the consequences and causes. Int J Endocrinol. (2014) 2014:309570. doi: 10.1155/2014/309570
315. Kim JE, Dunville K, Li J, Cheng JX, Conley TB, Couture CS, et al. Intermuscular adipose tissue content and intramyocellular lipid fatty acid saturation are associated with glucose homeostasis in middle-aged and older adults. Endocrinol Metab. (2017) 32:257–64. doi: 10.3803/EnM.2017.32.2.257
316. Goodpaster BH, Thaete FL, Kelley DE. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. Am J Clin Nutr. (2000) 71:885–92. doi: 10.1093/ajcn/71.4.885
317. Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen EW. Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes. (1997) 46:1768–74. doi: 10.2337/diabetes.46.11.1768
318. Goodpaster BH, Kelley DE. Role of muscle in triglyceride metabolism. Curr Opin Lipidol. (1998) 9:231–6. doi: 10.1097/00041433-199806000-00008
319. Granados A, Gebremariam A, Gidding SS, Terry JG, Carr JJ, Steffen LM, et al. Association of abdominal muscle composition with prediabetes and diabetes: the cardia study. Diabetes Obes Metab. (2019) 21:267–75. doi: 10.1111/dom.13513
320. Haykowsky MJ, Nicklas BJ, Brubaker PH, Hundley WG, Brinkley TE, Upadhya B, et al. Regional adipose distribution and its relationship to exercise intolerance in older obese patients who have heart failure with preserved ejection fraction. JACC Heart Fail. (2018) 6:640–9. doi: 10.1016/j.jchf.2018.06.002
321. Terry JG, Shay CM, Schreiner PJ, Jacobs DR, Sanchez OA, Reis JP, et al. Intermuscular adipose tissue and subclinical coronary artery calcification in midlife: the cardia study (coronary artery risk development in young adults). Arterioscler Thromb Vasc Biol. (2017) 37:2370–8. doi: 10.1161/ATVBAHA.117.309633
322. Yim JE, Heshka S, Albu J, Heymsfield S, Kuznia P, Harris T, et al. Intermuscular adipose tissue rivals visceral adipose tissue in independent associations with cardiovascular risk. Int J Obes. (2007) 31:1400–5. doi: 10.1038/sj.ijo.0803621
323. Ferrara D, Montecucco F, Dallegri F, Carbone F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J Cell Physiol. (2019) 234:21630–41. doi: 10.1002/jcp.28821
324. Yafei S, Elsewy F, Youssef E, Ayman M, Elshafei M, Abayazeed R. Echocardiographic association of epicardial fat with carotid intima-media thickness in patients with type 2 diabetes. Diab Vasc Dis Res. (2019) 16:378–84. doi: 10.1177/1479164119827602
325. Calcaterra V, Cena H, Casali PM, Iacobellis G, Albertini R, De Amici M, et al. Epicardial fat thickness in non-obese neurologically impaired children: association with unfavorable cardiometabolic risk profile. Ann Nutr Metab. (2018) 72:96–103. doi: 10.1159/000484326
326. Rabkin SW. The relationship between epicardial fat and indices of obesity and the metabolic syndrome: a systematic review and meta-analysis. Metab Syndr Relat Disord. (2014) 12:31–42. doi: 10.1089/met.2013.0107
327. Christensen RH, von Scholten BJ, Hansen CS, Jensen MT, Vilsbøll T, Rossing P, et al. Epicardial adipose tissue predicts incident cardiovascular disease and mortality in patients with type 2 diabetes. Cardiovasc Diabetol. (2019) 18:114. doi: 10.1186/s12933-019-0917-y
328. Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. (2007) 101:335–47. doi: 10.1161/CIRCRESAHA.107.150417
329. Iozzo P. Myocardial, perivascular, and epicardial fat. Diabetes Care. (2011) 34(Suppl. 2):S371–9. doi: 10.2337/dc11-s250
330. Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol. (2017) 174:3496–513. doi: 10.1111/bph.13705
331. Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J, Czech MP. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am J Physiol Heart Circ Physiol. (2011) 301:H1425–37. doi: 10.1152/ajpheart.00376.2011
332. Xia N, Weisenburger S, Koch E, Burkart M, Reifenberg G, Förstermann U, et al. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br J Pharmacol. (2017) 174:3443–53. doi: 10.1111/bph.13703
333. Costa RM, Neves KB, Tostes RC, Lobato NS. Perivascular adipose tissue as a relevant fat depot for cardiovascular risk in obesity. Front Physiol. (2018) 9:253. doi: 10.3389/fphys.2018.00253
334. Takaoka M, Nagata D, Kihara S, Shimomura I, Kimura Y, Tabata Y, et al. Periadventitial adipose tissue plays a critical role in vascular remodeling. Circ Res. (2009) 105:906–11. doi: 10.1161/CIRCRESAHA.109.199653
335. Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, Leprince P, et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc Res. (2015) 108:62–73. doi: 10.1093/cvr/cvv208
336. Nasarre L, Juan-Babot O, Gastelurrutia P, Llucia-Valldeperas A, Badimon L, Bayes-Genis A, et al. Low density lipoprotein receptor-related protein 1 is upregulated in epicardial fat from type 2 diabetes mellitus patients and correlates with glucose and triglyceride plasma levels. Acta Diabetol. (2014) 51:23–30. doi: 10.1007/s00592-012-0436-8
337. Uygun A, Kadayifci A, Demirci H, Saglam M, Sakin YS, Ozturk K, et al. The effect of fatty pancreas on serum glucose parameters in patients with nonalcoholic steatohepatitis. Eur J Intern Med. (2015) 26:37–41. doi: 10.1016/j.ejim.2014.11.007
338. Heni M, Machann J, Staiger H, Schwenzer NF, Peter A, Schick F, et al. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: a nuclear magnetic resonance study. Diabetes Metab Res Rev. (2010) 26:200–5. doi: 10.1002/dmrr.1073
339. Steven S, Hollingsworth KG, Small PK, Woodcock SA, Pucci A, Aribisala B, et al. Weight loss decreases excess pancreatic triacylglycerol specifically in type 2 diabetes. Diabetes Care. (2016) 39:158–65. doi: 10.2337/dc15-0750
340. Wong VW, Wong GL, Yeung DK, Abrigo JM, Kong AP, Chan RS, et al. Fatty pancreas, insulin resistance, and β-cell function: a population study using fat-water magnetic resonance imaging. Am J Gastroenterol. (2014) 109:589–97. doi: 10.1038/ajg.2014.1
341. Wang CY, Ou HY, Chen MF, Chang TC, Chang CJ. Enigmatic ectopic fat: prevalence of nonalcoholic fatty pancreas disease and its associated factors in a chinese population. J Am Heart Assoc. (2014) 3:e000297. doi: 10.1161/JAHA.113.000297
342. Pinnick KE, Collins SC, Londos C, Gauguier D, Clark A, Fielding BA. Pancreatic ectopic fat is characterized by adipocyte infiltration and altered lipid composition. Obesity. (2008) 16:522–30. doi: 10.1038/oby.2007.110
343. Navina S, Acharya C, DeLany JP, Orlichenko LS, Baty CJ, Shiva SS, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med. (2011) 3:107ra110. doi: 10.1126/scitranslmed.3002573
344. Noel P, Patel K, Durgampudi C, Trivedi RN, de Oliveira C, Crowell MD, et al. Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut. (2016) 65:100–11. doi: 10.1136/gutjnl-2014-308043
345. Yoneshiro T, Aita S, Matsushita M, Okamatsu-Ogura Y, Kameya T, Kawai Y, et al. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity. (2011) 9:1755–60. doi: 10.1038/oby.2011.125
346. Wang Q, Zhang M, Xu M, Gu W, Xi Y, Qi L, et al. Brown adipose tissue activation is inversely related to central obesity and metabolic parameters in adult human. PLoS ONE. (2015) 10:e0123795. doi: 10.1371/journal.pone.0123795
347. Brendle C, Werner MK, Schmadl M, la Fougère C, Nikolaou K, Stefan N, et al. Correlation of brown adipose tissue with other body fat compartments and patient characteristics: a retrospective analysis in a large patient cohort using pet/ct. Acad Radiol. (2018) 25:102–10. doi: 10.1016/j.acra.2017.09.007
348. Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. (2009) 58:1526–31. doi: 10.2337/db09-0530
349. Matsushita M, Yoneshiro T, Aita S, Kameya T, Sugie H, Saito M. Impact of brown adipose tissue on body fatness and glucose metabolism in healthy humans. Int J Obes. (2014) 38:812–7. doi: 10.1038/ijo.2013.206
350. Hanssen MJ, Hoeks J, Brans B, van der Lans AA, Schaart G, van den Driessche JJ, et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med. (2015) 21:863–5. doi: 10.1038/nm.3891
351. Hanssen MJ, van der Lans AA, Brans B, Hoeks J, Jardon KM, Schaart G, et al. Short-term cold acclimation recruits brown adipose tissue in obese humans. Diabetes. (2015) 65:1179–89. doi: 10.2337/db15-1372
352. Villarroya F, Cereijo R, Gavaldà-Navarro A, Villarroya J, Giralt M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J Intern Med. (2018) 284:492–504. doi: 10.1111/joim.12803
353. Ferré P, Burnol AF, Leturque A, Terretaz J, Penicaud L, Jeanrenaud B, et al. Glucose utilization in vivo and insulin-sensitivity of rat brown adipose tissue in various physiological and pathological conditions. Biochem J. (1986) 233:249–52. doi: 10.1042/bj2330249
354. Orava J, Nuutila P, Noponen T, Parkkola R, Viljanen T, Enerbäck S, et al. Blunted metabolic responses to cold and insulin stimulation in brown adipose tissue of obese humans. Obesity. (2013) 21:2279–87. doi: 10.1002/oby.20456
355. Nedergaard J, Bengtsson T, Cannon B. New powers of brown fat: fighting the metabolic syndrome. Cell Metab. (2011) 13:238–40. doi: 10.1016/j.cmet.2011.02.009
356. Moonen MPB, Nascimento EBM, van Marken Lichtenbelt WD. Human brown adipose tissue: underestimated target in metabolic disease? Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:104–12. doi: 10.1016/j.bbalip.2018.05.012
357. Ouellet V, Labbé SM, Blondin DP, Phoenix S, Guérin B, Haman F, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest. (2012) 122:545–52. doi: 10.1172/JCI60433
358. Scheele C, Nielsen S. Metabolic regulation and the anti-obesity perspectives of human brown fat. Redox Biol. (2017) 12:770–5. doi: 10.1016/j.redox.2017.04.011
359. Sakamoto T, Nitta T, Maruno K, Yeh YS, Kuwata H, Tomita K, et al. Macrophage infiltration into obese adipose tissues suppresses the induction of ucp1 level in mice. Am J Physiol Endocrinol Metab. (2016) 310:E676–87. doi: 10.1152/ajpendo.00028.2015
360. Roberts-Toler C, O'Neill BT, Cypess AM. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity. (2015) 23:1765–70. doi: 10.1002/oby.21134
361. Bae J, Ricciardi CJ, Esposito D, Komarnytsky S, Hu P, Curry BJ, et al. Activation of pattern recognition receptors in brown adipocytes induces inflammation and suppresses uncoupling protein 1 expression and mitochondrial respiration. Am J Physiol Cell Physiol. (2014) 306:C918–30. doi: 10.1152/ajpcell.00249.2013
362. Martins FF, Bargut TCL, Aguila MB, Mandarim-de-Lacerda CA. Thermogenesis, fatty acid synthesis with oxidation, and inflammation in the brown adipose tissue of ob/ob (-/-) mice. Ann Anat. (2017) 210:44–51. doi: 10.1016/j.aanat.2016.11.013
363. Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase ikkepsilon regulates energy balance in obese mice. Cell. (2009) 138:961–75. doi: 10.1016/j.cell.2009.06.046
364. Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, et al. Irf3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest. (2016) 126:2839–54. doi: 10.1172/JCI86080
365. Estève D, Boulet N, Volat F, Zakaroff-Girard A, Ledoux S, Coupaye M, et al. Human white and brite adipogenesis is supported by msca1 and is impaired by immune cells. Stem Cells. (2015) 33:1277–91. doi: 10.1002/stem.1916
366. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. (2006) 444:840–6. doi: 10.1038/nature05482
367. Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, Kratz M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr Physiol. (2018) 9:1–58. doi: 10.1002/cphy.c170040
368. Raji A, Seely EW, Arky RA, Simonson DC. Body fat distribution and insulin resistance in healthy asian indians and caucasians. J Clin Endocrinol Metab. (2001) 86:5366–71. doi: 10.1210/jcem.86.11.7992
369. Paradisi G, Smith L, Burtner C, Leaming R, Garvey WT, Hook G, et al. Dual energy x-ray absorptiometry assessment of fat mass distribution and its association with the insulin resistance syndrome. Diabetes Care. (1999) 22:1310–7. doi: 10.2337/diacare.22.8.1310
370. Mittendorfer B, Magkos F, Fabbrini E, Mohammed BS, Klein S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity. (2009) 17:1872–7. doi: 10.1038/oby.2009.224
371. Björntorp P, Bergman H, Varnauskas E. Plasma free fatty acid turnover rate in obesity. Acta Med Scand. (1969) 185:351–6. doi: 10.1111/j.0954-6820.1969.tb07347.x
372. Jensen MD, Haymond MW, Rizza RA, Cryer PE, Miles JM. Influence of body fat distribution on free fatty acid metabolism in obesity. J Clin Invest. (1989) 83:1168–73. doi: 10.1172/JCI113997
373. Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with niddm. Diabetes. (1988) 37:1020–4. doi: 10.2337/diabetes.37.8.1020
374. Ferrannini E, Camastra S, Coppack SW, Fliser D, Golay A, Mitrakou A. Insulin action and non-esterified fatty acids. The European group for the study of insulin resistance (egir). Proc Nutr Soc. (1997) 56:753–61. doi: 10.1079/PNS19970076
375. Succurro E, Marini MA, Frontoni S, Hribal ML, Andreozzi F, Lauro R, et al. Insulin secretion in metabolically obese, but normal weight, and in metabolically healthy but obese individuals. Obesity. (2008) 16:1881–6. doi: 10.1038/oby.2008.308
376. Swarbrick MM, Havel PJ. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab Syndr Relat Disord. (2008) 6:87–102. doi: 10.1089/met.2007.0029
377. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. (2001) 7:941–6. doi: 10.1038/90984
378. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. (1999) 257:79–83. doi: 10.1006/bbrc.1999.0255
379. Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. (2000) 20:1595–9. doi: 10.1161/01.ATV.20.6.1595
380. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. (2001) 86:1930–5. doi: 10.1210/jcem.86.5.7463
381. Bobbert T, Schwarz F, Fischer-Rosinsky A, Pfeiffer AF, Möhlig M, Mai K, et al. Fibroblast growth factor 21 predicts the metabolic syndrome and type 2 diabetes in caucasians. Diabetes Care. (2013) 36:145–9. doi: 10.2337/dc12-0703
382. Markan KR, Potthoff MJ. Metabolic fibroblast growth factors (fgfs): mediators of energy homeostasis. Semin Cell Dev Biol. (2016) 53:85–93. doi: 10.1016/j.semcdb.2015.09.021
383. Li H, Wu G, Fang Q, Zhang M, Hui X, Sheng B, et al. Fibroblast growth factor 21 increases insulin sensitivity through specific expansion of subcutaneous fat. Nat Commun. (2018) 9:272. doi: 10.1038/s41467-017-02677-9
384. Hale C, Chen MM, Stanislaus S, Chinookoswong N, Hager T, Wang M, et al. Lack of overt fgf21 resistance in two mouse models of obesity and insulin resistance. Endocrinology. (2012) 153:69–80. doi: 10.1210/en.2010-1262
385. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. Fgf-21 as a novel metabolic regulator. J Clin Invest. (2005) 115:1627–35. doi: 10.1172/JCI23606
386. Markan KR. Defining “Fgf21 resistance” during obesity: controversy, criteria and unresolved questions. F1000Res. (2018) 7:289. doi: 10.12688/f1000research.14117.1
387. Gustafson B, Hedjazifar S, Gogg S, Hammarstedt A, Smith U. Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab. (2015) 26:193–200. doi: 10.1016/j.tem.2015.01.006
388. Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. (2019) 20:242–58. doi: 10.1038/s41580-018-0093-z
389. Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci. (2010) 67:1277–92. doi: 10.1007/s00018-010-0263-4
390. Huber J, Löffler M, Bilban M, Reimers M, Kadl A, Todoric J, et al. Prevention of high-fat diet-induced adipose tissue remodeling in obese diabetic mice by n-3 polyunsaturated fatty acids. Int J Obes. (2007) 31:1004–13. doi: 10.1038/sj.ijo.0803511
391. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen vi. Mol Cell Biol. (2009) 29:1575–91. doi: 10.1128/MCB.01300-08
392. Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE, Peterson CA, et al. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab. (2011) 96:E1990–8. doi: 10.1210/jc.2011-1567
393. Roedig H, Nastase MV, Wygrecka M, Schaefer L. Breaking down chronic inflammatory diseases: the role of biglycan in promoting a switch between inflammation and autophagy. FEBS J. (2019) 286:2965–79. doi: 10.1111/febs.14791
394. Wight TN, Kang I, Merrilees MJ. Versican and the control of inflammation. Matrix Biol. (2014) 35:152–61. doi: 10.1016/j.matbio.2014.01.015
395. Sun K, Park J, Gupta OT, Holl WL, Auerbach P, Zhang N, et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun. (2014) 5:3485. doi: 10.1038/ncomms4485
396. Leibel RL, Edens NK, Fried SK. Physiologic basis for the control of body fat distribution in humans. Annu Rev Nutr. (1989) 9:417–43. doi: 10.1146/annurev.nu.09.070189.002221
397. Bonora E. Relationship between regional fat distribution and insulin resistance. Int J Obes Relat Metab Disord. (2000) 24(Suppl. 2):S32–5. doi: 10.1038/sj.ijo.0801274
398. Araneta MR, Barrett-Connor E. Ethnic differences in visceral adipose tissue and type 2 diabetes: filipino, African-American, and white women. Obes Res. (2005) 13:1458–65. doi: 10.1038/oby.2005.176
399. Kotronen A, Yki-Järvinen H, Sevastianova K, Bergholm R, Hakkarainen A, Pietiläinen KH, et al. Comparison of the relative contributions of intra-abdominal and liver fat to components of the metabolic syndrome. Obesity. (2011) 19:23–8. doi: 10.1038/oby.2010.137
400. Blüher M. Are there still healthy obese patients? Curr Opin Endocrinol Diabetes Obes. (2012) 19:341–6. doi: 10.1097/MED.0b013e328357f0a3
401. Thomas EL, Parkinson JR, Frost GS, Goldstone AP, Doré CJ, McCarthy JP, et al. The missing risk: mri and mrs phenotyping of abdominal adiposity and ectopic fat. Obesity. (2012) 20:76–87. doi: 10.1038/oby.2011.142
402. Zhang M, Hu T, Zhang S, Zhou L. Associations of different adipose tissue depots with insulin resistance: a systematic review and meta-analysis of observational studies. Sci Rep. (2015) 5:18495. doi: 10.1038/srep18495
403. Lopes HF, Corrêa-Giannella ML, Consolim-Colombo FM, Egan BM. Visceral adiposity syndrome. Diabetol Metab Syndr. (2016) 8:40. doi: 10.1186/s13098-016-0156-2
404. Manolopoulos KN, Karpe F, Frayn KN. Gluteofemoral body fat as a determinant of metabolic health. Int J Obes. (2010) 34:949–59. doi: 10.1038/ijo.2009.286
405. Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, et al. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. (2013) 17:644–56. doi: 10.1016/j.cmet.2013.03.008
406. McLaughlin T, Lamendola C, Liu A, Abbasi F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J Clin Endocrinol Metab. (2011) 96:E1756–60. doi: 10.1210/jc.2011-0615
407. Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, et al. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. (2004) 350:2549–57. doi: 10.1056/NEJMoa033179
408. Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. (2008) 7:410–20. doi: 10.1016/j.cmet.2008.04.004
409. Liu X, Zheng Z, Zhu X, Meng M, Li L, Shen Y, et al. Brown adipose tissue transplantation improves whole-body energy metabolism. Cell Res. (2013) 23:851–4. doi: 10.1038/cr.2013.64
410. Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. (2013) 123:215–23. doi: 10.1172/JCI62308
411. Zhu Z, Spicer EG, Gavini CK, Goudjo-Ako AJ, Novak CM, Shi H. Enhanced sympathetic activity in mice with brown adipose tissue transplantation (transbatation). Physiol Behav. (2014) 125:21–9. doi: 10.1016/j.physbeh.2013.11.008
412. Ma SW, Foster DO. Uptake of glucose and release of fatty acids and glycerol by rat brown adipose tissue in vivo. Can J Physiol Pharmacol. (1986) 64:609–14. doi: 10.1139/y86-101
413. Loyd C, Obici S. Brown fat fuel use and regulation of energy homeostasis. Curr Opin Clin Nutr Metab Care. (2014) 17:368–72. doi: 10.1097/MCO.0000000000000063
414. Lee P, Greenfield JR, Ho KK, Fulham MJ. A critical appraisal of the prevalence and metabolic significance of brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. (2010) 299:E601–6. doi: 10.1152/ajpendo.00298.2010
415. Orava J, Nuutila P, Lidell ME, Oikonen V, Noponen T, Viljanen T, et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. (2011) 14:272–9. doi: 10.1016/j.cmet.2011.06.012
416. Dallner OS, Chernogubova E, Brolinson KA, Bengtsson T. Beta3-adrenergic receptors stimulate glucose uptake in brown adipocytes by two mechanisms independently of glucose transporter 4 translocation. Endocrinology. (2006) 147:5730–9. doi: 10.1210/en.2006-0242
417. Mössenböck K, Vegiopoulos A, Rose AJ, Sijmonsma TP, Herzig S, Schafmeier T. Browning of white adipose tissue uncouples glucose uptake from insulin signaling. PLoS ONE. (2014) 9:e110428. doi: 10.1371/journal.pone.0110428
418. Olsen JM, Sato M, Dallner OS, Sandström AL, Pisani DF, Chambard JC, et al. Glucose uptake in brown fat cells is dependent on mtor complex 2-promoted glut1 translocation. J Cell Biol. (2014) 207:365–74. doi: 10.1083/jcb.201403080
419. Peirce V, Vidal-Puig A. Regulation of glucose homoeostasis by brown adipose tissue. Lancet Diabetes Endocrinol. (2013) 1:353–60. doi: 10.1016/S2213-8587(13)70055-X
420. Kaisanlahti A, Glumoff T. Browning of white fat: agents and implications for beige adipose tissue to type 2 diabetes. J Physiol Biochem. (2019) 75:1–10. doi: 10.1007/s13105-018-0658-5
421. Chondronikola M, Volpi E, Børsheim E, Porter C, Annamalai P, Enerbäck S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. (2014) 63:4089–99. doi: 10.2337/db14-0746
422. Eckel RH, Krauss RM. American heart association call to action: obesity as a major risk factor for coronary heart disease. Aha nutrition committee. Circulation. (1998) 97:2099–100. doi: 10.1161/01.CIR.97.21.2099
423. Stefan N, Häring HU, Schulze MB. Metabolically healthy obesity: the low-hanging fruit in obesity treatment? Lancet Diabetes Endocrinol. (2018) 6:249–58. doi: 10.1016/S2213-8587(17)30292-9
424. Wang ZJ, Zhou YJ, Galper BZ, Gao F, Yeh RW, Mauri L. Association of body mass index with mortality and cardiovascular events for patients with coronary artery disease: a systematic review and meta-analysis. Heart. (2015) 101:1631–8. doi: 10.1136/heartjnl-2014-307119
425. Horwich TB, Fonarow GC, Clark AL. Obesity and the obesity paradox in heart failure. Prog Cardiovasc Dis. (2018) 61:151–6. doi: 10.1016/j.pcad.2018.05.005
426. Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol. (2009) 53:1925–32. doi: 10.1016/j.jacc.2008.12.068
427. St-Pierre J, Lemieux I, Vohl MC, Perron P, Tremblay G, Després JP, et al. Contribution of abdominal obesity and hypertriglyceridemia to impaired fasting glucose and coronary artery disease. Am J Cardiol. (2002) 90:15–8. doi: 10.1016/S0002-9149(02)02378-0
428. Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C, Association AH, et al. Definition of metabolic syndrome: Report of the national heart, lung, and blood institute/American heart association conference on scientific issues related to definition. Circulation. (2004) 109:433–8. doi: 10.1161/01.CIR.0000111245.75752.C6
429. Haffner SM. Relationship of metabolic risk factors and development of cardiovascular disease and diabetes. Obesity. (2006) 14(Suppl. 3):121S−7S. doi: 10.1038/oby.2006.291
430. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; American heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation. (2009) 120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644
431. Paolisso G, Tataranni PA, Foley JE, Bogardus C, Howard BV, Ravussin E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of niddm. Diabetologia. (1995) 38:1213–7. doi: 10.1007/s001250050414
432. Santomauro AT, Boden G, Silva ME, Rocha DM, Santos RF, Ursich MJ, et al. Overnight lowering of free fatty acids with acipimox improves insulin resistance and glucose tolerance in obese diabetic and nondiabetic subjects. Diabetes. (1999) 48:1836–41. doi: 10.2337/diabetes.48.9.1836
433. Dutheil F, Gordon BA, Naughton G, Crendal E, Courteix D, Chaplais E, et al. Cardiovascular risk of adipokines: a review. J Int Med Res. (2018) 46:2082–95. doi: 10.1177/0300060517706578
434. Matsuzawa Y. The metabolic syndrome and adipocytokines. FEBS Lett. (2006) 580:2917–21. doi: 10.1016/j.febslet.2006.04.028
435. Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res. (2007) 74:11–8. doi: 10.1016/j.cardiores.2006.10.009
436. Qiao L, Zou C, van der Westhuyzen DR, Shao J. Adiponectin reduces plasma triglyceride by increasing vldl triglyceride catabolism. Diabetes. (2008) 57:1824–33. doi: 10.2337/db07-0435
437. Aldhahi W, Hamdy O. Adipokines, inflammation, and the endothelium in diabetes. Curr Diab Rep. (2003) 3:293–8. doi: 10.1007/s11892-003-0020-2
438. Yuhki K, Kawabe J, Ushikubi F. Fat, keeping the heart healthy? Nat Med. (2005) 11:1048–9. doi: 10.1038/nm1005-1048
439. Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. (2003) 278:45021–6. doi: 10.1074/jbc.M307878200
440. Du Y, Li R, Lau WB, Zhao J, Lopez B, Christopher TA, et al. Adiponectin at physiologically relevant concentrations enhances the vasorelaxative effect of acetylcholine via cav-1/adipor-1 signaling. PLoS ONE. (2016) 11:e0152247. doi: 10.1371/journal.pone.0152247
441. Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol. (2003) 24:225–53. doi: 10.1016/j.yfrne.2003.10.001
442. Söderberg S, Ahrén B, Stegmayr B, Johnson O, Wiklund PG, Weinehall L, et al. Leptin is a risk marker for first-ever hemorrhagic stroke in a population-based cohort. Stroke. (1999) 30:328–37. doi: 10.1161/01.STR.30.2.328
443. Söderberg S, Ahrén B, Jansson JH, Johnson O, Hallmans G, Asplund K, et al. Leptin is associated with increased risk of myocardial infarction. J Intern Med. (1999) 246:409–18. doi: 10.1046/j.1365-2796.1999.00571.x
444. Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A, et al. Plasma leptin and the risk of cardiovascular disease in the west of scotland coronary prevention study (woscops). Circulation. (2001) 104:3052–6. doi: 10.1161/hc5001.101061
445. Schulze PC, Kratzsch J, Linke A, Schoene N, Adams V, Gielen S, et al. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. Eur J Heart Fail. (2003) 5:33–40. doi: 10.1016/S1388-9842(02)00177-0
446. Paolisso G, Tagliamonte MR, Galderisi M, Zito GA, Petrocelli A, Carella C, et al. Plasma leptin level is associated with myocardial wall thickness in hypertensive insulin-resistant men. Hypertension. (1999) 34:1047–52. doi: 10.1161/01.HYP.34.5.1047
447. Lehrke M, Reilly MP, Millington SC, Iqbal N, Rader DJ, Lazar MA. An inflammatory cascade leading to hyperresistinemia in humans. PLoS Med. (2004) 1:e45. doi: 10.1371/journal.pmed.0010045
448. Verma S, Li SH, Wang CH, Fedak PW, Li RK, Weisel RD, et al. Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation. (2003) 108:736–40. doi: 10.1161/01.CIR.0000084503.91330.49
449. Calabro P, Samudio I, Willerson JT, Yeh ET. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. (2004) 110:3335–40. doi: 10.1161/01.CIR.0000147825.97879.E7
450. Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. (2005) 111:932–9. doi: 10.1161/01.CIR.0000155620.10387.43
451. Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. (1999) 100:2473–6. doi: 10.1161/01.CIR.100.25.2473
452. Fatkhullina AR, Peshkova IO, Koltsova EK. The role of cytokines in the development of atherosclerosis. Biochemistry (Mosc). (2016) 81:1358–70. doi: 10.1134/S0006297916110134
453. Burstein SA, Peng J, Friese P, Wolf RF, Harrison P, Downs T, et al. Cytokine-induced alteration of platelet and hemostatic function. Stem Cells. (1996) 14(Suppl. 1):154–62. doi: 10.1002/stem.5530140720
454. Bhagat K, Vallance P. Inflammatory cytokines impair endothelium-dependent dilatation in human veins in vivo. Circulation. (1997) 96:3042–7. doi: 10.1161/01.CIR.96.9.3042
455. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. (2000) 342:836–43. doi: 10.1056/NEJM200003233421202
456. Ong KL, Januszewski AS, O'Connell R, Jenkins AJ, Xu A, Sullivan DR, et al. The relationship of fibroblast growth factor 21 with cardiovascular outcome events in the fenofibrate intervention and event lowering in diabetes study. Diabetologia. (2015) 58:464–73. doi: 10.1007/s00125-014-3458-7
457. Ong KL, McClell RL, Allison MA, Kokkinos J, Wu BJ, Barter PJ, et al. Association of elevated circulating fibroblast growth factor 21 levels with prevalent and incident metabolic syndrome: the multi-ethnic study of atherosclerosis. Atherosclerosis. (2018) 281:200–6 doi: 10.1016/j.atherosclerosis.2018.10.011
458. Domouzoglou EM, Naka KK, Vlahos AP, Papafaklis MI, Michalis LK, Tsatsoulis A, et al. Fibroblast growth factors in cardiovascular disease: the emerging role of fgf21. Am J Physiol Heart Circ Physiol. (2015) 309:H1029–38. doi: 10.1152/ajpheart.00527.2015
459. Lin Z, Wu Z, Yin X, Liu Y, Yan X, Lin S, et al. Serum levels of fgf-21 are increased in coronary heart disease patients and are independently associated with adverse lipid profile. PLoS ONE. (2010) 5:e15534. doi: 10.1371/journal.pone.0015534
460. Shen Y, Ma X, Zhou J, Pan X, Hao Y, Zhou M, et al. Additive relationship between serum fibroblast growth factor 21 level and coronary artery disease. Cardiovasc Diabetol. (2013) 12:124. doi: 10.1186/1475-2840-12-124
461. Ebert T, Gebhardt C, Scholz M, Wohl T, Schleinitz D, Fasshauer M, et al. Relationship between 12 adipocytokines and distinct components of the metabolic syndrome. J Clin Endocrinol Metab. (2018) 103:1015–23. doi: 10.1210/jc.2017-02085
462. Lee Y, Lim S, Hong ES, Kim JH, Moon MK, Chun EJ, et al. Serum fgf21 concentration is associated with hypertriglyceridaemia, hyperinsulinaemia and pericardial fat accumulation, independently of obesity, but not with current coronary artery status. Clin Endocrinol. (2014) 80:57–64. doi: 10.1111/cen.12134
463. Kharitonenkov A, Wroblewski VJ, Koester A, Chen YF, Clutinger CK, Tigno XT, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. (2007) 148:774–81. doi: 10.1210/en.2006-1168
464. Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holl WL, et al. The effects of ly2405319, an fgf21 analog, in obese human subjects with type 2 diabetes. Cell Metab. (2013) 18:333–40. doi: 10.1016/j.cmet.2013.08.005
465. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. (2005) 96:939–49. doi: 10.1161/01.RES.0000163635.62927.34
466. Forouzandeh F, Chang SM, Muhyieddeen K, Zaid RR, Trevino AR, Xu J, et al. Does quantifying epicardial and intrathoracic fat with noncontrast computed tomography improve risk stratification beyond calcium scoring alone? Circ Cardiovasc Imaging. (2013) 6:58–66. doi: 10.1161/CIRCIMAGING.112.976316
467. Iacobellis G, Leonetti F. Epicardial adipose tissue and insulin resistance in obese subjects. J Clin Endocrinol Metab. (2005) 90:6300–2. doi: 10.1210/jc.2005-1087
468. Iacobellis G, Gao YJ, Sharma AM. Do cardiac and perivascular adipose tissue play a role in atherosclerosis? Curr Diab Rep. (2008) 8:20–4. doi: 10.1007/s11892-008-0005-2
469. Verhagen SN, Vink A, van der Graaf Y, Visseren FL. Coronary perivascular adipose tissue characteristics are related to atherosclerotic plaque size and composition. A post-mortem study. Atherosclerosis. (2012) 225:99–104. doi: 10.1016/j.atherosclerosis.2012.08.031
470. Fox CS, Massaro JM, Schlett CL, Lehman SJ, Meigs JB, O'Donnell CJ, et al. Periaortic fat deposition is associated with peripheral arterial disease: the framingham heart study. Circ Cardiovasc Imaging. (2010) 3:515–9. doi: 10.1161/CIRCIMAGING.110.958884
471. Iacobellis G, Ribaudo MC, Assael F, Vecci E, Tiberti C, Zappaterreno A, et al. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: a new indicator of cardiovascular risk. J Clin Endocrinol Metab. (2003) 88:5163–8. doi: 10.1210/jc.2003-030698
472. Mookadam F, Goel R, Alharthi MS, Jiamsripong P, Cha S. Epicardial fat and its association with cardiovascular risk: a cross-sectional observational study. Heart Views. (2010) 11:103–8. doi: 10.4103/1995-705X.76801
473. Nelson MR, Mookadam F, Thota V, Emani U, Al Harthi M, Lester SJ, et al. Epicardial fat: an additional measurement for subclinical atherosclerosis and cardiovascular risk stratification? J Am Soc Echocardiogr. (2011) 24:339–45. doi: 10.1016/j.echo.2010.11.008
474. Gorter PM, de Vos AM, van der Graaf Y, Stella PR, Doevendans PA, Meijs MF, et al. Relation of epicardial and pericoronary fat to coronary atherosclerosis and coronary artery calcium in patients undergoing coronary angiography. Am J Cardiol. (2008) 102:380–5. doi: 10.1016/j.amjcard.2008.04.002
475. Mahabadi AA, Reinsch N, Lehmann N, Altenbernd J, Kälsch H, Seibel RM, et al. Association of pericoronary fat volume with atherosclerotic plaque burden in the underlying coronary artery: a segment analysis. Atherosclerosis. (2010) 211:195–9. doi: 10.1016/j.atherosclerosis.2010.02.013
476. Lim S, Meigs JB. Links between ectopic fat and vascular disease in humans. Arterioscler Thromb Vasc Biol. (2014) 34:1820–6. doi: 10.1161/ATVBAHA.114.303035
477. Verhagen SN, Visseren FL. Perivascular adipose tissue as a cause of atherosclerosis. Atherosclerosis. (2011) 214:3–10. doi: 10.1016/j.atherosclerosis.2010.05.034
478. Yudkin JS, Eringa E, Stehouwer CD. “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. (2005) 365:1817–20. doi: 10.1016/S0140-6736(05)66585-3
479. Sun X, Hou N, Han F, Guo Y, Hui Z, Du G, et al. Effect of high free fatty acids on the anti-contractile response of perivascular adipose tissue in rat aorta. J Mol Cell Cardiol. (2013) 63:169–74. doi: 10.1016/j.yjmcc.2013.07.018
480. Sacks HS, Fain JN. Human epicardial fat: what is new and what is missing? Clin Exp Pharmacol Physiol. (2011) 38:879–87. doi: 10.1111/j.1440-1681.2011.05601.x
481. Kralova Lesna I, Tonar Z, Malek I, Maluskova J, Nedorost L, Pirk J, et al. Is the amount of coronary perivascular fat related to atherosclerosis? Physiol Res. (2015) 64(Suppl. 3):S435–43.
