


Abstract
To elucidate the role of cyclooxygenase (COX)-2 in ulcer healing, we compared the effects of NS-398 (COX-2-selective inhibitor) and indomethacin (nonselective COX inhibitor) on the healing of acetic acid-induced gastric ulcers in rats. Prostaglandin E2(PGE2) production was elevated in ulcerated tissue, but remained unaffected in intact tissue. COX-2 mRNA was only detected in the ulcerated tissue, in which the COX-2 protein was found in fibroblasts, macrophages/monocytes and granulocytes. In contrast, COX-1 mRNA expression was not affected by ulceration. In an in vitro study, the increased PGE2 production was inhibited by NS-398; this had no effect on PGE2 production in the intact tissue. When NS-398 and indomethacin were administered to rats, 3 and 6 mg/kg NS-398 only reduced PGE2 production in the ulcerated tissue, but 10 mg/kg NS-398 and 0.5 to 2 mg/kg indomethacin inhibited the production in both the ulcerated and intact tissues. The healing of gastric ulcers was significantly impaired by 3 to 10 mg/kg NS-398 and 1 and 2 mg/kg indomethacin. The delay in ulcer healing was associated with the inhibition of PGE2production in the ulcerated tissue. As observed upon histological analysis, regeneration of the mucosa, maturation of the ulcer base and angiogenesis in the base were significantly prevented by 6 mg/kg NS-398 and 2 mg/kg indomethacin, although the inhibitory effect of NS-398 was weaker than that of indomethacin. These results clearly indicate that COX-2 plays an important role in the healing of gastric ulcers in rats.
NSAIDs inhibit the activity of COX, the key enzyme in PG production (Vane, 1971). COX exists in two isoforms, of which COX-1 is a constitutive enzyme expressed in many tissues including the stomach, and COX-2 is normally undetectable in most tissues, its expression being induced at inflammatory sites (Mitchell et al., 1995; Herschman, 1996). Conventional NSAIDs such as indomethacin inhibit both COX-1 and COX-2 activities, although COX-2 selective inhibitors such as NS-398 have been developed as new NSAIDs (Arai et al., 1993; Futakiet al., 1993, 1994; Vane, 1994; Pairet and Engelhardt, 1996). It is well known that treatment with conventional NSAIDs causes a delay in the healing of gastric ulcers; this delay is associated with a reduction of the increased PG production in ulcerated tissues in rats and humans (Szelenyi et al., 1982; Wang et al.,1989; Levi et al., 1990; Lancaster-Smith et al.,1991). It follows that exogenous PGE2 prevents the indomethacin-induced delay in ulcer healing in rats (Wang et al., 1989).
Recently, Mizuno et al. (1997) reported that COX-2 is induced by gastric ulceration in mice, resulting in an increase in PG production. Consequently, it is thought that COXs and PGs play important roles in gastric ulcer healing. In addition, Mizuno et al. (1997) claimed that COX-2 may play an important role in ulcer healing, based on the finding that the ulcerated area is larger in NS-398-treated mice than controls. However, in their study, it was impossible to distinguish between the roles of COX-2 in ulcer development and ulcer healing, because NS-398 was administered from the day of ulcer induction. Furthermore, they only examined the effect of NS-398 at a single dose (10 mg/kg, i.p.), and did not determine PG production in the gastric tissue after the administration of NS-398.Futaki et al. (1993) reported that orally administered NS-398 at 10 mg/kg significantly reduces PGE2 production in the gastric mucosa to about 40% of the control level, suggesting that NS-398 administered at >10 mg/kg may also inhibit COX-1 activity in the gastric tissue, as do conventional NSAIDs. Accordingly, it remains unknown whether or not the impairment of ulcer healing can be caused by the inhibition of COX-2 activity alone. Therefore, we closely examined the effect of NS-398 on PGE2 production in gastric tissues and the healing of acetic acid ulcers in rats, obtained corresponding data for indomethacin, and analyzed the dissimilarities to elucidate the role of COX-2 in gastric ulcer healing.
Materials and Methods
Production of gastric ulcers.
