


Abstract
3,5-Seco-4-nor-cholestan-5-one oxime-3-ol (TRO40303) is a new cardioprotective compound coming from a chemical series identified initially for neuroprotective properties. TRO40303 binds specifically to the mitochondrial translocator protein 18 kDa (TSPO) at the cholesterol site. After intravenous administration, TRO40303 tissue distribution was comparable to that of TSPO, and, in particular, the drug accumulated rapidly in the heart. In a model of 35 min of myocardial ischemia/24 h of reperfusion in rats, TRO40303 (2.5 mg/kg) reduced infarct size by 38% (p < 0.01 versus control), when administered 10 min before reperfusion, which was correlated with reduced release of apoptosis-inducing factor from mitochondria to the cytoplasm in the ischemic area at risk. Although TRO40303 had no effect on the calcium retention capacity of isolated mitochondria, unlike cyclosporine A, the drug delayed mitochondrial permeability transition pore (mPTP) opening and cell death in isolated adult rat cardiomyocytes subjected to 2 h of hypoxia followed by 2 h of reoxygenation and inhibited mPTP opening in neonatal rat cardiomyocytes treated with hydrogen peroxide. The effects of TRO40303 on mPTP in cell models of oxidative stress are correlated with a significant reduction in reactive oxygen species production and subsequent calcium overload. TRO40303 is a new mitochondrial-targeted drug and inhibits mPTP triggered by oxidative stress. Its mode of action differs from that of other mPTP inhibitors such as cyclosporine A, thus providing a new pharmacological approach to study mPTP regulation. Its efficacy in an animal model of myocardial infarctions makes TRO40303 a promising new drug for the reduction of cardiac ischemia-reperfusion injury.
Opening of the mitochondrial permeability transition pore (mPTP) within the few minutes after reperfusion is now well established as a key event contributing to myocardial ischemia-reperfusion (I/R) injury. Mitochondrial permeability transition is triggered by the burst of reactive oxygen species (ROS) combined with calcium overload, lack of ATP, and recovery of cytoplasmic pH that follows tissue reperfusion. It is characterized by matrix swelling and rupture of the outer mitochondrial membrane (Griffiths and Halestrap, 1995). The mPTP opening is followed by mitochondrial leakage of protein factors inducing apoptosis or necrosis, depending on the ATP level and whether all or only a subpopulation of mitochondria undergo permeability transition (Halestrap, 2006). The mPTP concept first proposed by Haworth and Hunter (1979) gained prominence as an important factor in I/R and a target for cardioprotective agents when mice devoid of cyclophilin D (CyD) were found to have significantly reduced infarct size compared with wild-type mice (Baines et al., 2005). Furthermore, drugs such as cyclosporine A (CsA) that delay mPTP opening through binding, inhibition, and displacement of CyD provide cardioprotection after myocardial I/R in various animal models (Massoudy et al., 1997; Squadrito et al., 1999; Argaud et al., 2005; Gomez et al., 2007). Other procedures such as pre- and postconditioning have also been shown to reduce I/R via inhibition of mPTP opening (Hausenloy et al., 2009). More recently, postconditioning and CsA were shown to reduce infarct size in patients undergoing percutaneous coronary intervention to establish reperfusion after an acute myocardial infarction (Staat et al., 2005; Piot et al., 2008), providing the strong clinical proof of concept that modulation of mPTP is a validated strategy to reduce myocardial damage at the time of reperfusion in humans.
Several models of the mPTP have been described previously (Baines, 2009; Javadov et al., 2009). Proteins implicated in mPTP formation include the matrix CyD, the inner membrane adenine nucleotide translocase and the outer membrane voltage-dependent anion channel (VDAC). However, none of these proteins is mandatory for mPTP opening as permeability transition, leading to mitochondrial-mediated cell death, even though altered for some cases, still persists after each of these proteins has been knocked-out (Kokoszka et al., 2004; Basso et al., 2005; Baines et al., 2007). Additional proteins such as the translocator protein 18 kDa (TSPO), formerly known as the peripheral benzodiazepine receptor, have been shown to interact with proteins implicated in mPTP formation and potentially regulate the pore (Veenman et al., 2007). Selective TSPO ligands, such as chlorodiazepam (Ro5-4864) (Obame et al., 2007) or 7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide (SSR180575) (Leducq et al., 2003), were reported to provide cardioprotection in preclinical models of I/R. The pro- and antiapoptotic Bcl-2 family proteins regulate mitochondrial permeability transition either by directly and independently forming a membrane channel or by interaction with mitochondrial membrane proteins such as VDAC (Shimizu et al., 1999) or adenine nucleotide translocase (Marzo et al., 1998). Other mPTP modulators include glycogen synthase kinase-3β inhibitors (Obame et al., 2008), protein kinase C-ε (Inagaki et al., 2005), and hexokinase I and II (Pastorino and Hoek, 2008). More recently, the phosphate carrier has also been shown to play an important role in mPTP regulation (Leung et al., 2008), and, finally, others have proposed that mPTP opening is the result of protein oxidation (Hossain et al., 2009) or accumulation of unfolded proteins in the outer mitochondrial membrane (He and Lemasters, 2002).
In the present study, we investigated the cardioprotective effects of 3,5-seco-4-nor-cholestan-5-one oxime-3-ol (TRO40303), which was originally selected using a cell-based screening assay aimed to identify small molecules that maintain survival of trophic factor-deprived rat motor neurons (Bordet et al., 2007). TRO40303 binds specifically to the cholesterol site of TSPO and exhibits cytoprotective properties in various cell types correlated with prevention of mPTP opening. Here we present evidence that TRO40303 rapidly accumulates in the heart after intravenous administration and prevents cardiomyocyte death after I/R, both in vitro and in vivo. To better understand how TRO40303 provides protection against oxidative damage associated with I/R injury, we investigated its effects on isolated cardiac mitochondria and isolated cardiomyocytes subjected to oxidative stress monitoring calcium retention capacity, ROS accumulation, Ca2+ overload, mPTP formation, loss of mitochondrial membrane potential (Δψm), and cell death. These studies show that TRO40303 has a novel mode of mPTP modulation as it does not inhibit the calcium retention capacity of isolated mitochondria, but it reduces mPTP opening in cells, probably via reduction in ROS production.
Materials and Methods
Drugs.
All compounds were purchased from Sigma-Aldrich (St. Louis, MO) unless specified. TRO40303 (Fig. 1A) was synthesized by Synkem (Dijon, France). The radiolabeled [14C]TRO40303 was synthesized by GE Healthcare (Little Chalfont, Buckinghamshire, UK) with a specific activity of 2.11 GBq/mmol. For the in vitro tests, drugs such as CsA and TRO40303 were dissolved in dimethyl sulfoxide (DMSO) to prepare 10−2 M stock solutions that were diluted to their final concentration in the appropriate buffer or medium. For in vivo studies, TRO40303 was dissolved in a solution of 30% hydroxypropyl-β-cyclodextrin in phosphate-buffered saline (HPBCD).
Animals.