482. Ketonen J, Shi J, Martonen E, Mervaala E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese c57bl/6 mice. Circ J. (2010) 74:1479–87. doi: 10.1253/circj.CJ-09-0661
483. Spiroglou SG, Kostopoulos CG, Varakis JN, Papadaki HH. Adipokines in periaortic and epicardial adipose tissue: differential expression and relation to atherosclerosis. J Atheroscler Thromb. (2010) 17:115–30. doi: 10.5551/jat.1735
484. Arangalage D, Mathieu T, Nguyen V, Cimadevilla C, Kerneis C, Duval X, et al. Epicardial adipose tissue volume is associated with left ventricular remodelling in calcific aortic valve stenosis. Arch Cardiovasc Dis. (2019) 112:594–603. doi: 10.1016/j.acvd.2019.06.005
485. Fernández-Alfonso MS, Gil-Ortega M, García-Prieto CF, Aranguez I, Ruiz-Gayo M, Somoza B. Mechanisms of perivascular adipose tissue dysfunction in obesity. Int J Endocrinol. (2013) 2013:402053. doi: 10.1155/2013/402053
486. Berbée JF, Boon MR, Khedoe PP, Bartelt A, Schlein C, Worthmann A, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun. (2015) 6:6356. doi: 10.1038/ncomms7356
487. Hung CM, Calejman CM, Sanchez-Gurmaches J, Li H, Clish CB, Hettmer S, et al. Rictor/mtorc2 loss in the myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep. (2014) 8:256–71. doi: 10.1016/j.celrep.2014.06.007
488. Bray GA, Heisel WE, Afshin A, Jensen MD, Dietz WH, Long M, et al. The science of obesity management: an endocrine society scientific statement. Endocr Rev. (2018) 39:79–132. doi: 10.1210/er.2017-00253
489. Clifton PM, Keogh JB. Effects of different weight loss approaches on cvd risk. Curr Atheroscler Rep. (2018) 20:27. doi: 10.1007/s11883-018-0728-8
490. Harrington M, Gibson S, Cottrell RC. A review and meta-analysis of the effect of weight loss on all-cause mortality risk. Nutr Res Rev. (2009) 22:93–108. doi: 10.1017/S0954422409990035
491. Ross R. Effects of diet- and exercise-induced weight loss on visceral adipose tissue in men and women. Sports Med. (1997) 24:55–64. doi: 10.2165/00007256-199724010-00005
492. van Gemert WA, Peeters PH, May AM, Doornbos AJH, Elias SG, van der Palen J, et al. Effect of diet with or without exercise on abdominal fat in postmenopausal women - a randomised trial. BMC Public Health. (2019) 19:174. doi: 10.1186/s12889-019-6510-1
493. Borges JH, Carter SJ, Bryan DR, Hunter GR. Exercise training and/or diet on reduction of intra-abdominal adipose tissue and risk factors for cardiovascular disease. Eur J Clin Nutr. (2019) 73:1063–8. doi: 10.1038/s41430-018-0318-4
494. Ross R, Dagnone D, Jones PJ, Smith H, Paddags A, Hudson R, et al. Reduction in obesity and related comorbid conditions after diet-induced weight loss or exercise-induced weight loss in men. A randomized, controlled trial. Ann Intern Med. (2000) 133:92–103. doi: 10.7326/0003-4819-133-2-200007180-00008
495. Janssen I, Ross R. Effects of sex on the change in visceral, subcutaneous adipose tissue and skeletal muscle in response to weight loss. Int J Obes Relat Metab Disord. (1999) 23:1035–46. doi: 10.1038/sj.ijo.0801038
496. Ryan AS, Ge S, Blumenthal JB, Serra MC, Prior SJ, Goldberg AP. Aerobic exercise and weight loss reduce vascular markers of inflammation and improve insulin sensitivity in obese women. J Am Geriatr Soc. (2014) 62:607–14. doi: 10.1111/jgs.12749
497. Janssen I, Fortier A, Hudson R, Ross R. Effects of an energy-restrictive diet with or without exercise on abdominal fat, intermuscular fat, and metabolic risk factors in obese women. Diabetes Care. (2002) 25:431–8. doi: 10.2337/diacare.25.3.431
498. Gregg EW, Jakicic JM, Blackburn G, Bloomquist P, Bray GA, Clark JM, et al. Association of the magnitude of weight loss and changes in physical fitness with long-term cardiovascular disease outcomes in overweight or obese people with type 2 diabetes: a post-hoc analysis of the look ahead randomised clinical trial. Lancet Diabetes Endocrinol. (2016) 4:913–21. doi: 10.1016/S2213-8587(16)30162-0
499. Baum A, Scarpa J, Bruzelius E, Tamler R, Basu S, Faghmous J. Targeting weight loss interventions to reduce cardiovascular complications of type 2 diabetes: a machine learning-based post-hoc analysis of heterogeneous treatment effects in the look ahead trial. Lancet Diabetes Endocrinol. (2017) 5:808–15. doi: 10.1016/S2213-8587(17)30176-6
500. Wing RR, Group LAR. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four-year results of the look ahead trial. Arch Intern Med. (2010) 170:1566–75. doi: 10.1001/archinternmed.2010.334
501. Després JP, Bouchard C, Savard R, Tremblay A, Allard C. Lack of relationship between changes in adiposity and plasma lipids following endurance training. Atherosclerosis. (1985) 54:135–43. doi: 10.1016/0021-9150(85)90173-X
502. Simonsen L, Henriksen O, Enevoldsen LH, Bülow J. The effect of exercise on regional adipose tissue and splanchnic lipid metabolism in overweight type 2 diabetic subjects. Diabetologia. (2004) 47:652–9. doi: 10.1007/s00125-004-1374-y
503. Klein S, Coyle EF, Wolfe RR. Fat metabolism during low-intensity exercise in endurance-trained and untrained men. Am J Physiol. (1994) 267:E934–40. doi: 10.1152/ajpendo.1994.267.6.E934
504. Gollisch KS, Brandauer J, Jessen N, Toyoda T, Nayer A, Hirshman MF, et al. Effects of exercise training on subcutaneous and visceral adipose tissue in normal- and high-fat diet-fed rats. Am J Physiol Endocrinol Metab. (2009) 297:E495–504. doi: 10.1152/ajpendo.90424.2008
505. Poirier P, Després JP. Exercise in weight management of obesity. Cardiol Clin. (2001) 19:459–70. doi: 10.1016/S0733-8651(05)70229-0
506. O'Donovan G, Thomas EL, McCarthy JP, Fitzpatrick J, Durighel G, Mehta S, et al. Fat distribution in men of different waist girth, fitness level and exercise habit. Int J Obes. (2009) 33:1356–62. doi: 10.1038/ijo.2009.189
507. Lee S, Kuk JL, Davidson LE, Hudson R, Kilpatrick K, Graham TE, et al. Exercise without weight loss is an effective strategy for obesity reduction in obese individuals with and without type 2 diabetes. J Appl Physiol. (2005) 99:1220–5. doi: 10.1152/japplphysiol.00053.2005
508. Stewart KJ, Bacher AC, Turner K, Lim JG, Hees PS, Shapiro EP, et al. Exercise and risk factors associated with metabolic syndrome in older adults. Am J Prev Med. (2005) 28:9–18. doi: 10.1016/j.amepre.2004.09.006
509. Lee S, Norheim F, Langleite TM, Gulseth HL, Birkel KI, Drevon CA. Effects of long-term exercise on plasma adipokine levels and inflammation-related gene expression in subcutaneous adipose tissue in sedentary dysglycaemic, overweight men and sedentary normoglycaemic men of healthy weight. Diabetologia. (2019) 62:1048–64. doi: 10.1007/s00125-019-4866-5
510. Lehnig AC, Dewal RS, Baer LA, Kitching KM, Munoz VR, Arts PJ, et al. Exercise training induces depot-specific adaptations to white and brown adipose tissue. Science. (2019) 11:425–39. doi: 10.1016/j.isci.2018.12.033
511. Sakurai T, Ogasawara J, Kizaki T, Sato S, Ishibashi Y, Takahashi M, et al. The effects of exercise training on obesity-induced dysregulated expression of adipokines in white adipose tissue. Int J Endocrinol. (2013) 2013:801743. doi: 10.1155/2013/801743
512. Zachwieja JJ, Hendry SL, Smith SR, Harris RB. Voluntary wheel running decreases adipose tissue mass and expression of leptin mRNA in osborne-mendel rats. Diabetes. (1997) 46:1159–66. doi: 10.2337/diabetes.46.7.1159
513. Berggren JR, Hulver MW, Houmard JA. Fat as an endocrine organ: influence of exercise. J Appl Physiol. (2005) 99:757–64. doi: 10.1152/japplphysiol.00134.2005
514. Halle M, Berg A, Garwers U, Grathwohl D, Knisel W, Keul J. Concurrent reductions of serum leptin and lipids during weight loss in obese men with type ii diabetes. Am J Physiol. (1999) 277:E277–82. doi: 10.1152/ajpendo.1999.277.2.E277
515. Miyatake N, Takahashi K, Wada J, Nishikawa H, Morishita A, Suzuki H, et al. Changes in serum leptin concentrations in overweight japanese men after exercise. Diabetes Obes Metab. (2004) 6:332–7. doi: 10.1111/j.1462-8902.2004.00351.x
516. Polak J, Klimcakova E, Moro C, Viguerie N, Berlan M, Hejnova J, et al. Effect of aerobic training on plasma levels and subcutaneous abdominal adipose tissue gene expression of adiponectin, leptin, interleukin 6, and tumor necrosis factor alpha in obese women. Metabolism. (2006) 55:1375–81. doi: 10.1016/j.metabol.2006.06.008
517. Hulver MW, Zheng D, Tanner CJ, Houmard JA, Kraus WE, Slentz CA, et al. Adiponectin is not altered with exercise training despite enhanced insulin action. Am J Physiol Endocrinol Metab. (2002) 283:E861–5. doi: 10.1152/ajpendo.00150.2002
518. Boudou P, Sobngwi E, Mauvais-Jarvis F, Vexiau P, Gautier JF. Absence of exercise-induced variations in adiponectin levels despite decreased abdominal adiposity and improved insulin sensitivity in type 2 diabetic men. Eur J Endocrinol. (2003) 149:421–4. doi: 10.1530/eje.0.1490421
519. O'Leary VB, Marchetti CM, Krishnan RK, Stetzer BP, Gonzalez F, Kirwan JP. Exercise-induced reversal of insulin resistance in obese elderly is associated with reduced visceral fat. J Appl Physiol. (2006) 100:1584–9. doi: 10.1152/japplphysiol.01336.2005
520. Kondo T, Kobayashi I, Murakami M. Effect of exercise on circulating adipokine levels in obese young women. Endocr J. (2006) 53:189–95. doi: 10.1507/endocrj.53.189
521. Fatouros IG, Tournis S, Leontsini D, Jamurtas AZ, Sxina M, Thomakos P, et al. Leptin and adiponectin responses in overweight inactive elderly following resistance training and detraining are intensity related. J Clin Endocrinol Metab. (2005) 90:5970–7. doi: 10.1210/jc.2005-0261
522. Blüher M, Bullen JW, Lee JH, Kralisch S, Fasshauer M, Klöting N, et al. Circulating adiponectin and expression of adiponectin receptors in human skeletal muscle: associations with metabolic parameters and insulin resistance and regulation by physical training. J Clin Endocrinol Metab. (2006) 91:2310–6. doi: 10.1210/jc.2005-2556
523. Christiansen T, Paulsen SK, Bruun JM, Pedersen SB, Richelsen B. Exercise training versus diet-induced weight-loss on metabolic risk factors and inflammatory markers in obese subjects: a 12-week randomized intervention study. Am J Physiol Endocrinol Metab. (2010) 298:E824–31. doi: 10.1152/ajpendo.00574.2009
524. Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab. (2006) 290:E961–7. doi: 10.1152/ajpendo.00506.2005
525. Miyazaki S, Izawa T, Ogasawara JE, Sakurai T, Nomura S, Kizaki T, et al. Effect of exercise training on adipocyte-size-dependent expression of leptin and adiponectin. Life Sci. (2010) 86:691–8. doi: 10.1016/j.lfs.2010.03.004
526. García-Hermoso A, Ceballos-Ceballos RJ, Poblete-Aro CE, Hackney AC, Mota J, Ramírez-Vélez R. Exercise, adipokines and pediatric obesity: a meta-analysis of randomized controlled trials. Int J Obes. (2017) 41:475–82. doi: 10.1038/ijo.2016.230
527. Yang SJ, Hong HC, Choi HY, Yoo HJ, Cho GJ, Hwang TG, et al. Effects of a three-month combined exercise programme on fibroblast growth factor 21 and fetuin-a levels and arterial stiffness in obese women. Clin Endocrinol. (2011) 75:464–9. doi: 10.1111/j.1365-2265.2011.04078.x