Male Donryu rats (Nihon SLC, Hamamatsu, Japan), weighing 250 to 300 g, were used. Under ether anesthesia, gastric ulcers were induced by submucosal injection of 20% acetic acid (0.04 ml) into the border between the antrum and the fundus on the anterior wall of the stomach (Takagi et al., 1969). After closure of the abdomen, the rats were maintained in the usual manner. Because deep, well-defined ulcers were observed 5 days after the acid injection, we defined the 5th day as the day of ulceration (day 0). At the indicated times, rats were killed and their stomachs were excised. Subsequently, the stomachs were incised along the greater curvature and the ulcerated area (mm2) was determined under a dissecting microscope (×10; Olympus, Tokyo, Japan). The investigator (S.O.) determining the ulcer size was unaware of the treatment given the animals. The preventive rates for ulcer healing with COX inhibitors were calculated as follows: Preventive rate (%) = {[(ulcerated area on day X in the drug group) − (ulcerated area on day X in the control group)]/[(ulcerated area on day 0 in the control group) − (ulcerated area on day X in the control group)]} × 100.
Northern blot analysis of COX mRNA expression.
Rat COX-1 and COX-2 cDNA probes were prepared by means of the reverse transcription polymerase chain reaction, as described previously (Feng et al., 1993). Total RNA for amplification of these cDNAs was isolated from the spleen of a lipopolysaccharide-infused rat. The primers used were as follows: 5′-AACCGTGTGTGTGACTTGCTGAA-3′ and 5′-AGAAGGAGCCCCTCAGAGCTCAGTG-3′ for COX-1 and 5′-TGATGACTGCCCAACTCCCATG-3′ and 5′-AATGTTGAAGGTGTCCGGCAGC-3′ for COX-2. The products corresponding to COX-1 (887 base pairs) and COX-2 (702 base pairs) were purified from polyacrylamide gels, and used after their sequences had been confirmed to be completely identical to known ones (with reference to the databases of GenBank and EMBL). The cDNA probes were 32P-labeled by the random primer method (Ready-To-Go; Pharmacia Biotec, Uppsala, Sweden). Gastric specimens were taken from both intact (posterior side) and ulcerated tissues of stomachs with ulcers and from stomachs without ulcers. Total RNAs were extracted by means of the acid-guanidinium thiocyanate-phenol-chloroform method (Chomczynski and Sacchi, 1987), using TRIZOL (GIBCO BRL, Gaithersburg, MD). Poly (A)+ RNAs were purified with Oligotex dT30 (TaKaRa, Kyoto, Japan). Poly (A)+ RNAs (0.2 μg) were separated by electrophoresis on 1.2% agarose gels, transferred to nylon membranes (Gene Screen Plus; New England Nuclear, Boston, MA), and then hybridized with32P-labeled cDNA probes (Sambrook et al., 1989). The detection and quantification of hybridized mRNAs were carried out with an imaging analyzer (BAS-5000Mac; Fuji Film, Tokyo, Japan). The levels of the mRNAs were expressed as the ratio to glyceraldehyde-3-phosphate dehydrogenase mRNA.
Immunohistochemical detection of the COX-2 protein.
Gastric specimens were taken from the ulcerated tissue. After they had been fixed with 4% paraformaldehyde in phosphate-buffered saline, frozen sections (14-μm thick) were prepared. The sections were incubated with anti-COX-2 antibody (Cayman Chemicals, Ann Arbor, MI) after deactivation of endogenous peroxidase with 0.3% H2O2 and blockage of nonspecific binding sites. The COX-2 protein was visualized by the avidin-biotin-peroxidase complex method using a Vectastain ABC-peroxidase kit (Vector Laboratories, Burlingame, CA) and 3, 3′-diaminobenzidine tetrahydrochloride (Dojindo Laboratories, Kumamoto, Japan). The sections were successively stained with hematoxylin. Macrophages/monocytes and granulocytes were identified by staining with macrophage-specific and granulocyte-specific esterase activities (Sigma Chemical Co., St. Louis, MO), respectively.
Determination of PGE2 production in gastric tissues.