All animal procedures used in this study were in strict accordance with the European Community Council Directive (86-609/87-848 EEC) and recommendations of the French Ministère de l'Agriculture or in accordance with the American Veterinary Medical Association guidelines, consistent with United States federal regulations and approved by the University of Louisville Institutional Animal Care and Use Committee. Male Wistar rats (250–280 g; Janvier, Le Genest Saint Isle, France) were used for in vivo myocardial infarction studies and for preparation of adult cardiomyocytes and cardiac mitochondria. For tissue distribution studies, male Sprague-Dawley rats (250–330 g; Janvier) were used. Animals were maintained in a room with controlled temperature (21–25°C) and a reverse 12-h light/dark cycle with food and water available ad libitum.
Cell Culture and Fluorescent Probes.
All cell culture medium and supplements, fetal bovine serum, Calcium Green-5N, calcein-AM, 5(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acyl ester (DCF), tetramethylrhodamine methyl ester (TMRM), MitoTracker Red, and propidium iodide were purchased from Invitrogen (Carlsbad, CA) unless specified.
In Vitro Binding to TSPO.
The affinity of TRO40303 for TSPO was evaluated by measuring its ability to compete with [3H]1-(2-chlorophenyl)-N-methyl-N-(1-methyl-propyl)-3-isoquinoline carboxamide (PK11195) or [3H]cholesterol binding to recombinant mouse TSPO reconstituted in proteoliposomes (Lacapère et al., 2001). TSPO-containing liposomes (2 μg of protein) were incubated at room temperature for 30 min in the presence of 3 nM [3H]PK11195 (100% initial PK11195 binding; specific activity, 83.5 Ci/mmol) or for 60 min in the presence of 3 nM [3H]cholesterol (100% initial cholesterol binding; specific activity, 60 Ci/mmol). Radioactive ligands were displaced by incubation in the presence of increasing concentrations of cold ligands (PK11195, cholesterol, or TRO40303). Vesicle suspensions were filtered on Whatman GF/C filters and washed, and radioactivity was measured by liquid scintillation. Nonspecific binding was estimated as the remaining radioactivity after washing in the presence of excess cold ligand. Binding data were fitted to a simple sigmoid curve using the following equation: Y = 100 − amplitude × S/(Kd + S).
Tissue Distribution.
Tissue distribution of TRO40303 was evaluated by quantitative whole-body autoradiography after intravenous administration of 2 mg/kg [14C]TRO40303, corresponding to 3.7 MBq/kg, to six fasted male rats (Avogadro, Fontenilles, France). Blood samples were taken 1, 2, 4, 8, 24, and 48 h postdose. The total radioactivity was determined in whole blood by liquid scintillation counting. After each blood sampling, one animal was sacrificed with thiopental, quickly frozen in dry ice, and stored at −20°C until slicing and whole-body autoradiography (Biotec Center, Orléans, France).
Myocardial Infarction Model.
Using a protocol described by Obame et al. (2007), rats were anesthetized with sodium pentobarbital (60 mg/kg i.p.), intubated, and ventilated. A fluid-filled Tygon catheter was placed in the right carotid artery for measurement of arterial blood pressure with a Statham P23ID strain-gauge transducer. Heart rate was calculated by integration over time of systolic pulses issued from the arterial blood pressure signal. Tightening the snare under the left main coronary artery induced coronary artery occlusion (CAO) and releasing the ends of the suture initiated coronary artery reperfusion (CAR). In all groups of rats the coronary artery was occluded during 35 min and released for reperfusion, except in the so-called sham group in which the surgical procedure was identical to others, but the coronary artery was not occluded. In the other groups of treated rats, the vehicle (HPBCD 30% in PBS) and TRO40303 at increasing doses (0.5, 1.25, and 2.5 mg/kg) were administered at random as a 3-min infusion through the jugular vein starting 10 min before CAR. Rats were allowed to recover for 24 h before measurement of infarct size. Infarct size was determined as a percentage of the area at risk (AAR) by triphenyltetrazolium chloride staining as described previously (Obame et al., 2007). In experiments on isolated mitochondria obtained from heart tissue after I/R, rats were sacrificed after 10 min of reperfusion. To analyze the release of cytochrome c and apoptosis-inducing factor (AIF) from mitochondria, cytosolic fractions were prepared from heart tissue after 1 h of reperfusion.
Isolation of Mitochondria and Cytosolic Fractions from Rat Heart.
To prepare mitochondria for Ca2+ retention capacity (CRC) experiments, the myocardium from healthy rats (noninfarcted) or from the AAR of rats that underwent I/R was excised and rapidly minced followed by homogenization with a Polytron homogenizer (low setting 3) and then with a Potter homogenizer (5 up and down strokes at 1500 rpm) in 15 ml of a cold buffer containing 220 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mM EGTA, and 0.04 mM fatty acid-free bovine serum albumin, pH = 7.4 at 4°C. Homogenates were centrifuged at 1000g for 5 min, and supernatants were centrifuged again at 10,000g for 10 min. The final pellets were resuspended in the homogenization buffer including 0.1 mM EGTA without bovine serum albumin to obtain a protein concentration of approximately 15 mg/ml.
To measure cytochrome c and AIF release from mitochondria, cardiac homogenates were prepared according to the same protocol except that bovine serum albumin was omitted from the buffer. Homogenates were centrifuged at 1000g for 5 min, and supernatants were centrifuged at 10,000g for 10 min. The final supernatants, corresponding to the cytosolic fractions, were collected, and 20 μl of a protease inhibitor cocktail (Sigma-Aldrich) and 20 μl of 1 mM phenylmethanesulfonyl fluoride (Sigma-Aldrich) were added. The supernatants were immediately frozen at −80°C until determination of protein concentration and analysis of cytochrome c and AIF by Western blot.
Evaluation of CRC of Isolated Mitochondria.
mPTP opening was assessed by monitoring mitochondrial CRC. Mitochondria were loaded with increasing concentrations of Ca2+ until the load reached a threshold at which mitochondria underwent a fast process of Ca2+ release, which was due to mPTP opening. Cardiac mitochondria (1 mg/ml), energized with 5 mM pyruvate/malate, were incubated in a buffer containing 50 mM sucrose, 100 mM KCl, 5 mM KH2PO4, 10 mM HEPES, and 1 μM Calcium Green-5N fluorescent probe. The concentration of Ca2+ in the extramitochondrial medium was monitored by means of an LS 50B spectrofluorimeter (PerkinElmer Life and Analytical Sciences, Waltham, MA) at excitation and emission wavelengths of 506 and 532 nm, respectively. The Ca2+ signal was calibrated by addition to the medium of known Ca2+ amounts.
Western Blot Experiments.