528. Yang W, Liu L, Wei Y, Fang C, Zhou F, Chen J, et al. Exercise ameliorates the fgf21-adiponectin axis impairment in diet-induced obese mice. Endocr Connect. (2019) 8:596–604. doi: 10.1530/EC-19-0034
529. Stanford KI, Middelbeek RJ, Townsend KL, Lee MY, Takahashi H, So K, et al. A novel role for subcutaneous adipose tissue in exercise-induced improvements in glucose homeostasis. Diabetes. (2015) 64:2002–14. doi: 10.2337/db14-0704
530. Sutherl LN, Bomhof MR, Capozzi LC, Basaraba SA, Wright DC. Exercise and adrenaline increase pgc-1{alpha} mRNA expression in rat adipose tissue. J Physiol. (2009) 587:1607–17. doi: 10.1113/jphysiol.2008.165464
531. Trevellin E, Scorzeto M, Olivieri M, Granzotto M, Valerio A, Tedesco L, et al. Exercise training induces mitochondrial biogenesis and glucose uptake in subcutaneous adipose tissue through enos-dependent mechanisms. Diabetes. (2014) 63:2800–11. doi: 10.2337/db13-1234
532. De Matteis R, Lucertini F, Guescini M, Polidori E, Zeppa S, Stocchi V, et al. Exercise as a new physiological stimulus for brown adipose tissue activity. Nutr Metab Cardiovasc Dis. (2013) 23:582–90. doi: 10.1016/j.numecd.2012.01.013
533. Stinkens R, Brouwers B, Jocken JW, Blaak EE, Teunissen-Beekman KF, Hesselink MK, et al. Exercise training-induced effects on the abdominal subcutaneous adipose tissue phenotype in humans with obesity. J Appl Physiol. (2018) 125:1585–93. doi: 10.1152/japplphysiol.00496.2018
534. Vosselman MJ, Hoeks J, Brans B, Pallubinsky H, Nascimento EB, van der Lans AA, et al. Low brown adipose tissue activity in endurance-trained compared with lean sedentary men. Int J Obes (Lond). (2015) 39:1696–702. doi: 10.1038/ijo.2015.130
535. Otero-Díaz B, Rodríguez-Flores M, Sánchez-Muñoz V, Monraz-Preciado F, Ordoñez-Ortega S, Becerril-Elias V, et al. Exercise induces white adipose tissue browning across the weight spectrum in humans. Front Physiol. (2018) 9:1781. doi: 10.3389/fphys.2018.01781
536. Dewal RS, Stanford KI. Effects of exercise on brown and beige adipocytes. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:71–8. doi: 10.1016/j.bbalip.2018.04.013
537. Forsythe LK, Wallace JM, Livingstone MB. Obesity and inflammation: the effects of weight loss. Nutr Res Rev. (2008) 21:117–33. doi: 10.1017/S0954422408138732
538. Selvin E, Paynter NP, Erlinger TP. The effect of weight loss on c-reactive protein: a systematic review. Arch Intern Med. (2007) 167:31–9. doi: 10.1001/archinte.167.1.31
539. Heilbronn LK, Noakes M, Clifton PM. Energy restriction and weight loss on very-low-fat diets reduce c-reactive protein concentrations in obese, healthy women. Arterioscler Thromb Vasc Biol. (2001) 21:968–70. doi: 10.1161/01.ATV.21.6.968
540. Madsen EL, Rissanen A, Bruun JM, Skogstr K, Tonstad S, Hougaard DM, et al. Weight loss larger than 10% is needed for general improvement of levels of circulating adiponectin and markers of inflammation in obese subjects: a 3-year weight loss study. Eur J Endocrinol. (2008) 158:179–87. doi: 10.1530/EJE-07-0721
541. Ellsworth DL, Mamula KA, Blackburn HL, McDyer FA, Jellema GL, van Laar R, et al. Importance of substantial weight loss for altering gene expression during cardiovascular lifestyle modification. Obesity. (2015) 23:1312–9. doi: 10.1002/oby.21079
542. You T, Arsenis NC, Disanzo BL, Lamonte MJ. Effects of exercise training on chronic inflammation in obesity : current evidence and potential mechanisms. Sports Med. (2013) 43:243–56. doi: 10.1007/s40279-013-0023-3
543. Giugliano G, Nicoletti G, Grella E, Giugliano F, Esposito K, Scuderi N, et al. Effect of liposuction on insulin resistance and vascular inflammatory markers in obese women. Br J Plast Surg. (2004) 57:190–4. doi: 10.1016/j.bjps.2003.12.010
544. Gabriely I, Ma XH, Yang XM, Atzmon G, Rajala MW, Berg AH, et al. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes. (2002) 51:2951–8. doi: 10.2337/diabetes.51.10.2951
545. Illán-Gómez F, Gonzálvez-Ortega M, Orea-Soler I, Alcaraz-Tafalla MS, Aragón-Alonso A, Pascual-Díaz M, et al. Obesity and inflammation: change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes Surg. (2012) 22:950–5. doi: 10.1007/s11695-012-0643-y
546. Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H, et al. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut. (2010) 59:1259–64. doi: 10.1136/gut.2010.214577
547. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. (2005) 54:2277–86. doi: 10.2337/diabetes.54.8.2277
548. Trachta P, Dostálová I, Haluzíková D, Kasalický M, Kaválková P, Drápalová J, et al. Laparoscopic sleeve gastrectomy ameliorates mRNA expression of inflammation-related genes in subcutaneous adipose tissue but not in peripheral monocytes of obese patients. Mol Cell Endocrinol. (2014) 383:96–102. doi: 10.1016/j.mce.2013.11.013
549. Hagman DK, Larson I, Kuzma JN, Cromer G, Makar K, Rubinow KB, et al. The short-term and long-term effects of bariatric/metabolic surgery on subcutaneous adipose tissue inflammation in humans. Metabolism. (2017) 70:12–22. doi: 10.1016/j.metabol.2017.01.030
550. Kratz M, Hagman DK, Kuzma JN, Foster-Schubert KE, Chan CP, Stewart S, et al. Improvements in glycemic control after gastric bypass occur despite persistent adipose tissue inflammation. Obesity. (2016) 24:1438–45. doi: 10.1002/oby.21524
551. Šrámková V, Rossmeislová L, Krauzová E, Kračmerová J, Koc M, Langin D, et al. Comparison of early (2 days) and later (28 days) response of adipose tissue to very low-calorie diet in obese women. J Clin Endocrinol Metab. (2016) 101:5021–9. doi: 10.1210/jc.2016-2161
552. Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. (2016) 23:591–601. doi: 10.1016/j.cmet.2016.02.005
553. Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. (2014) 20:103–18. doi: 10.1016/j.cmet.2014.05.005
554. Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez-Cuervo C, Berger Z, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. (2016) 164:740–51. doi: 10.7326/M15-2650
555. Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. (2013) 56:1898–906. doi: 10.1007/s00125-013-2991-0
556. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. (2017) 60:1577–85. doi: 10.1007/s00125-017-4342-z
557. Yerevanian A, Soukas AA. Metformin: mechanisms in human obesity and weight loss. Curr Obes Rep. (2019) 8:156–64. doi: 10.1007/s13679-019-00335-3
558. Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. (1995) 333:550–4. doi: 10.1056/NEJM199508313330903
559. Aghili R, Malek M, Valojerdi AE, Banazadeh Z, Najafi L, Khamseh ME. Body composition in adults with newly diagnosed type 2 diabetes: effects of metformin. J Diabetes Metab Disord. (2014) 13:88. doi: 10.1186/s40200-014-0088-z
560. Feng WH, Bi Y, Li P, Yin TT, Gao CX, Shen SM, et al. Effects of liraglutide, metformin and gliclazide on body composition in patients with both type 2 diabetes and non-alcoholic fatty liver disease: a randomized trial. J Diabetes Investig. (2019) 10:399–407. doi: 10.1111/jdi.12888
561. Kim HJ, Park EY, Oh MJ, Park SS, Shin KH, Choi SH, et al. Central administration of metformin into the third ventricle of c57bl/6 mice decreases meal size and number and activates hypothalamic s6 kinase. Am J Physiol Regul Integr Comp Physiol. (2013) 305:R499–505. doi: 10.1152/ajpregu.00099.2013
562. Helvaci MR, Kaya H, Borazan A, Ozer C, Seyhanli M, Yalcin A. Metformin and parameters of physical health. Intern Med. (2008) 47:697–703. doi: 10.2169/internalmedicine.47.0787
563. Lee AJ. Metformin in noninsulin-dependent diabetes mellitus. Pharmacotherapy. (1996) 16:327–51.
564. Tokubuchi I, Tajiri Y, Iwata S, Hara K, Wada N, Hashinaga T, et al. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE. (2017) 12:e0171293. doi: 10.1371/journal.pone.0171293
565. Geerling JJ, Boon MR, van der Zon GC, van den Berg SA, van den Hoek AM, Lombès M, et al. Metformin lowers plasma triglycerides by promoting vldl-triglyceride clearance by brown adipose tissue in mice. Diabetes. (2014) 63:880–91. doi: 10.2337/db13-0194
566. Watanabe T, Watanabe-Kominato K, Takahashi Y, Kojima M, Watanabe R. Adipose tissue-derived omentin-1 function and regulation. Compr Physiol. (2017) 7:765–81. doi: 10.1002/cphy.c160043
567. Lin MJ, Dai W, Scott MJ, Li R, Zhang YQ, Yang Y, et al. Metformin improves nonalcoholic fatty liver disease in obese mice via down-regulation of apolipoprotein a5 as part of the ampk/lxrα signaling pathway. Oncotarget. (2017) 8:108802–9. doi: 10.18632/oncotarget.22163
568. Zhu X, Yan H, Xia M, Chang X, Xu X, Wang L, et al. Metformin attenuates triglyceride accumulation in hepg2 cells through decreasing stearyl-coenzyme a desaturase 1 expression. Lipids Health Dis. (2018) 17:114. doi: 10.1186/s12944-018-0762-0
569. Fung CS, Wan EY, Wong CK, Jiao F, Chan AK. Effect of metformin monotherapy on cardiovascular diseases and mortality: a retrospective cohort study on Chinese type 2 diabetes mellitus patients. Cardiovasc Diabetol. (2015) 14:137. doi: 10.1186/s12933-015-0304-2
570. Griffin SJ, Leaver JK, Irving GJ. Impact of metformin on cardiovascular disease: a meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia. (2017) 60:1620–9. doi: 10.1007/s00125-017-4337-9
571. Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med. (2008) 121:149–57.e142. doi: 10.1016/j.amjmed.2007.09.016
572. Adeshirlarijaney A, Zou J, Tran HQ, Chassaing B, Gewirtz AT. Amelioration of metabolic syndrome by metformin associates with reduced indices of low-grade inflammation independently of the gut microbiota. Am J Physiol Endocrinol Metab. (2019) 317:E1121–30. doi: 10.1152/ajpendo.00245.2019
573. Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr Metab Immune Disord Drug Targets. (2015) 15:196–205. doi: 10.2174/1871530315666150316124019
574. Li SN, Wang X, Zeng QT, Feng YB, Cheng X, Mao XB, et al. Metformin inhibits nuclear factor kappab activation and decreases serum high-sensitivity c-reactive protein level in experimental atherogenesis of rabbits. Heart Vessels. (2009) 24:446–53. doi: 10.1007/s00380-008-1137-7
575. Ruggiero-Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol. (1999) 58:1765–73. doi: 10.1016/S0006-2952(99)00263-4
576. Beisswenger P, Ruggiero-Lopez D. Metformin inhibition of glycation processes. Diabetes Metab. (2003) 29:6S95–103. doi: 10.1016/S1262-3636(03)72793-1
577. Burmeister MA, Ayala JE, Smouse H, Landivar-Rocha A, Brown JD, Drucker DJ, et al. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes. (2017) 66:372–84. doi: 10.2337/db16-1102
578. López-Ferreras L, Richard JE, Noble EE, Eerola K, Anderberg RH, Olandersson K, et al. Lateral hypothalamic glp-1 receptors are critical for the control of food reinforcement, ingestive behavior and body weight. Mol Psychiatry. (2018) 23:1157–68. doi: 10.1038/mp.2017.187
579. Garber A, Henry RR, Ratner R, Hale P, Chang CT, Bode B, et al. Liraglutide, a once-daily human glucagon-like peptide 1 analogue, provides sustained improvements in glycaemic control and weight for 2 years as monotherapy compared with glimepiride in patients with type 2 diabetes. Diabetes Obes Metab. (2011) 13:348–56. doi: 10.1111/j.1463-1326.2010.01356.x