PGE2 production was assayed according to the method of Lee and Feldman (1994). Unless otherwise stated, 3 hr after the administration of the drugs, gastric specimens were taken from both intact (posterior side) and ulcerated tissues of stomachs with ulcers, and from stomachs without ulcers. The specimens were placed in 50 mM Tris-HCl (pH 8.4) buffer and then finely minced. After the tissues had been washed and resuspended in 1 ml of buffer, they were subjected to vortex mixing at room temperature for 1 min to stimulate PGE2 production, followed by centrifugation at 10,000 × g for 15 sec. To examine the in vitro effects of COX inhibitors, the tissues were preincubated with the drugs or the vehicle on ice for 10 min before stimulation. The amounts of PGE2 in the resulting supernatants were determined by enzyme-immunoassaying (PGE2 EIA kit; Cayman Chemicals). PGE2 production was expressed as pg PGE2/mg tissue/min.
Histological evaluation of ulcer healing.
Gastric specimens were taken from ulcerated tissues, and then fixed in 10% formalin and embedded in paraffin. Six-μm sections were prepared, and then stained with hematoxylin and eosin. As described previously by our group (Tsukimi et al., 1996), the length of the regenerated mucosa on the ulcer base and the thickness of the base were measured under a light microscope. These measurements are presented, respectively, as regeneration of the mucosa and maturation of the ulcer base.
For evaluation of angiogenesis in the ulcer base, gastric specimens were fixed in 4% paraformaldehyde and 14-μm frozen sections were prepared. The sections were incubated with the antibody against von Willebrand factor (factor VIII-related endothelial antigen) (DAKO, Glostrop, Denmark) after deactivation of endogenous peroxidase with 0.3% H2O2 and blockage of nonspecific binding sites. Microvessels were visualized by the avidin-biotin-peroxidase complex method, as described above. The number of microvessels in the ulcer base was determined in three randomly chosen 1 mm2fields. The microvessel density was expressed as the number of microvessels per mm2 of the ulcer base.