Samples of cytosolic proteins, 5 μg for cytochrome c and 15 μg for AIF, were boiled at 95°C in a buffer containing 20% sucrose, 2.4% SDS, 5% β-mercaptoethanol, and 5% bromphenol blue. They were subjected to electrophoresis on 4 to 15% (cytochrome c) and 4 to 10% (AIF) gradient SDS-polyacrylamide gel electrophoresis gels and then transferred on polyvinylidene difluoride membranes. Membranes were blocked with 5% nonfat dry milk in a Tris buffer (10 mM Tris and 100 mM NaCl, pH 7.5) containing 0.05% Tween 20 overnight at 4°C. Subsequently, membranes were exposed for 1 h to either mouse cytochrome c antibody (1:1000; BD Biosciences, San Jose, CA) or goat polyclonal anti-AIF antibody (1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). After incubation with horseradish peroxidase-coupled goat anti-mouse immunoglobulins (Santa Cruz Biotechnology, Inc.) or donkey anti-goat immunoglobulins (Santa Cruz Biotechnology, Inc.), proteins were revealed by enhanced chemiluminescence reaction (ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK) after exposure to X-ray films (GE Healthcare). Band intensities were analyzed by densitometry using ImageJ software (National Institutes of Health, Bethesda, MD).
Measurement of mPTP Opening in Primary Adult Rat Cardiomyocytes Subjected to Hypoxia/Reoxygenation.
Ventricular cardiomyocytes were isolated from male Wistar rats as described previously (Obame et al., 2008). The cardiomyocytes were placed into a thermostated (37°C) chamber (Warner Instruments, Hamden, CT), which was mounted on the stage of a IX81 Olympus microscope and were perfused with a Tyrode's solution (130 mM NaCl, 5 mM KCl, 10 mM HEPES, 1 mM MgCl2, and 1.8 mM CaCl2; pH = 7.4 at 37°C) at a rate of 0.5 ml/min. The chamber was connected to a gas bottle diffusing a constant stream of O2 (21%), N2 (74%), and CO2 (5%), maintaining a partial O2 pressure of 21%. Oxygen in the perfusate was measured in the chamber using a fiberoptic sensor system (Ocean Optics, Inc., Dunedin, FL). Myocytes were paced to beat by field stimulation (5 ms, 0.5 Hz). To simulate ischemia, the perfusion was stopped, and cardiomyocytes were exposed for 2 h to a hypoxic medium maintaining a partial O2 pressure of 2 to 3%. This medium was the Tyrode's solution supplemented with 20 mM 2-deoxyglucose and subjected to a constant stream of N2 (100%). At the end of the ischemic period, reoxygenation was induced by rapidly restoring the Tyrode's solution flow containing 21% O2 into the chamber.
Direct assessment of mPTP opening in cardiomyocytes was made by means of the established loading procedure of the cells with calcein-AM and CoCl2, resulting in mitochondrial localization of calcein fluorescence (Petronilli et al., 1999). Cells were loaded with 1 μM calcein-AM for 30 min at 37°C in 2 ml of M199, pH = 7.4, supplemented with 1 mM CoCl2 after 20 min of incubation. They were then washed free of calcein-AM and CoCl2 and incubated in the Tyrode's solution. To determine cell death, cells were coloaded in all experiments with propidium iodide (5 μM), which permeates only the damaged cells. Cardiomyocytes were imaged with an Olympus IX-81 motorized inverted microscope equipped with a mercury lamp as a source of light for epifluorescence illumination and with a cooled charge-coupled device Hamamatsu ORCA ER digital camera. For detection of calcein fluorescence 460- to 490-nm excitation and 510-nm emission filters were used. Propidium iodide fluorescence was excited at 520 to 550 nm and recorded at 580 nm. Images were acquired every 60 s after an illumination time of 21 ms (calcein) and 80 ms (propidium iodide) per image using a digital epifluorescence imaging software (Cell∧M; Olympus, Rungis, France).
Fluorescence was integrated over a region of interest (≈80 μm2) for each cardiomyocyte and a fluorescence background corresponding to an area without cells was subtracted. For comparative purposes, the fluorescence intensity minus background was normalized according to the maximal fluorescence value (initial value for calcein and final value for propidium iodide).
At first, for each experiment, the response was observed from a single cardiomyocyte and the time of reoxygenation necessary to induce a 50% decrease in calcein fluorescence [time to 50% mPTP opening (TmPTP50)] was determined. Then, the global response was analyzed by averaging the fluorescence changes obtained from all the cardiomyocytes contained in a single field.
Neonatal Rat Cardiomyocyte Isolation and Culture.
Neonatal rat cardiomyocytes (NRCMs) were isolated from 1- to 2-day-old Sprague-Dawley rats and cultured according to a well characterized protocol (Jones et al., 2008; Ngoh et al., 2009). All of the NRCM studies adhered to the American Physiological Society's Guiding Principles in the Care and Use of Animals. NRCMs were plated on 35-mm glass-bottom culture dishes at 75% confluence. During the first 4 days of culture, medium contained the antimitotic, bromodeoxyuridine (0.1 mM), to inhibit fibroblast growth in addition to 5% fetal bovine serum, penicillin/streptomycin, and vitamin B12. Twenty-four hours before experimentation, medium was changed to serum-free Dulbecco's modified Eagle's medium (HEPES-free). All experiments were performed on at least five separate NRCM cultures.
Induction of Oxidative Stress by H2O2, Drug Treatment, and Time-Lapse Fluorescence Microscopy.
After loading with the various fluorescent dyes as described below, NRCMs were washed and medium was replaced with imaging medium (Dulbecco's modified Eagle's medium with 25 mM HEPES and minus phenol red and pyruvate). Cells were treated with either DMSO (0.1%), TRO40303 (3 μM), or CsA (1 μM) just before induction of oxidative stress by addition of 100 μM H2O2. Imaging was initiated immediately after pipetting up and down three times to assure mixing. Images were captured using a Photometrics CoolSNAP ES camera attached to a Nikon-TE2000E2 fluorescence microscope with a T-PFS Perfect Focus Unit all controlled with MetaMorph 6.3r2 software. The Perfect Focus System was used to prevent minute defocusing caused by changes over time during time-lapse imaging. An X-Cite 120 Fluor light source (level of 12%) was used and a Plan Apo 60xA/oil (numerical aperture = 1.4) objective was used for magnification. Neutral density filter setting was set at ND4 and binning of 2 × for all image acquisition. Images were captured every 90 s for up to 90 min.
Assessment of mPTP Opening in NRCMs after H2O2 Intoxication.
Formation of mPTP was assessed by following the changes in calcein fluorescence using a modified cold and warm loading protocol (Nieminen et al., 1995). NRCMs were loaded with 1 μM calcein-AM for 150 min at 4°C. Medium was changed to imaging medium, and NRCMs were loaded with 2 μM MitoTracker Red FM at room temperature for 90 min. Excitation for calcein was through a 470/40-nm bandpass filter, and emission was through a 522/40-nm bandpass filter, whereas excitation for MitoTracker Red was through a 560/28-nm bandpass filter and emission was through 646/38-nm bandpass filter. Exposure duration was set at 50 ms for calcein and 100 ms for MitoTracker Red. Mitochondrial calcein fluorescence was determined using MetaMorph software in regions devoid of MitoTracker Red fluorescence to detect nonmitochondrial calcein fluorescence and then subtracting nonmitochondrial calcein fluorescence from the total calcein fluorescence. Calcein localization was assessed by measuring the S.D. of the intensity of all green fluorescent pixels within the same region of interest. A region of interest was defined by drawing a circle (43.61 μm2) enclosing several mitochondria. The S.D. is high when fluorescence is punctate and low when fluorescence is diffuse. Fluorescence is normalized to time 0 for each treatment.