580. Nauck M, Frid A, Hermansen K, Thomsen AB, During M, Shah N, et al. Long-term efficacy and safety comparison of liraglutide, glimepiride and placebo, all in combination with metformin in type 2 diabetes: 2-year results from the lead-2 study. Diabetes Obes Metab. (2013) 15:204–12. doi: 10.1111/dom.12012
581. Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the lead (liraglutide effect and action in diabetes)-2 study. Diabetes Care. (2009) 32:84–90. doi: 10.2337/dc08-1355
582. Jendle J, Nauck MA, Matthews DR, Frid A, Hermansen K, Düring M, et al. Weight loss with liraglutide, a once-daily human glucagon-like peptide-1 analogue for type 2 diabetes treatment as monotherapy or added to metformin, is primarily as a result of a reduction in fat tissue. Diabetes Obes Metab. (2009) 11:1163–72. doi: 10.1111/j.1463-1326.2009.01158.x
583. Inoue K, Maeda N, Kashine S, Fujishima Y, Kozawa J, Hiuge-Shimizu A, et al. Short-term effects of liraglutide on visceral fat adiposity, appetite, and food preference: a pilot study of obese japanese patients with type 2 diabetes. Cardiovasc Diabetol. (2011) 10:109. doi: 10.1186/1475-2840-10-109
584. Hansotia T, Maida A, Flock G, Yamada Y, Tsukiyama K, Seino Y, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest. (2007) 117:143–52. doi: 10.1172/JCI25483
585. Kim Chung lT, Hosaka T, Yoshida M, Harada N, Sakaue H, Sakai T, et al. Exendin-4, a glp-1 receptor agonist, directly induces adiponectin expression through protein kinase a pathway and prevents inflammatory adipokine expression. Biochem Biophys Res Commun. (2009) 390:613–8. doi: 10.1016/j.bbrc.2009.10.015
586. Ejarque M, Guerrero-Pérez F, de la Morena N, Casajoana A, Virgili N, López-Urdiales R, et al. Role of adipose tissue glp-1r expression in metabolic improvement after bariatric surgery in patients with type 2 diabetes. Sci Rep. (2019) 9:6274. doi: 10.1038/s41598-019-42770-1
587. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2016) 375:311–22. doi: 10.1056/NEJMoa1603827
588. Ansar S, Koska J, Reaven PD. Postprandial hyperlipidemia, endothelial dysfunction and cardiovascular risk: focus on incretins. Cardiovasc Diabetol. (2011) 10:61. doi: 10.1186/1475-2840-10-61
589. Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. (2010) 59:1030–7. doi: 10.2337/db09-1694
590. Gaspari T, Liu H, Welungoda I, Hu Y, Widdop RE, Knudsen LB, et al. A glp-1 receptor agonist liraglutide inhibits endothelial cell dysfunction and vascular adhesion molecule expression in an apoe-/- mouse model. Diab Vasc Dis Res. (2011) 8:117–24. doi: 10.1177/1479164111404257
591. Iacobellis G, Mohseni M, Bianco SD, Banga PK. Liraglutide causes large and rapid epicardial fat reduction. Obesity. (2017) 25:311–6. doi: 10.1002/oby.21718
592. Gallo LA, Wright EM, Vallon V. Probing sglt2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res. (2015) 12:78–89. doi: 10.1177/1479164114561992
593. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
594. Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, et al. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab. (2015) 17:1180–93. doi: 10.1111/dom.12572
595. Tikkanen I, Narko K, Zeller C, Green A, Salsali A, Broedl UC, et al. Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care. (2015) 38:420–8. doi: 10.2337/dc14-1096
596. Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, et al. Effects of sglt2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol. (2013) 715:246–55. doi: 10.1016/j.ejphar.2013.05.014
597. Honda Y, Imajo K, Kato T, Kessoku T, Ogawa Y, Tomeno W, et al. The selective sglt2 inhibitor ipragliflozin has a therapeutic effect on nonalcoholic steatohepatitis in mice. PLoS ONE. (2016) 11:e0146337. doi: 10.1371/journal.pone.0146337
598. Qiang S, Nakatsu Y, Seno Y, Fujishiro M, Sakoda H, Kushiyama A, et al. Treatment with the sglt2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol Metab Syndr. (2015) 7:104. doi: 10.1186/s13098-015-0102-8
599. Osataphan S, Macchi C, Singhal G, Chimene-Weiss J, Sales V, Kozuka C, et al. Sglt2 inhibition reprograms systemic metabolism via fgf21-dependent and -independent mechanisms. JCI Insight. (2019) 4:1–17. doi: 10.1172/jci.insight.123130
600. Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (e-lift trial). Diabetes Care. (2018) 41:1801–8. doi: 10.2337/dc18-0165
601. Häring HU, Merker L, Seewaldt-Becker E, Weimer M, Meinicke T, Woerle HJ, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. (2013) 36:3396–404. doi: 10.2337/dc12-2673
602. Kovacs CS, Seshiah V, Swallow R, Jones R, Rattunde H, Woerle HJ, et al. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: A 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. (2014) 16:147–58. doi: 10.1111/dom.12188
603. Rosenstock J, Jelaska A, Frappin G, Salsali A, Kim G, Woerle HJ, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. (2014) 37:1815–23. doi: 10.2337/dc13-3055
604. Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, et al. Sglt2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing m2 macrophages in diet-induced obese mice. EBiomedicine. (2017) 20:137–49. doi: 10.1016/j.ebiom.2017.05.028
605. Obata A, Kubota N, Kubota T, Iwamoto M, Sato H, Sakurai Y, et al. Tofogliflozin improves insulin resistance in skeletal muscle and accelerates lipolysis in adipose tissue in male mice. Endocrinology. (2016) 157:1029–42. doi: 10.1210/en.2015-1588
606. Angrisani L, Santonicola A, Iovino P, Formisano G, Buchwald H, Scopinaro N. Bariatric surgery worldwide 2013. Obes Surg. (2015) 25:1822–32. doi: 10.1007/s11695-015-1657-z
607. Galanakis CG, Daskalakis M, Manios A, Xyda A, Karantanas AH, Melissas J. Computed tomography-based assessment of abdominal adiposity changes and their impact on metabolic alterations following bariatric surgery. World J Surg. (2015) 39:417–23. doi: 10.1007/s00268-014-2826-2
608. Keidar A, Appelbaum L, Schweiger C, Hershkop K, Matot I, Constantini N, et al. Baseline abdominal lipid partitioning is associated with the metabolic response to bariatric surgery. Obes Surg. (2014) 24:1709–16. doi: 10.1007/s11695-014-1249-3
609. Faria G, Pestana D, Aral M, Preto J, Norberto S, Calhau C, et al. Metabolic score: insights on the development and prediction of remission of metabolic syndrome after gastric bypass. Ann Surg. (2014) 260:279–86. doi: 10.1097/SLA.0000000000000686
610. Toro-Ramos T, Goodpaster BH, Janumala I, Lin S, Strain GW, Thornton JC, et al. Continued loss in visceral and intermuscular adipose tissue in weight-stable women following bariatric surgery. Obesity. (2015) 23:62–9. doi: 10.1002/oby.20932
611. Yoon DY, Kim HK, Kim JA, Choi CS, Yun EJ, Chang SK, et al. Changes in the abdominal fat distribution after gastrectomy: computed tomography assessment. ANZ J Surg. (2007) 77:121–5. doi: 10.1111/j.1445-2197.2006.03990.x
612. Kim MK, Kim W, Kwon HS, Baek KH, Kim EK, Song KH. Effects of bariatric surgery on metabolic and nutritional parameters in severely obese Korean patients with type 2 diabetes: a prospective 2-year follow up. J Diabetes Investig. (2014) 5:221–7. doi: 10.1111/jdi.12137
613. Singh RG, Yoon HD, Wu LM, Lu J, Plank LD, Petrov MS. Ectopic fat accumulation in the pancreas and its clinical relevance: a systematic review, meta-analysis, and meta-regression. Metabolism. (2017) 69:1–13. doi: 10.1016/j.metabol.2016.12.012
614. Gaborit B, Abdesselam I, Kober F, Jacquier A, Ronsin O, Emungania O, et al. Ectopic fat storage in the pancreas using 1h-mrs: importance of diabetic status and modulation with bariatric surgery-induced weight loss. Int J Obes. (2015) 39:480–7. doi: 10.1038/ijo.2014.126
615. Cancello R, Zulian A, Gentilini D, Mencarelli M, Della Barba A, Maffei M, et al. Permanence of molecular features of obesity in subcutaneous adipose tissue of ex-obese subjects. Int J Obes. (2013) 37:867–73. doi: 10.1038/ijo.2013.7
616. Andersson DP, Eriksson Hogling D, Thorell A, Toft E, Qvisth V, Näslund E, et al. Changes in subcutaneous fat cell volume and insulin sensitivity after weight loss. Diabetes Care. (2014) 37:1831–6. doi: 10.2337/dc13-2395
617. Chen J, Pamuklar Z, Spagnoli A, Torquati A. Serum leptin levels are inversely correlated with omental gene expression of adiponectin and markedly decreased after gastric bypass surgery. Surg Endosc. (2012) 26:1476–80. doi: 10.1007/s00464-011-2059-5
618. Auguet T, Terra X, Hernández M, Sabench F, Porras JA, Orellana-Gavaldà JM, et al. Clinical and adipocytokine changes after bariatric surgery in morbidly obese women. Obesity. (2014) 22:188–94. doi: 10.1002/oby.20470
619. Sams VG, Blackledge C, Wijayatunga N, Barlow P, Mancini M, Mancini G, et al. Effect of bariatric surgery on systemic and adipose tissue inflammation. Surg Endosc. (2016) 30:3499–504. doi: 10.1007/s00464-015-4638-3
620. Miller GD, Nicklas BJ, Fernandez A. Serial changes in inflammatory biomarkers after roux-en-y gastric bypass surgery. Surg Obes Relat Dis. (2011) 7:618–24. doi: 10.1016/j.soard.2011.03.006
621. Aghamohammadzadeh R, Greenstein AS, Yadav R, Jeziorska M, Hama S, Soltani F, et al. Effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J Am Coll Cardiol. (2013) 62:128–35. doi: 10.1016/S0140-6736(13)60458-4
622. Emery CF, Fondow MD, Schneider CM, Christofi FL, Hunt C, Busby AK, et al. Gastric bypass surgery is associated with reduced inflammation and less depression: a preliminary investigation. Obes Surg. (2007) 17:759–63. doi: 10.1007/s11695-007-9140-0
623. Gumbau V, Bruna M, Canelles E, Guaita M, Mulas C, Basés C, et al. A prospective study on inflammatory parameters in obese patients after sleeve gastrectomy. Obes Surg. (2014) 24:903–8. doi: 10.1007/s11695-014-1186-1
624. Holdstock C, Lind L, Engstrom BE, Ohrvall M, Sundbom M, Larsson A, et al. Crp reduction following gastric bypass surgery is most pronounced in insulin-sensitive subjects. Int J Obes. (2005) 29:1275–80. doi: 10.1038/sj.ijo.0803000
625. Iannelli A, Anty R, Piche T, Dahman M, Gual P, Tran A, et al. Impact of laparoscopic roux-en-y gastric bypass on metabolic syndrome, inflammation, and insulin resistance in super versus morbidly obese women. Obes Surg. (2009) 19:577–82. doi: 10.1007/s11695-008-9764-8
626. Iannelli A, Martini F, Rodolphe A, Schneck AS, Gual P, Tran A, et al. Body composition, anthropometrics, energy expenditure, systemic inflammation, in premenopausal women 1 year after laparoscopic roux-en-y gastric bypass. Surg Endosc. (2014) 28:500–7. doi: 10.1007/s00464-013-3191-1
627. Monte SV, Caruana JA, Ghanim H, Sia CL, Korzeniewski K, Schentag JJ, et al. Reduction in endotoxemia, oxidative and inflammatory stress, and insulin resistance after roux-en-y gastric bypass surgery in patients with morbid obesity and type 2 diabetes mellitus. Surgery. (2012) 151:587–93. doi: 10.1016/j.surg.2011.09.038
628. Viardot A, Lord RV, Samaras K. The effects of weight loss and gastric banding on the innate and adaptive immune system in type 2 diabetes and prediabetes. J Clin Endocrinol Metab. (2010) 95:2845–50. doi: 10.1210/jc.2009-2371
629. Aron-Wisnewsky J, Tordjman J, Poitou C, Darakhshan F, Hugol D, Basdevant A, et al. Human adipose tissue macrophages: M1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. (2009) 94:4619–23. doi: 10.1210/jc.2009-0925