Drugs.
NS-398 (kindly provided by Taisho Pharmaceutical Co., Tokyo, Japan) and indomethacin (Sigma) were suspended in a trace of Tween 80 and saline. The drugs were administered s.c. in a volume of 5 ml/kg body weight. In the case of repeated administration, the drugs were administered once daily for the indicated periods starting from day 0. Control animals received the vehicle alone.
Statistical analysis.
The data are presented as means ± S.E. Statistical differences were evaluated using Student’st test or Dunnett’s multiple comparison test, with P < .05 being regarded as significant.
Results
Expression of COX-2 in ulcerated gastric tissue.
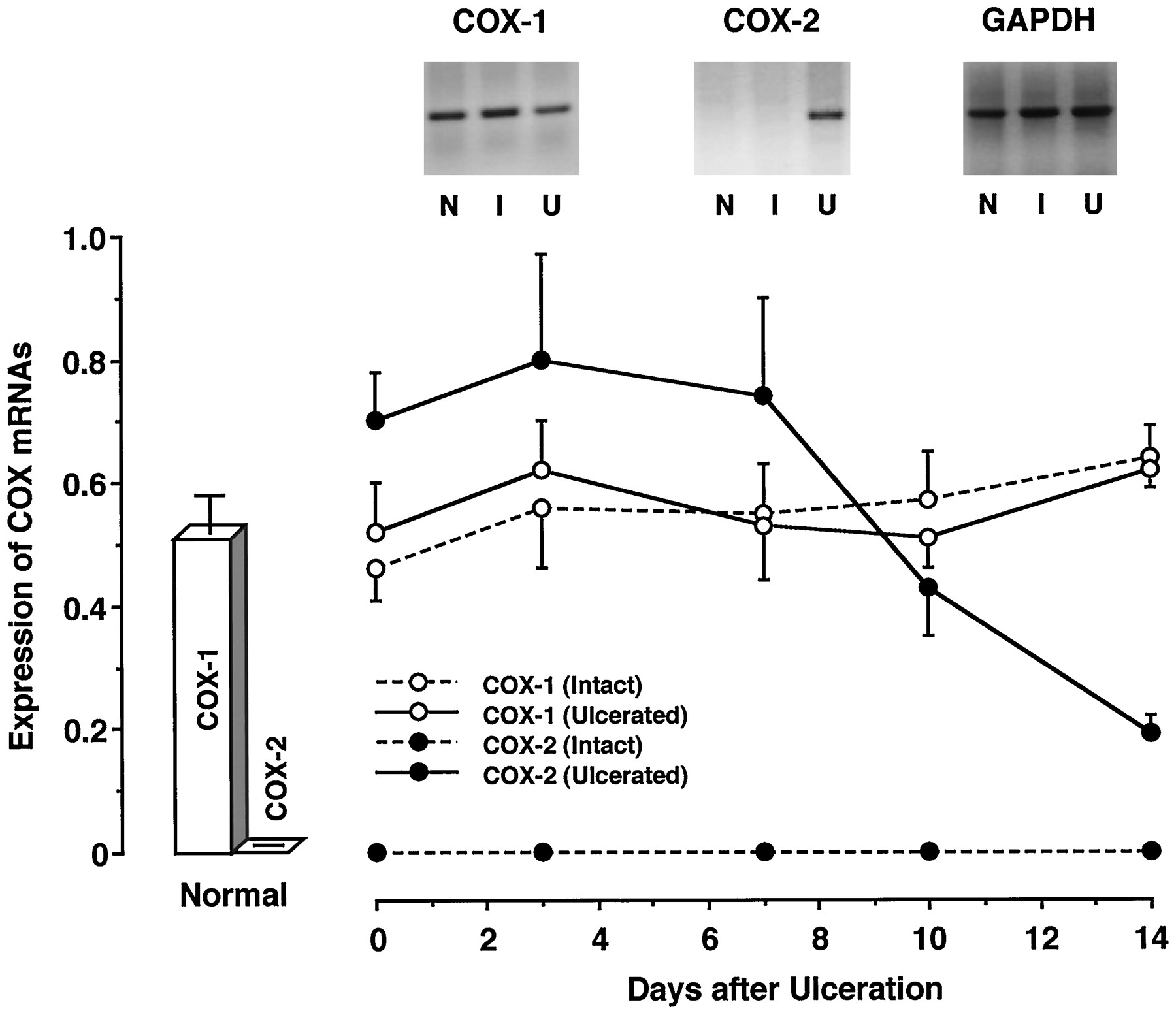
We examined the expression of COX-1 and COX-2 mRNAs in gastric tissues by Northern blotting (fig. 1). The gastric mucosa of normal stomachs contained COX-1 mRNA, but not COX-2 mRNA. However, COX-2 mRNA expression was found in ulcerated tissue. In the ulcerated tissue, the expression of COX-2 mRNA was already detectable on day 0, and its level remained constant to day 7. Thereafter, COX-2 mRNA expression decreased with time. This change in COX-2 mRNA expression was well associated with those in the ulcerated area and PGE2 production in the ulcerated tissue, as shown in figure8. Despite the presence of ulcers, COX-2 mRNA was not expressed in the intact tissue of stomachs with ulcers. However, COX-1 mRNA was found in both the ulcerated and intact tissues. The levels of COX-1 mRNA in both tissues were similar to that in the gastric mucosa of normal stomachs, and these levels did not change throughout the course of the experiment.

Expression of COX-2 mRNA in the gastric tissues of rats. At the indicated times, poly (A)+ RNAs were isolated and then subjected to Northern blot analysis. N, I and U represent the gastric tissue of normal rats, and the intact and ulcerated gastric tissues of rats having ulcers, respectively. The data are presented as means ± S.E. (n = 6).