Assessment of ROS Production.
ROS levels were assessed by following the changes in DCF fluorescence (Teshima et al., 2003b). The DCF used for these experiments is improved compared with 2′,7′-dichlorodihydrofluorescein diacetate because of enhanced retention in cells. According to the manufacturer, it has been shown to detect H2O2 generation in the cells as well as other reactive oxygen intermediates such as OH−, HOO, and ONOO−.
NRCMs were loaded with 2 μM DCF for 30 min at 37°C. Medium was changed to imaging medium as mentioned above. DCF fluorescence was recorded through a 470/40-nm bandpass filter and emission through a 522/40-nm bandpass filter. Exposure duration was set at 100 ms. Fluorescence is normalized to time 0 for each treatment.
Assessment of Calcium Overload.
Calcium levels were assessed by following the changes in Rhod-2 (Ngoh et al., 2009) and Fluo-4 fluorescence (Teshima et al., 2003a). NRCMs were loaded with 2 μM Rhod-2AM (used to assess mitochondrial calcium) or 1 μM Fluo-4AM (used to assess cytosolic calcium) for 30 min at 37°C, and media were changed to imaging media. BAPTA-AM (50 μM), a membrane permeable intracellular calcium chelator, was used as a control. Fluorescence was recorded by sequentially exciting Rhod-2 and Fluo-4 through 560/28- and 470/40-nm bandpass filters, respectively. Emission was sequentially assessed through 646/38-nm (for Rhod-2) and 522/40-nm (for Fluo-4) bandpass filters. Exposure duration was set at 100 ms for both Rhod-2 and Fluo-4.
Assessment of Mitochondrial Membrane Potential.
Mitochondrial membrane potential changes was detected by following changes in TMRM fluorescence, which is rapidly and reversibly accumulated in the mitochondrial matrix in proportion to the mitochondrial membrane potential. NRCMs were loaded with 100 nM TMRM 30 min before imaging. Imaging was initiated by exciting TMRM through a 546/11-nm bandpass filter, and emission was assessed through a 567/15-nm bandpass filter. Exposure duration was set at 200 ms. Fluorescence is normalized to time 0 for each treatment.
Data Analysis.
The data are reported as means ± S.E.M. Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Dunnett's post-test. Significance was accepted when p < 0.05.
Results
TRO40303 Specifically Binds to the TSPO Protein.
TRO40303 (Fig. 1A) is a new chemical entity that was derived from a chemistry optimization program based on the structure of cholest-4-en-3-one, oxime (TRO19622), also called olesoxime (Bordet et al., 2007). TRO40303 was able to reduce cell death of trophic factor-deprived motor neurons as effectively as olesoxime (not shown). Because olesoxime showed specific binding to TSPO and VDAC (Bordet et al., 2007), we investigated whether TRO40303 also binds to these two proteins.

Chemical structure of TRO40303 (3,5-seco-4-nor-cholestan-5-one oxime-3-ol) (A), binding of TRO40303 to cholesterol but not PK11195 sites on TSPO (B and C), and tissue distribution of TRO40303 (D and E). Recombinant mouse TSPO was reconstituted into liposomes and binding was measured by the ability to displace specific TSPO radioligands with TRO40303. B, [3H]cholesterol displaced by TRO40303 (▵) or unlabeled cholesterol (■). C, [3H]PK11195 displaced by TRO40303 (○) or unlabeled PK11195 (●). Data points are representative of four to five independent experiments, each performed in triplicate. Blood levels and tissue distribution of TRO40303 was measured in rats after intravenous administration of 2 mg/kg [14C]TRO40303. D, tissue distribution pattern of the drug measured by whole-body autoradiography 1 h after its administration. E, relative distribution of TRO40303 in whole blood and heart with time (▴, myocardium; ■, blood).
TRO40303 was studied for its ability to interact with either the PK11195- or cholesterol-binding sites on purified recombinant mouse TSPO reconstituted into proteoliposomes (Jamin et al., 2005). As shown for olesoxime, TRO40303 significantly displaced [3H]cholesterol binding to recombinant TSPO (Ki = 150 nM) (Fig. 1B) but did not displace [3H]PK11195 (Fig. 1C). These results demonstrate that this cholesterol-oxime family interacts with TSPO specifically at the cholesterol-binding site probably because of their cholesterol-like structure. TRO40303 was also tested for its ability to bind to the neurosteroid binding site on VDAC (Darbandi-Tonkabon et al., 2003). Unlike olesoxime, TRO40303 did not significantly displace [3H]6-azi-pregnanolone binding to VDAC in rat brain membrane preparations (not shown).
To identify other potential molecular targets of TRO40303, it was screened on a large panel of pharmacologically important targets (Diversity profile; CEREP, Poitiers, France) at concentrations up to 30 μM. Among the 69 receptors, channels, or transporters and 16 enzymes tested, TRO40303 did not show any reproducible or specific binding or functional interference (not shown). Thus, the binding of TRO40303 to the cholesterol site of the TSPO protein is extremely specific, and this protein might represent its cellular target.
Tissue Distribution of TRO40303.
Tissue distribution of TRO40303 was evaluated after a single intravenous administration of 2 mg/kg [14C]TRO40303 to fasted male rats. After 1 h [14C]TRO40303 was highly distributed throughout the body with the highest affinities for the liver, lungs, hypophysis, kidneys, adrenals, intestines/contents, bone marrow, and spleen and lowest for brain (Fig. 1D). TRO40303 accumulated in the heart by a factor of 2 to 4 times that in total blood, depending on time (Fig. 1E). The distribution pattern of TRO40303 was similar to that of TSPO described for ligands such as chlorodiazepam or PK11195, which showed the highest levels in steroidogenic tissues such as adrenal cortex but also substantial levels in tissues with a high concentration of mitochondria such as heart, brown fat, and regions of the kidney, lung, liver, and spleen but only low levels in brain (Anholt et al., 1985).
Effects of TRO40303 in a Myocardial Infarction Model in Rats.
Because TRO40303 accumulates in heart and possesses cytoprotective activity similar to that of olesoxime, which appears to involve a reduction in mitochondrial permeability transition (Bordet et al., 2007), and because mPTP appears to be a key step in I/R cardiotoxicity, TRO40303 was tested for its effect on infarct size in a model of I/R in rats. As shown in Fig. 2A, TRO40303 at 2.5 mg/kg, administered just before reperfusion, reduced infarct size by 38% (p < 0.01), whereas the doses of 0.5 and 1.25 mg/kg were not active. At 2.5 mg/kg, TRO40303 had no effects on either heart rate or mean arterial pressure (not shown).