630. Pardina E, Ferrer R, Baena-Fustegueras JA, Rivero J, Lecube A, Fort JM, et al. Only c-reactive protein, but not tnf-α or il6, reflects the improvement in inflammation after bariatric surgery. Obes Surg. (2012) 22:131–9. doi: 10.1007/s11695-011-0546-3
631. Lips MA, de Groot GH, Berends FJ, Wiezer R, van Wagensveld BA, Swank DJ, et al. Calorie restriction and roux-en-y gastric bypass have opposing effects on circulating fgf21 in morbidly obese subjects. Clin Endocrinol. (2014) 81:862–70. doi: 10.1111/cen.12496
632. Jansen PL, van Werven J, Aarts E, Berends F, Janssen I, Stoker J, et al. Alterations of hormonally active fibroblast growth factors after roux-en-y gastric bypass surgery. Dig Dis. (2011) 29:48–51. doi: 10.1159/000324128
633. Pei E, Liu Y, Jiang W, Lin S, Huang L, Lin M, et al. Sleeve gastrectomy attenuates high fat diet-induced non-alcoholic fatty liver disease. Lipids Health Dis. (2018) 17:243. doi: 10.1186/s12944-018-0875-5
634. Gómez-Ambrosi J, Gallego-Escuredo JM, Catalán V, Rodríguez A, Domingo P, Moncada R, et al. Fgf19 and fgf21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clin Nutr. (2017) 36:861–8. doi: 10.1016/j.clnu.2016.04.027
635. Haluzíková D, Lacinová Z, Kaválková P, Drápalová J, KríŽová J, Bártlová M, et al. Laparoscopic sleeve gastrectomy differentially affects serum concentrations of fgf-19 and fgf-21 in morbidly obese subjects. Obesity. (2013) 21:1335–42. doi: 10.1002/oby.20208
636. Angelin B, Larsson TE, Rudling M. Circulating fibroblast growth factors as metabolic regulators–a critical appraisal. Cell Metab. (2012) 16:693–705. doi: 10.1016/j.cmet.2012.11.001
637. Sheng B, Truong K, Spitler H, Zhang L, Tong X, Chen L. The long-term effects of bariatric surgery on type 2 diabetes remission, microvascular and macrovascular complications, and mortality: a systematic review and meta-analysis. Obes Surg. (2017) 27:2724–32. doi: 10.1007/s11695-017-2866-4
638. Sjöström L, Peltonen M, Jacobson P, Ahlin S, Andersson-Assarsson J, Anveden Å, et al. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA. (2014) 311:2297–304. doi: 10.1001/jama.2014.5988
639. Sjöström L. Review of the key results from the Swedish obese subjects (sos) trial - a prospective controlled intervention study of bariatric surgery. J Intern Med. (2013) 273:219–34. doi: 10.1111/joim.12012
640. Sjöström L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. (2004) 351:2683–93. doi: 10.1056/NEJMoa035622
641. Diamant M, Heine RJ. Thiazolidinediones in type 2 diabetes mellitus: current clinical evidence. Drugs. (2003) 63:1373–405. doi: 10.2165/00003495-200363130-00004
642. Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet Med. (1999) 16:179–92. doi: 10.1046/j.1464-5491.1999.00023.x
643. Mauvais-Jarvis F, Andreelli F, Hanaire-Broutin H, Charbonnel B, Girard J. Therapeutic perspectives for type 2 diabetes mellitus: molecular and clinical insights. Diabetes Metab. (2001) 27:415–23.
644. Fonseca V. Effect of thiazolidinediones on body weight in patients with diabetes mellitus. Am J Med. (2003) 115 Suppl 8A:42S−8S. doi: 10.1016/j.amjmed.2003.09.005
645. Adams M, Montague CT, Prins JB, Holder JC, Smith SA, Sanders L, et al. Activators of peroxisome proliferator-activated receptor gamma have depot-specific effects on human preadipocyte differentiation. J Clin Invest. (1997) 100:3149–53. doi: 10.1172/JCI119870
646. Nichols GA, Gomez-Caminero A. Weight changes following the initiation of new anti-hyperglycaemic therapies. Diabetes Obes Metab. (2007) 9:96–102. doi: 10.1111/j.1463-1326.2006.00580.x
647. Boden G, Zhang M. Recent findings concerning thiazolidinediones in the treatment of diabetes. Expert Opin Investig Drugs. (2006) 15:243–50. doi: 10.1517/13543784.15.3.243
648. Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. (2001) 50:2094–9. doi: 10.2337/diabetes.50.9.2094
649. Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia. (2016) 59:1075–88. doi: 10.1007/s00125-016-3933-4
650. Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. (2002) 87:2784–91. doi: 10.1210/jcem.87.6.8567
651. Dietze-Schroeder D, Sell H, Uhlig M, Koenen M, Eckel J. Autocrine action of adiponectin on human fat cells prevents the release of insulin resistance-inducing factors. Diabetes. (2005) 54:2003–11. doi: 10.2337/diabetes.54.7.2003
652. Zoico E, Garbin U, Olioso D, Mazzali G, Fratta Pasini AM, Di Francesco V, et al. The effects of adiponectin on interleukin-6 and mcp-1 secretion in lipopolysaccharide-treated 3t3-l1 adipocytes: role of the nf-kappab pathway. Int J Mol Med. (2009) 24:847–51. doi: 10.3892/ijmm_00000302
653. Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, et al. Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3t3-l1 adipocytes. Biochem Biophys Res Commun. (2003) 301:1045–50. doi: 10.1016/S0006-291X(03)00090-1
654. Kappes A, Löffler G. Influences of ionomycin, dibutyryl-cycloamp and tumour necrosis factor-alpha on intracellular amount and secretion of apm1 in differentiating primary human preadipocytes. Horm Metab Res. (2000) 32:548–54. doi: 10.1055/s-2007-978684
655. Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. (2005) 28:1547–54. doi: 10.2337/diacare.28.7.1547
656. Erdmann E, Charbonnel B, Wilcox R. Thiazolidinediones and cardiovascular risk - a question of balance. Curr Cardiol Rev. (2009) 5:155–65. doi: 10.2174/157340309788970333
657. van Wijk JP, de Koning EJ, Martens EP, Rabelink TJ. Thiazolidinediones and blood lipids in type 2 diabetes. Arterioscler Thromb Vasc Biol. (2003) 23:1744–9. doi: 10.1161/01.ATV.0000090521.25968.4D
658. Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the proactive study (prospective pioglitazone clinical trial in macrovascular events): a randomised controlled trial. Lancet. (2005) 366:1279–89. doi: 10.1016/S0140-6736(05)67528-9
659. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
660. Tardif JC, Kouz S, Waters DD, Bertr OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
661. Blanco-Colio LM, Tuñón J, Martín-Ventura JL, Egido J. Anti-inflammatory and immunomodulatory effects of statins. Kidney Int. (2003) 63:12–23. doi: 10.1046/j.1523-1755.2003.00744.x
662. Zeiser R. Immune modulatory effects of statins. Immunology. (2018) 154:69–75. doi: 10.1111/imm.12902
663. Jialal I, Stein D, Balis D, Grundy SM, Adams-Huet B, Devaraj S. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive c-reactive protein levels. Circulation. (2001) 103:1933–5. doi: 10.1161/01.CIR.103.15.1933
664. Shovman O, Levy Y, Gilburd B, Shoenfeld Y. Antiinflammatory and immunomodulatory properties of statins. Immunol Res. (2002) 25:271–85. doi: 10.1385/IR:25:3:271
665. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. (2005) 366:1267–78. doi: 10.1016/S0140-6736(05)67394-1
666. Baigent C, Blackwell L, Emberson J, Holl LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of ldl cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. (2010) 376:1670–81. doi: 10.1016/S0140-6736(10)61350-5
667. Soucek F, Covassin N, Singh P, Ruzek L, Kara T, Suleiman M, et al. Effects of atorvastatin (80 mg) therapy on quantity of epicardial adipose tissue in patients undergoing pulmonary vein isolation for atrial fibrillation. Am J Cardiol. (2015) 116:1443–6. doi: 10.1016/j.amjcard.2015.07.067
668. Takei A, Nagashima S, Takei S, Yamamuro D, Murakami A, Wakabayashi T, et al. Myeloid hmg-coa (3-hydroxy-3-methylglutaryl-coenzyme a) reductase determines adipose tissue inflammation, insulin resistance and hepatic steatosis in diet-induced obese mice. Diabetes. (2019) 69:158–64. doi: 10.2337/db19-0076
669. Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. (2008) 3:213–23. doi: 10.1016/j.chom.2008.02.015
670. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. (2006) 444:1022–3. doi: 10.1038/4441022a
671. Drissi F, Raoult D, Merhej V. Metabolic role of lactobacilli in weight modification in humans and animals. Microb Pathog. (2016) 106:182–94. doi: 10.1016/j.micpath.2016.03.006
672. Patil DP, Dhotre DP, Chavan SG, Sultan A, Jain DS, Lanjekar VB, et al. Molecular analysis of gut microbiota in obesity among indian individuals. J Biosci. (2012) 37:647–57. doi: 10.1007/s12038-012-9244-0
673. Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med. (2017) 23:859–68. doi: 10.1038/nm.4358
674. Bik EM. You lose some, you win some: weight loss induces microbiota and metabolite shifts. EBioMedicine. (2015) 2:806–7. doi: 10.1016/j.ebiom.2015.07.037
675. Damms-Machado A, Mitra S, Schollenberger AE, Kramer KM, Meile T, Königsrainer A, et al. Effects of surgical and dietary weight loss therapy for obesity on gut microbiota composition and nutrient absorption. Biomed Res Int. (2015) 2015:806248. doi: 10.1155/2015/806248
676. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
677. Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. (2007) 104:979–84. doi: 10.1073/pnas.0605374104
678. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
679. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes. (2012) 3:279–88. doi: 10.4161/gmic.19625
680. Lau K, Srivatsav V, Rizwan A, Nashed A, Liu R, Shen R, et al. Bridging the gap between gut microbial dysbiosis and cardiovascular diseases. Nutrients. (2017) 9:1–17. doi: 10.3390/nu9080859
681. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. (2008) 57:1470–81. doi: 10.2337/db07-1403
682. Moreira AP, Texeira TF, Ferreira AB, Peluzio MoC, Alfenas ReC. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr. (2012) 108:801–9. doi: 10.1017/S0007114512001213
683. de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol. (2010) 299:G440–8. doi: 10.1152/ajpgi.00098.2010
684. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
685. Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. (2011) 34:1809–15. doi: 10.2337/dc10-2197
686. Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G518–25. doi: 10.1152/ajpgi.00024.2006
687. Jiao N, Baker SS, Chapa-Rodriguez A, Liu W, Nugent CA, Tsompana M, et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in nafld. Gut. (2018) 67:1881–91. doi: 10.1136/gutjnl-2017-314307
688. Pierre JF, Martinez KB, Ye H, Nadimpalli A, Morton TC, Yang J, et al. Activation of bile acid signaling improves metabolic phenotypes in high-fat diet-induced obese mice. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G286–304. doi: 10.1152/ajpgi.00202.2016
689. Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci USA. (2011) 108(Suppl. 1):4592–8. doi: 10.1073/pnas.1011383107
690. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. (2014) 30:332–8. doi: 10.1097/MOG.0000000000000057
691. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. (2014) 11:55–67. doi: 10.1038/nrgastro.2013.151
692. Cornejo-Pareja I, Muñoz-Garach A, Clemente-Postigo M, Tinahones FJ. Importance of gut microbiota in obesity. Eur J Clin Nutr. (2019) 72:26–37. doi: 10.1038/s41430-018-0306-8
693. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
694. Cariou B, Chetiveaux M, Zaïr Y, Pouteau E, Disse E, Guyomarc'h-Delasalle B, et al. Fasting plasma chenodeoxycholic acid and cholic acid concentrations are inversely correlated with insulin sensitivity in adults. Nutr Metab. (2011) 8:48. doi: 10.1186/1743-7075-8-48