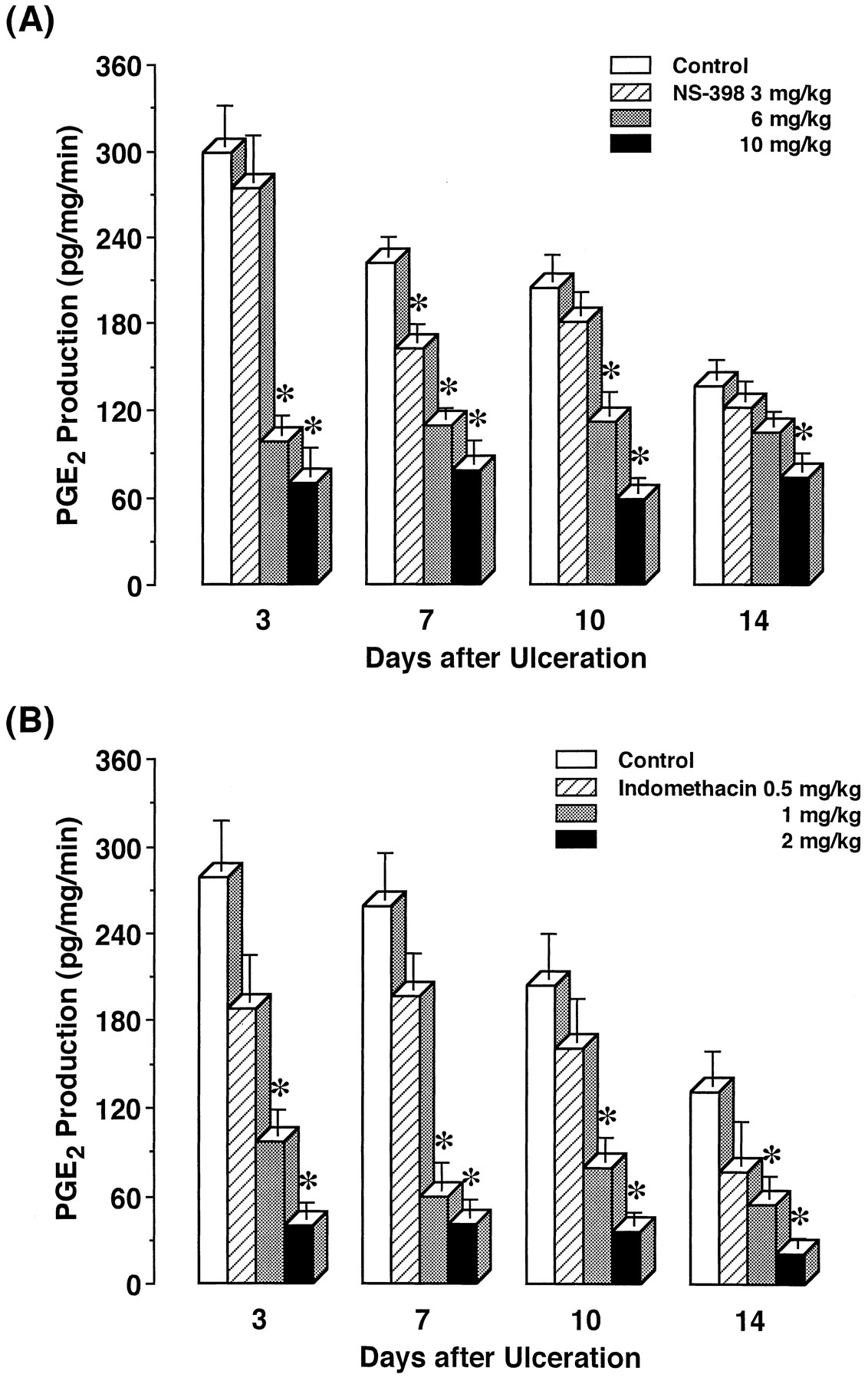
Effects of repeated administration of NS-398 and indomethacin on PGE2 production in ulcerated gastric tissue of rats. The vehicle (control), NS-398 or indomethacin was repeatedly administered starting from day 0, and then PGE2 production in the ulcerated gastric tissue was determined on the indicated days. The data are presented as means ± S.E. (n = 6–8). * Significantly different from the corresponding control.