TRO40303 reduces infarct size in a model of myocardial infarction in rats. TRO40303 formulated in HPBCD was administrated by intravenous bolus to rats 10 min before CAR after 35 min of CAO. A, infarct sizes were quantified at 24 h of reperfusion and expressed for each group as a percentage of the AAR. The corresponding AAR of each group of treated rats was expressed as a percentage of the left ventricular weight (LVW). n, number of treated rats per group; ○, individual values; ●, mean ± S.E.M. for each group. The dose of 2.5 mg/kg provided significant protection in this model of myocardial infarction (**, p < 0.01 versus control I/R rats treated with HPBCD as vehicle by ANOVA followed by Dunnett's post-test), whereas lower doses were not active. B and C, Western blot of cytosolic fractions issued from the AAR isolated 1 h after CAR or a similar region of sham-operated rats. B, typical Western blot of cytochrome c (top image) and quantification of the cytochrome c band in cytosolic fractions (bottom graph). C, typical Western blot of AIF (top image), indicated by the arrow, and quantification of the AIF band (bottom graph). The numbers of fractions analyzed were collected from three to seven animals as indicated. **, p < 0.01 versus the corresponding I/R value by ANOVA followed by Dunnett's post-test. A.U., arbitrary units.
TRO40303 Did Not Modify CRC of Isolated Cardiac Mitochondria but Inhibited Apoptotic Factor Release after I/R In Vivo.
In a first step, we investigated whether TRO40303 could increase CRC in isolated cardiac mitochondria from a normal myocardium. As seen in Fig. 3, 12.5 ± 1.1 pulses of 10 μM Ca2+ (125 nmol/mg mitochondrial protein) were required to induce mPTP opening in control mitochondria. The presence of 3 μM TRO40303 in the medium did not increase CRC (11.0 ± 1.5 pulses: 110 nmol/mg mitochondrial protein), whereas 1 μM CsA, a known mPTP and CRC inhibitor, significantly increased it (16.3 ± 0.4 pulses: 163 nmol/mg mitochondrial protein).
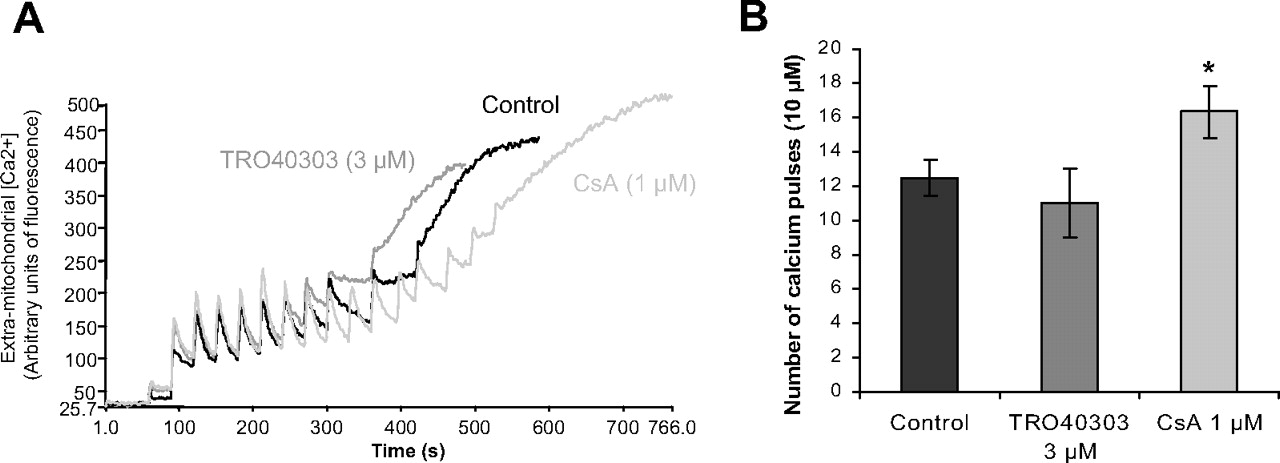
TRO40303 does not inhibit mPTP opening in isolated mitochondria. The reaction was started by addition of successive 10 μM Ca2+ pulses. After each addition, a rapid uptake was observed followed by a dynamic steady state corresponding to the equilibrium between the influx and the efflux of Ca2+. When the maximal Ca2+ loading threshold was reached, this equilibrium was disrupted and Ca2+ was released. A, in this representative experiment, 11 pulses of 10 μM Ca2+ were required to induce mPTP opening in control mitochondria, 10 pulses in the presence of TRO40303 (3 μM), and 16 pulses in the presence of CsA (1 μM). B, average of pulses required for mPTP opening in n = 3 experiments. *, p < 0.05 versus control by ANOVA followed by Dunnett's post-test.
In a second step, to determine whether the effects of TRO40303 on CRC would only be observed when mitochondria were challenged in vivo with a more complex mitochondrial stress, the effects of TRO40303 on CRC were then evaluated on cardiac mitochondria isolated from rats subjected to 35 min of CAO and 10 min of CAR treated or not with 2.5 mg/kg TRO40303, 10 min before CAR. In addition, administration of TRO40303 in vivo might allow the compound to interact with its physiological target more easily than when incubated in vitro with isolated mitochondria from a normal heart. The amount of Ca2+ required to trigger mPTP opening in mitochondria isolated from sham-operated rats reached 92.5 ± 16.0 nmol/mg mitochondrial protein, similar to that seen in the previous study. After I/R, CRC was reduced (35.8 ± 9.0 nmol/mg mitochondrial protein) (p < 0.05), and this was unchanged by TRO40303 administration (38.9 ± 11.6 nmol/mg mitochondrial protein), again demonstrating that the drug did not modify isolated mitochondrial sensitivity to Ca2+-induced mPTP opening sensitized by I/R.
However, in the same series of experiments, mitochondria and cytosol were separated and analyzed by Western blot to observe the effect of TRO40303 treatment on the release of proapoptotic factors. As shown in Fig. 2, B and C, cytochrome c and AIF were increased in cytosolic fractions isolated from AAR of rats after 35 min of CAO and 1 h of CAR. Treatment with TRO40303 (2.5 mg/kg) 10 min before reperfusion limited the release of AIF (p < 0.05) although the release of cytochrome c was not significantly affected. These results show that although TRO40303 treatment had no effect on Ca2+-induced mPTP as measured by CRC in mitochondria once they have been isolated from their intracellular environment, TRO40303 reduced the release of AIF and further cell death in the AAR after I/R in rats.
TRO40303 Delayed mPTP Opening after Hypoxia/Reoxygenation in Isolated Cardiomyocytes.