695. Svensson PA, Olsson M, Andersson-Assarsson JC, Taube M, Pereira MJ, Froguel P, et al. The tgr5 gene is expressed in human subcutaneous adipose tissue and is associated with obesity, weight loss and resting metabolic rate. Biochem Biophys Res Commun. (2013) 433:563–6. doi: 10.1016/j.bbrc.2013.03.031
696. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. (2006) 439:484–9. doi: 10.1038/nature04330
697. Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, et al. The farnesoid x receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. (2006) 281:11039–49. doi: 10.1074/jbc.M510258200
698. Frikke-Schmidt H, O'Rourke RW, Lumeng CN, Sandoval DA, Seeley RJ. Does bariatric surgery improve adipose tissue function? Obes Rev. (2016) 17:795–809. doi: 10.1111/obr.12429
699. Keitel V, Donner M, Winandy S, Kubitz R, Häussinger D. Expression and function of the bile acid receptor tgr5 in kupffer cells. Biochem Biophys Res Commun. (2008) 372:78–84. doi: 10.1016/j.bbrc.2008.04.171
700. Lou G, Ma X, Fu X, Meng Z, Zhang W, Wang YD, et al. Gpbar1/tgr5 mediates bile acid-induced cytokine expression in murine kupffer cells. PLoS ONE. (2014) 9:e93567. doi: 10.1371/journal.pone.0093567
701. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid x receptor antagonizes nuclear factor kappab in hepatic inflammatory response. Hepatology. (2008) 48:1632–43. doi: 10.1002/hep.22519
702. De Giorgi S, Campos V, Egli L, Toepel U, Carrel G, Cariou B, et al. Long-term effects of roux-en-y gastric bypass on postprandial plasma lipid and bile acids kinetics in female non diabetic subjects: a cross-sectional pilot study. Clin Nutr. (2015) 34:911–7. doi: 10.1016/j.clnu.2014.09.018
703. Kohli R, Myronovych A, Tan BK, Salazar-Gonzalez RM, Miles L, Zhang W, et al. Bile acid signaling: mechanism for bariatric surgery, cure for nash? Dig Dis. (2015) 33:440–6. doi: 10.1159/000371699
704. Albaugh VL, Flynn CR, Cai S, Xiao Y, Tamboli RA, Abumrad NN. Early increases in bile acids post roux-en-y gastric bypass are driven by insulin-sensitizing, secondary bile acids. J Clin Endocrinol Metab. (2015) 100:E1225–33. doi: 10.1210/jc.2015-2467
705. Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. Fxr is a molecular target for the effects of vertical sleeve gastrectomy. Nature. (2014) 509:183–8. doi: 10.1038/nature13135
706. Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, Pellicciari R, et al. The farnesoid x receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Mol Pharmacol. (2006) 70:1164–73. doi: 10.1124/mol.106.023820
707. Abdelkarim M, Caron S, Duhem C, Prawitt J, Dumont J, Lucas A, et al. The farnesoid x receptor regulates adipocyte differentiation and function by promoting peroxisome proliferator-activated receptor-gamma and interfering with the wnt/beta-catenin pathways. J Biol Chem. (2010) 285:36759–67. doi: 10.1074/jbc.M110.166231
708. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. (2012) 490:55–60. doi: 10.1038/nature11450
709. Burcelin R. Gut microbiota and immune crosstalk in metabolic disease. Mol Metab. (2016) 5:771–81. doi: 10.1016/j.molmet.2016.05.016
710. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. (2018) 359:1151–6. doi: 10.1126/science.aao5774
711. Chan YK, Brar MS, Kirjavainen PV, Chen Y, Peng J, Li D, et al. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma a-fabp and cholesterol: a pilot study of high fat diet and its intervention with lactobacillus rhamnosus gg (lgg) or telmisartan in apoe-/- mice. BMC Microbiol. (2016) 16:264. doi: 10.1186/s12866-016-0883-4
712. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58:1509–17. doi: 10.2337/db08-1637
713. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor gpr43. Nat Commun. (2013) 4:1829. doi: 10.1038/ncomms2852
714. Tang WH, Hazen SL. The gut microbiome and its role in cardiovascular diseases. Circulation. (2017) 135:1008–10. doi: 10.1161/CIRCULATIONAHA.116.024251
715. Kimura I, Inoue D, Hirano K, Tsujimoto G. The scfa receptor gpr43 and energy metabolism. Front Endocrinol. (2014) 5:85. doi: 10.3389/fendo.2014.00085
716. Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. (2011) 22:849–5. doi: 10.1016/j.jnutbio.2010.07.009
717. Vinolo MA, Rodrigues HG, Festuccia WT, Crisma AR, Alves VS, Martins AR, et al. Tributyrin attenuates obesity-associated inflammation and insulin resistance in high-fat-fed mice. Am J Physiol Endocrinol Metab. (2012) 303:E272–82. doi: 10.1152/ajpendo.00053.2012
718. Kim CH, Park J, Kim M. Gut microbiota-derived short-chain fatty acids, t cells, and inflammation. Immune Netw. (2014) 14:277–88. doi: 10.4110/in.2014.14.6.277
719. Asarat M, Apostolopoulos V, Vasiljevic T, Donkor O. Short-chain fatty acids produced by synbiotic mixtures in skim milk differentially regulate proliferation and cytokine production in peripheral blood mononuclear cells. Int J Food Sci Nutr. (2015) 66:755–65. doi: 10.3109/09637486.2015.1088935
720. Fabian E, Elmadfa I. Influence of daily consumption of probiotic and conventional yoghurt on the plasma lipid profile in young healthy women. Ann Nutr Metab. (2006) 50:387–93. doi: 10.1159/000094304
721. Anderson JW, Gillil SE. Effect of fermented milk (yogurt) containing lactobacillus acidophilus l1 on serum cholesterol in hypercholesterolemic humans. J Am Coll Nutr. (1999) 18:43–50. doi: 10.1080/07315724.1999.10718826
722. Pindjakova J, Sartini C, Lo Re O, Rappa F, Coupe B, Lelouvier B, et al. Gut dysbiosis and adaptive immune response in diet-induced obesity vs. Systemic inflammation. Front Microbiol. (2017) 8:1157. doi: 10.3389/fmicb.2017.01157
723. Clarke G, Stilling RM, Kennedy PJ, Stanton C, Cryan JF, Dinan TG. Minireview: gut microbiota: the neglected endocrine organ. Mol Endocrinol. (2014) 28:1221–38. doi: 10.1210/me.2014-1108
724. Thaiss CA, Levy M, Suez J, Elinav E. The interplay between the innate immune system and the microbiota. Curr Opin Immunol. (2014) 26:41–8. doi: 10.1016/j.coi.2013.10.016
725. Kato LM, Kawamoto S, Maruya M, Fagarasan S. The role of the adaptive immune system in regulation of gut microbiota. Immunol Rev. (2014) 260:67–75. doi: 10.1111/imr.12185
726. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. (2016) 535:75–84. doi: 10.1038/nature18848
727. Ohira H, Fujioka Y, Katagiri C, Mamoto R, Aoyama-Ishikawa M, Amako K, et al. Butyrate attenuates inflammation and lipolysis generated by the interaction of adipocytes and macrophages. J Atheroscler Thromb. (2013) 20:425–42. doi: 10.5551/jat.15065
728. Wang X, He G, Peng Y, Zhong W, Wang Y, Zhang B. Sodium butyrate alleviates adipocyte inflammation by inhibiting nlrp3 pathway. Sci Rep. (2015) 5:12676. doi: 10.1038/srep12676
729. Alemán JO, Bokulich NA, Swann JR, Walker JM, De Rosa JC, Battaglia T, et al. Fecal microbiota and bile acid interactions with systemic and adipose tissue metabolism in diet-induced weight loss of obese postmenopausal women. J Transl Med. (2018) 16:244. doi: 10.1186/s12967-018-1619-z
730. Janssens S, Beyaert R. Role of toll-like receptors in pathogen recognition. Clin Microbiol Rev. (2003) 16:637–46. doi: 10.1128/CMR.16.4.637-646.2003
731. Pineiro M, Asp NG, Reid G, Macfarlane S, Morelli L, Brunser O, et al. Fao technical meeting on prebiotics. J Clin Gastroenterol. (2008) 42(Suppl. 3 Pt 2):S156–9. doi: 10.1097/MCG.0b013e31817f184e
732. Ettinger G, MacDonald K, Reid G, Burton JP. The influence of the human microbiome and probiotics on cardiovascular health. Gut Microbes. (2014) 5:719–28. doi: 10.4161/19490976.2014.983775
733. Kim YA, Keogh JB, Clifton PM. Probiotics, prebiotics, synbiotics and insulin sensitivity. Nutr Res Rev. (2018) 31:35–51. doi: 10.1017/S095442241700018X
734. Karamali M, Dadkhah F, Sadrkhanlou M, Jamilian M, Ahmadi S, Tajabadi-Ebrahimi M, et al. Effects of probiotic supplementation on glycaemic control and lipid profiles in gestational diabetes: a randomized, double-blind, placebo-controlled trial. Diabetes Metab. (2016) 42:234–41. doi: 10.1016/j.diabet.2016.04.009
735. Crommen S, Simon MC. Microbial regulation of glucose metabolism and insulin resistance. Genes. (2017) 9:1–16. doi: 10.3390/genes9010010
736. Yoo SR, Kim YJ, Park DY, Jung UJ, Jeon SM, Ahn YT, et al. Probiotics l. Plantarum and l. Curvatus in combination alter hepatic lipid metabolism and suppress diet-induced obesity. Obesity. (2013) 21:2571–8. doi: 10.1002/oby.20428
737. Wang J, Tang H, Zhang C, Zhao Y, Derrien M, Rocher E, et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. (2015) 9:1–15. doi: 10.1038/ismej.2014.99
738. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. (2013) 110:9066–71. doi: 10.1073/pnas.1219451110
739. Mischke M, Arora T, Tims S, Engels E, Sommer N, van Limpt K, et al. Specific synbiotics in early life protect against diet-induced obesity in adult mice. Diabetes Obes Metab. (2018) 20:1408–18. doi: 10.1111/dom.13240
740. Aronsson L, Huang Y, Parini P, Korach-André M, Håkansson J, Gustafsson J, et al. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (angptl4). PLoS ONE. (2010) 5: e13087. doi: 10.1371/journal.pone.0013087
741. Park DY, Ahn YT, Park SH, Huh CS, Yoo SR, Yu R, et al. Supplementation of Lactobacillus curvatus hy7601 and Lactobacillus plantarum ky1032 in diet-induced obese mice is associated with gut microbial changes and reduction in obesity. PLoS ONE. (2013) 8:e59470. doi: 10.1371/journal.pone.0059470
742. Ukibe K, Miyoshi M, Kadooka Y. Administration of lactobacillus gasseri sbt2055 suppresses macrophage infiltration into adipose tissue in diet-induced obese mice. Br J Nutr. (2015) 114:1180–7. doi: 10.1017/S0007114515002627
743. Miyoshi M, Ogawa A, Higurashi S, Kadooka Y. Anti-obesity effect of lactobacillus gasseri sbt2055 accompanied by inhibition of pro-inflammatory gene expression in the visceral adipose tissue in diet-induced obese mice. Eur J Nutr. (2014) 53:599–606. doi: 10.1007/s00394-013-0568-9
744. Kadooka Y, Sato M, Imaizumi K, Ogawa A, Ikuyama K, Akai Y, et al. Regulation of abdominal adiposity by probiotics (lactobacillus gasseri sbt2055) in adults with obese tendencies in a randomized controlled trial. Eur J Clin Nutr. (2010) 64:636–43. doi: 10.1038/ejcn.2010.19
745. Gibson GR. Dietary modulation of the human gut microflora using the prebiotics oligofructose and inulin. J Nutr. (1999) 129:1438S−41S. doi: 10.1093/jn/129.7.1438S
Keywords: adipokines, subcutaneous white adipose tissue, visceral white adipose tissue, brown adipose tissue, beige adipose tissue, metabolic syndrome, insulin resistance
Citation: Chait A and den Hartigh LJ (2020) Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 7:22. doi: 10.3389/fcvm.2020.00022
Received: 23 December 2019; Accepted: 10 February 2020;
Published: 25 February 2020.
Edited by:
Catherine A. Reardon, University of Chicago, United StatesReviewed by:
Rei Shibata, Nagoya University Hospital, JapanYuichi Akasaki, Kagoshima University Medical and Dental Hospital, Japan
Copyright © 2020 Chait and den Hartigh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura J. den Hartigh, lauradh@u.washington.edu