We determined the localization of the COX-2 protein in the stomachs with ulcers by immunohistochemical staining (fig.2). Strong COX-2-immunoreactivity was abundant in the upper portion of the ulcer base. Immunoreactive signals were found in spindle-shaped cells, mononuclear cells and polymorphonuclear cells. Considering the existence in granulation tissue, these cell types were morphologically identified to be fibroblasts, macrophages/monocytes and granulocytes. In addition, COX-2-expressing mononuclear cells and polymorphonuclear cells were positive to macrophage-specific and granulocyte-specific esterases, respectively. The numbers of COX-2-expressing cells decreased with ulcer healing. However, immunoreactivity for the COX-2 protein was not detected in the intact gastric mucosa. When anti-COX-2 antibody was inactivated by heating, no immunoreactive signals were observed.

Immunohistological detection of the COX-2 protein in the gastric tissue of rats having ulcers. COX-2-immunoreactivity was only found in the upper portion of the ulcer base. (A) × 25, (B) × 100.
To clarify the relationship between COX-2 expression and PGE2 production, we determined the PGE2production in gastric tissues that had been incubated with NS-398, indomethacin or the vehicle (fig. 3). PGE2 production was significantly higher in the ulcerated tissue than in the mucosa of normal stomachs. The production in the intact tissue of stomachs with ulcers did not differ from that in the normal gastric tissue. NS-398, even at 50 μM, did not affect PGE2 production in either the intact tissue of stomachs with ulcers or normal gastric tissue. However, NS-398 dose-dependently