TRO40303 binds to TSPO, reduces infarct size, and limits apoptosis by reducing the release of AIF from the mitochondria to the cytosol after in vivo ischemia reperfusion, but TRO40303 showed no effect on the CRC of isolated mitochondria. To reconcile these effects that could seem paradoxical, we hypothesized that TRO40303 might inhibit mPTP opening only in a whole-cell environment, because it has an effect via the TSPO located at the outer membrane of the mitochondria. To investigate this hypothesis, we used a model of adult rat cardiomyocytes challenged by hypoxia and reoxygenation in vitro, and mPTP opening was followed using the calcein-cobalt staining method. As shown in Fig. 4 and Table 1, 3 μM TRO40303 delayed mPTP opening, as measured by an increase in the time to detect a 50% decrease in mitochondrial calcein fluorescence, in a manner similar to that of 1 μM CsA. Two types of fluorescent signal kinetics were observed: either a gradual decrease or a complete and rapid drop in calcein fluorescence. These phenomena probably correspond to the transient (reversible) and long-lasting (irreversible) opening of the mPTP as described previously (Sharov et al., 2007). TRO40303 greatly increased the percentage of cardiomyocytes that displayed gradual mPTP openings compared with that in control cells (Table 1). Concomitantly, TRO40303 delayed nuclear labeling by propidium iodide, indicating that the compound also delayed the hypoxia/reoxygenation-induced cardiomyocyte cell death. In conclusion, although TRO40303 had no effect on Ca2+-induced mPTP opening in isolated mitochondria, TRO40303 reduced mPTP opening in cardiomyocytes stressed by hypoxia/reoxygenation in vitro.

TRO40303 delays mPTP opening and cell death after hypoxia/reoxygenation in isolated adult rat cardiomyocytes. The time course of mPTP opening and cell death triggered by reoxygenation of hypoxic cardiomyocytes was evaluated in the absence or presence of 3 μM TRO40303. Cardiomyocytes were coloaded with calcein, CoCl2, and propidium iodide (PI). Images were collected at 1-min intervals. The fluorescence was normalized to 100% of the initial value for calcein and 100% of the final value for PI. Traces from three to five experiments were averaged.
Effects of TRO40303 and CsA on mPTP opening after hypoxia/reoxygenation
Cardiomyocytes were coloaded with calcein and CoCl2, and mPTP opening was induced by in vitro hypoxia/reoxygenation. The percentage of cells undergoing rapid or gradual opening and average time to mPTP opening for the cell population in which mPTP opened after reoxygenation was calculated. Statistical analysis was performed by one-way ANOVA versus control followed by Dunnett's post-test.
TRO40303 Inhibited H2O2-Induced mPTP Opening.
To better understand how TRO40303 mediates protection against oxidative damage present in I/R injury, we focused on oxidative stress-induced mitochondrial death signals using H2O2 intoxication in newborn rat cardiomyocytes, which is a well studied model of ROS-induced cardiomyocyte death in which various cardioprotective compounds and mechanisms have been studied in detail (Teshima et al., 2003a; Jones et al., 2008; Ngoh et al., 2009). We first assessed whether TRO40303 affected mPTP opening using calcein fluorescence in NRCMs (Petronilli et al., 1999). Fluorescent calcein concentrates in the mitochondrial matrix, and H2O2-induced mPTP formation resulted in changing the punctate calcein fluorescence colocalized with MitoTracker Red to a more diffuse fluorescence (Fig. 5A). Diminishing punctate calcein fluorescence is reflected by a decrease in the S.D. around the mean pixel fluorescence, and this was used to follow mPTP opening over time in NRCMs in response to treatment with H2O2 in the presence or absence of either TRO40303 or CsA (Fig. 5B). Measuring the absolute change in the S.D. showed that treatment with H2O2 initiates an immediate and sustained decrease in S.D. compared with that for control, untreated cells. At the same time, no change in mitochondria distribution was detected using MitoTracker Red fluorescence (Fig. 5A). By these criteria, pretreatment with 3 μM TRO40303 or 1 μM CsA significantly delayed H2O2-mediated mPTP formation as evaluated 60 min after H2O2 intoxication (Fig. 5, A–C; Table 2). These results demonstrate the similarity of the effects of TRO40303 and CsA on H2O2-induced mPTP opening in whole cells.

TRO40303 inhibits mPTP opening in newborn rat cardiomyocytes intoxicated with H2O2. Assessment of sensitivity to formation of mitochondrial permeability transition pore (mPTP) in NRCMs treated with DMSO (0.1%, control), TRO40303 (3 μM), or CsA (1 μM) before H2O2 treatment using calcein and MitoTracker Red. Loss of calcein fluorescence was used to indicate mPTP (n = 6 per group). H2O2 treatment induced mPTP opening, which was significantly attenuated by TRO40303 and CsA. A, representative montages showing change in calcein and MitoTracker Red fluorescence over time. B, time-dependent changes in S.D. of mean calcein fluorescence intensity in NRCMs exposed to nothing (control, □), H2O2 (■), H2O2 + TRO40303 (○), and H2O2 + CsA (gray circles) for one representative experiment (from n = 6). C, quantitative bar graph showing the change in S.D. of mean calcein fluorescence intensity between 0 and 60 min. Average of n = 6 experiments. *, p < 0.05; **, p < 0.01 versus H2O2 by ANOVA followed by Dunnett's post-test.
Quantitative effects of TRO40303 and CsA at 60 min after H2O2 exposition in newborn rat cardiomyocytes on ROS production (DCF), Ca2+ overload in cytosol (Fluo-4) and mitochondria (Rhod-2), mPTP opening (Δ calcein S.D.), and loss of mitochondrial membrane potential (TMRM)
Data are expressed in arbitrary units of fluorescence or percent variation ± S.E.M. Statistical analysis was performed by one-way ANOVA versus H2O2 treatment followed by Dunnett's post-test.
TRO40303 Reduced H2O2-Induced ROS Production, Ca2+ Overload, and Loss of Mitochondrial Membrane Potential.
ROS and Ca2+ overload are key contributors to I/R injury. We used the H2O2 intoxication model to investigate the mechanism of action of TRO40303 through the assessment of its effects on ROS production and Ca2+, as well as the sequence of these events in comparison with mPTP opening induction.
DCF was used to measure ROS production. H2O2 induced an immediate significant increase in ROS generation in NRCMs as reflected by increased DCF fluorescence (Fig. 6A; Table 2). Pretreatment of NRCMs with 3 μM TRO40303 before H2O2 reduced the induction of ROS production starting between 10 and 20 min after H2O2 exposition and reaching a significant reduction 60 min after the addition of H2O2. Pretreatment with 1 μM CsA slightly decreased the rate of H2O2-induced ROS production, but the effect on the DCF fluorescence decrement 60 min after addition of H2O2 was not statistically significant.

Evaluation of the effects of TRO40303 on the ROS, mitochondrial and cytosolic Ca2+, and ΔΨm in NRCMs after H2O2 treatment. NRCMs were treated with DMSO (0.1%, control), TRO40303 (3 μM) or CsA (1 μM) before H2O2 treatment. A, ROS was evaluated using DCF (n = 6/group). B, cytosolic calcium using Fluo-4 (n = 6 per group). C, mitochondrial calcium using Rhod-2 (n = 5/group). D, TMRM fluorescence was used to indicate mitochondrial membrane potential (n = 6/group). H2O2 treatment induced ROS production, mitochondrial and cytosolic Ca2+ overload, and loss of ΔΨm, events that were all inhibited by TRO40303 and CsA with various efficacy and kinetics. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus H2O2 by ANOVA followed by Dunnett's post-test for values at 60 min for DCF, Fluo-4, and Rhod-2 and at 90 min for TMRM.