and significantly reduced the increased PGE2 production in the ulcerated tissue. The inhibition by 50 μM NS-398 reached 50%. Indomethacin also dose-dependently inhibited PGE2production in both ulcerated tissue, intact tissue in stomachs with ulcers and normal gastric tissue. The inhibition by 50 μM indomethacin was around 80% in all groups.

In vitro effects of NS-398 and indomethacin on PGE2 production in rat gastric tissues. PGE2 production in the gastric tissue of normal rats, and in both the intact and ulcerated gastric tissues of rats with ulcers (day 3) was determined in the presence of the vehicle (control), NS-398 or indomethacin. The data are presented as means ± S.E. (n = 6). * Significantly different from the corresponding control.
PGE2 production in gastric tissues after single administration of NS-398 and indomethacin.
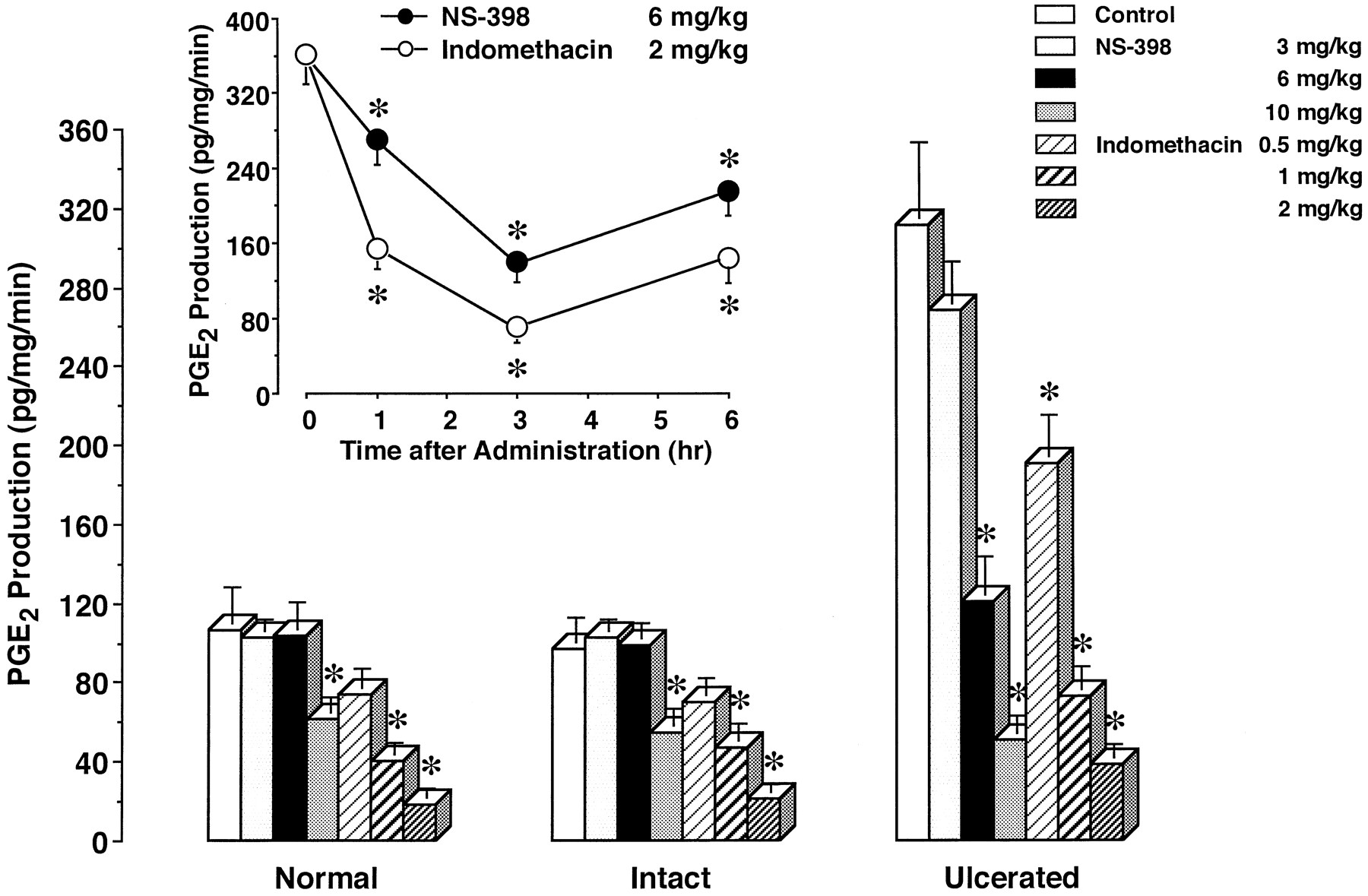
To properly assess thein vivo effects of NS-398 and indomethacin on PGE2 production in gastric tissues, we first considered the strength of their effects over time (fig. 4, inset). After 6 mg/kg NS-398 and 2 mg/kg indomethacin had been administered to rats with ulcers, their maximal effects were observed at 3 hr. Based on this finding, we examined thein vivo effects of NS-398 and indomethacin on PGE2 production at 3 hr after their administration.
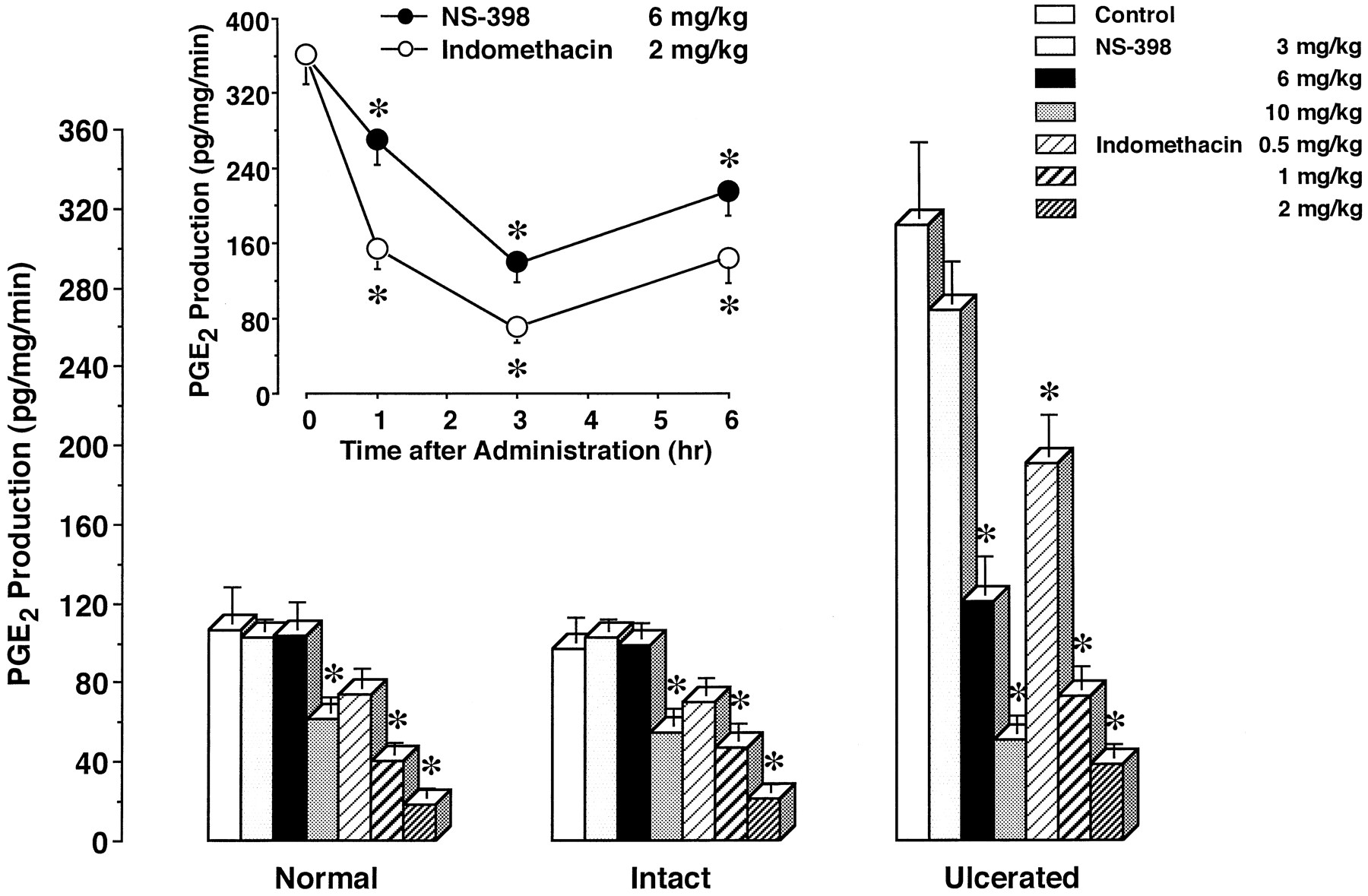
Effects of single administration of NS-398 and indomethacin on PGE2 production in rat gastric tissues. PGE2 production in the gastric tissue of normal rats, and in both the intact and ulcerated gastric tissues of rats with ulcers (day 3) was determined 3 hr after the administration