To determine whether TRO40303 had an effect on H2O2-induced Ca2+ overload, we measured cytoplasmic Ca2+ levels using Fluo-4 fluorescence and mitochondrial Ca2+ levels using Rhod-2 fluorescence. Treatment with 100 μM H2O2 caused a significant rise in cytosolic and mitochondria Ca2+ levels as measured by Fluo-4 and Rhod-2 fluorescence, respectively (Fig. 6, B and C), with the onset of the rise between 20 and 30 min after H2O2 addition. The increase in both cytosolic and mitochondrial Ca2+ were significantly attenuated by pretreatment with 3 μM TRO40303 or 1 μM CsA (Table 2). BAPTA-AM, an intracellular Ca2+ chelator, completely blocked the H2O2-mediated increase in both Fluo-4 and Rhod-2 fluorescence, confirming that these increases in fluorescence were due to increased [Ca2+]i (not shown).
We examined the effects of TRO40303 treatment on oxidative stress-induced loss of mitochondrial membrane potential using TMRM fluorescence. H2O2 treatment significantly reduced TMRM fluorescence by 90 min, and this decrease was reduced by 3 μM TRO40303, although not significantly, indicating a tendency for preservation of Δψm (Fig. 6D; Table 2). CsA pretreatment significantly attenuated the H2O2-mediated decrease in TMRM fluorescence and loss of Δψm at 90 min.
Discussion
We report here the identification and characterization of a new cardioprotective compound, TRO40303, selected from a chemical optimization program beginning with olesoxime based on the ability to protect trophic factor-deprived primary rat motor neurons. TRO40303 retains a cholesterol-like structure explaining its binding to the cholesterol site on TSPO. The compound has a tissue distribution that corresponds very well to the known expression pattern of TSPO, most probably accounting for its uptake and higher relative concentration in heart compared with plasma.
Binding of TRO40303 to TSPO was very specific as no displacement or competition with ligands or substrates was found for a large panel of physiologically important receptors, enzymes, transporters, and ion channels that could mediate its pharmacological action or interfere with further in vivo studies. Although TSPO is the target of a number of compounds previously shown to have neuroprotective and/or cardioprotective properties such as PK11195, chlorodiazepam (Obame et al., 2007), or SSR180575 (Leducq et al., 2003), these compounds bind to a promiscuous drug-binding site on TSPO that appears to influence cholesterol uptake and steroidogenesis, which is initiated by CYP11A1 (or P450scc), located in the inner mitochondrial membrane (Papadopoulos et al., 2006). Even though TRO40303 competes for cholesterol binding to TSPO, the drug had no major effect on plasma cholesterol levels in preclinical safety and toxicology studies (not shown) and so is unlikely to have an effect on cholesterol metabolism or steroidogenesis. This may be explained by the fact that TRO40303 has a lower affinity than cholesterol for TSPO. The full significance of the interaction of TRO40303 with the cholesterol site of TSPO remains to be investigated; however, it might account for the targeting of TRO40303 to the mitochondria as for cholesterol and olesoxime.
A tissue distribution study of TRO40303 in rat showed that it had a relatively high concentration in the heart, whereas it penetrated poorly in brain and that its distribution was similar to that of TSPO, highlighting the interest to test this cytoprotective compound in a myocardial infarction model in which the mitochondrial permeability transition has been shown to play a major role in I/R injury. We show that TRO40303 reduced myocardial infarct size in rats and delayed mPTP opening and mortality triggered by hypoxia reoxygenation of adult primary rat cardiomyocytes in vitro. Cardiac protection by TRO40303 was correlated with a reduction in AIF release in cytosolic fractions of cardiac tissue isolated from the ischemic area, whereas the effect on cytochrome c release was variable and not significant. Independent release of AIF versus cytochrome c has been shown in isolated heart after hypoxia/reoxygenation, and this phenomenon was blocked by CsA (Kim et al., 2003). In addition, an antibody against TSPO has also been shown to selectively prevent AIF release but not that of cytochrome c after Ca2+-induced mPTP opening in rat brain mitochondria (Azarashvili et al., 2007), providing evidence that the release of these two proapoptotic factors can be dissociated. Although the effect of TRO40303 on AIF alone might be sufficient to account for the 38% reduction in infarct size in rats treated with 2.5 mg/kg TRO40303, it must also be noted that AIF and cytochrome c release were measured 1 h after reperfusion, and it cannot be ruled out that an effect on cytochrome c might be more apparent at another time point.
The specific binding of TRO40303 to TSPO, a protein structurally related to TspO, a bacterial oxygen sensor (Yeliseev et al., 1997), may play a role in the modulation of ROS-induced mPTP opening. Indeed TSPO expression levels are positively correlated with survival of hematopoietic cells exposed to H2O2 (Carayon et al., 1996). To investigate this possibility, we have closely examined the effect of TRO40303 on oxidative stress-induced mPTP opening using H2O2 intoxication of newborn rat cardiomyocytes. When looking carefully at the onset of events and effects of both TRO40303 and CsA as well as the extent of these effects in these experiments, it seems that both compounds have slightly different effects: the effects of TRO40303 on ROS and mPTP opening occurs slightly earlier than the effects of CsA on these two events, whereas CsA inhibition of mPTP opening appears to coincide with its effects on ROS production and calcium overload. Interestingly, the effects of TRO40303 and CsA on both mitochondrial and cytosolic calcium increases are nearly identical. This finding suggests that the massive calcium increase is a secondary effect after mPTP opening, which is in agreement with the effect observed by Lemasters et al. (2009) in studying hypoxia reoxygenation in adult cardiomyocytes. The different modes of mPTP inhibition by TRO40303 and CsA could correspond to the ROS and calcium triggers of mPTP opening as described by Hossain et al. (2009). Interestingly, whereas TRO40303 significantly reduced ROS production, at 60 min H2O2-induced ROS production in the presence of CsA showed only a trend to reduction, yet both TRO40303 and CsA reduced mPTP opening by approximately 50%. This could be understood as follows: the inhibitory effects of TRO40303 on mPTP may be mediated primarily through reduction of ROS production, whereas CsA inhibits mPTP opening mainly by binding to CyD and inhibiting its prolyl-isomerase activity, resulting in the displacement of CyD from the mPTP.
Together, these studies demonstrate the ability of TRO40303 to modulate mPTP opening in intact cells after hypoxia/reoxygenation or H2O2 intoxication. Yet, unlike CsA, TRO40303 has no effect on CRC, a measure of Ca2+-triggered mPTP in isolated mitochondria, highlighting the different mechanisms that can be modulated to inhibit mPTP in cells. This finding is consistent with the different proteins targeted by CsA and TRO40303. CsA targets CyD, located in the mitochondrial matrix where Ca2+ overload triggers mPTP opening. TRO40303 binds to TSPO in the outer mitochondrial membrane where it prevents mPTP opening in intact cardiomyocytes induced by I/R and by H2O2-activated ROS production. The difference between TRO40303 and classic TSPO ligands such as PK11195, chlorodiazepam, or SSR180575 is that TRO40303 binds to the cholesterol site and is potentially a substrate of TSPO, whereas classic TSPO ligands modulate cholesterol uptake and steroidogenesis, which may mediate the role of TSPO in protection from oxidative stress. The effects of TRO40303 on mitochondrial permeability transition could also be due to an effect on the interaction of TSPO with other putative members or modulators of the mPTP such as VDAC (McEnery et al., 1992). Alternatively, because of its cholesterol-like structure, TRO40303 may, like cholesterol, favor the VDAC-hexokinase interaction, increasing glycolysis and reducing the need for oxidative phosphorylation to maintain ATP levels (Pastorino and Hoek, 2008). Such an effect would be beneficial under ischemic conditions and protect tissue by reducing ROS formation via the electron transport chain once oxygen supply is restored. These indirect effects may prevent or delay mPTP opening in cardiomyocytes subjected to I/R or H2O2 intoxication and explain the cytoprotective effects of TRO40303 in intact cells even though it has no effect on CRC as a measure of Ca2+-induced mPTP opening in isolated mitochondria. It must be noted that even CsA is unable to increase CRC in the absence of phosphate and does not inhibit ROS-triggered mPTP opening in mitochondrial assay systems (He and Lemasters, 2002; Basso et al., 2008; Hossain et al., 2009). Thus, the fact that a compound does not inhibit CRC under particular conditions does not mean that it is not an inhibitor of mPTP opening in a cellular context; and this is what is observed with TRO40303.
In conclusion, we have identified TRO40303 as a new cardioprotective molecule that binds to TSPO, inhibits the mitochondrial permeability transition in intact cells, and reduces infarct size in a rat model of myocardial infarction when administered at reperfusion. Therefore, TRO40303 represents both a promising new tool to further investigate mechanisms involved in mPTP opening as well as a new class of drugs to treat reperfusion injury to the heart.
Acknowledgments
We thank collaborators at Trophos for their contributions to this work: Isabel Clémançon de Bellefois, Mélanie Gabriac, and Caroline Gourné for their technical support and advice on in vitro studies; Virginie Latyszenok and Magali Michaud for performing the pharmacokinetics experiments; the screening and pharmacology department for their initial work identifying TRO40303, in particular Pascal Galéa, Gwenaëlle Tardif, and Esther-Marie Steidl; the analytical chemistry department for performing bioanalysis of TRO40303, in particular Jean Afxantidis, Delphine Maux, Prisca Lonski, and Florence Ransilhac; and finally Antoine Béret, Damian Marron, Patrick Berna, and Jean-Louis Abitbol for their continuous suggestions and encouragement to the project.
Footnotes
This work was supported in part by the French Agence Nationale pour la Recherche [Grant ANR-07-RIB-IRI stop]. S.P. and R.A. were supported by doctoral grants from the Ministère de la Recherche et de la Technologie and the Région Ile de France, respectively. A.B. has served as a consultant and received an honorarium from Trophos. S.P.J. and A.B. received research grants from TROPHOS to support this project.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.167486.
-
ABBREVIATIONS:
- mPTP
- mitochondrial permeability transition pore
- I/R
- ischemia-reperfusion
- ROS
- reactive oxygen species
- CsA
- cyclosporine A [(2S,5S,8S,11S,14R,17S,20S,23S,26S,32S)-32-ethyl-2-[(E,1R,2R)-1-hydroxy-2-methylpent-3-enyl]-3,6,9,12,14,17,21,27,30-nonamethyl-5,8, 11,20,26-pentakis(2-methylpropyl)-23-propan-2-yl-3,6,9,12,15,18,21,24,27,30,33-undecazacyclotritriacontane-1,4,7,10,13,16,19,22,25,28, 31-undecone]
- VDAC
- voltage-dependent anion channel
- TSPO
- translocator protein 18 kDa
- Ro5-4864
- 7-chloro-5-(4-chlorophenyl)-1,3-dihydro-1-methyl-2H-1,4-benzodiazepine-2-one
- SSR180575
- 7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide
- TRO40303
- 3,5-seco-4-nor-cholestan-5-one oxime-3-ol
- DMSO
- dimethyl sulfoxide
- HPBCD
- hydroxypropyl-β-cyclodextrin
- calcein-AM
- calcein acetoxymethyl ester
- DCF
- 5(and 6-)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acyl ester
- TMRM
- tetramethylrhodamine methyl ester
- PK11195
- 1-(2-chlorophenyl)-N-methyl-N-(1-methyl-propyl)-3-isoquinoline carboxamide
- CAO
- coronary artery occlusion
- CAR
- coronary artery reperfusion
- AAR
- area at risk
- AIF
- apoptosis inducing factor
- CRC
- calcium retention capacity
- NRCM
- neonatal rat cardiomyocyte
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- ANOVA
- one-way analysis of variance
- TRO19622
- cholest-4-en-3-one, oxime.
- Received February 19, 2010.
- Accepted March 9, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics
References
- Anholt et al., 1985.↵
- Argaud et al., 2005.↵
- Azarashvili et al., 2007.↵
- Baines, 2009.↵
- Baines et al., 2005.↵
- Baines et al., 2007.↵
- Basso et al., 2005.↵
- Basso et al., 2008.↵
- Bordet et al., 2007.↵
- Carayon et al., 1996.↵
- Darbandi-Tonkabon et al., 2003.↵
- Gomez et al., 2007.↵
- Griffiths and Halestrap, 1995.↵
- Halestrap, 2006.↵
- Hausenloy et al., 2009.↵
- Haworth and Hunter, 1979.↵
- He and Lemasters, 2002.↵
- Hossain et al., 2009.↵
- Inagaki et al., 2005.↵
- Jamin et al., 2005.↵
- Javadov et al., 2009.↵
- Jones et al., 2008.↵
- Kim et al., 2003.↵
- Kokoszka et al., 2004.↵
- Lacapère et al., 2001.↵
- Leducq et al., 2003.↵
- Lemasters et al., 2009.↵
- Leung et al., 2008.↵
- Marzo et al., 1998.↵
- Massoudy et al., 1997.↵
- McEnery et al., 1992.↵
- Ngoh et al., 2009.↵
- Nieminen et al., 1995.↵
- Obame et al., 2008.↵
- Obame et al., 2007.↵
- Papadopoulos et al., 2006.↵
- Pastorino and Hoek, 2008.↵
- Petronilli et al., 1999.↵
- Piot et al., 2008.↵
- Sharov et al., 2007.↵
- Shimizu et al., 1999.↵
- Squadrito et al., 1999.↵
- Staat et al., 2005.↵
- Teshima et al., 2003a.↵
- Teshima et al., 2003b.↵
- Veenman et al., 2007.↵
- Yeliseev et al., 1997.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}