of the vehicle (control), NS-398 or indomethacin. Inset, PGE2 production in the ulcerated gastric tissue (day 3) was determined at the indicated times after the administration of 6 mg/kg NS-398 or 2 mg/kg indomethacin. The data are presented as means ± S.E. (n = 6). * Significantly different from the corresponding control or the value at 0 hr.
We determined PGE2 production in gastric tissues after the drugs had been administered once to rats (fig. 4). In the gastric mucosa of normal rats, NS-398 at less than 6 mg/kg had no effect on PGE2 production, but the drug at 10 mg/kg significantly reduced it by 42.5%. Indomethacin at 0.5 to 2 mg/kg inhibited PGE2 production in a dose-dependent manner. The effect of indomethacin at more than 1 mg/kg was significant, and the inhibition at 1 and 2 mg/kg reached 62.7 and 82.7%, respectively. Similar results were obtained for the intact tissue of stomachs with ulcers. PGE2 production was significantly inhibited by NS-398 at 10 mg/kg, and by indomethacin at 1 and 2 mg/kg. The inhibition by 10 mg/kg NS-398, and 1 and 2 mg/kg indomethacin was 43.5, 51.7 and 78.2%, respectively.
In the case of ulcerated tissue, PGE2 production was found to be significantly elevated. NS-398 as well as indomethacin dose-dependently inhibited PGE2 production in the ulcerated tissue. The increased PGE2 production was significantly reduced to the normal level by 6 mg/kg NS-398 (61.0% inhibition). Furthermore, NS-398 at 10 mg/kg, and indomethacin at 1 and 2 mg/kg potently inhibited the production by 83.7, 76.5 and 87.8%, respectively.
Effects of NS-398 and indomethacin on the healing of gastric ulcers.
We examined the effects of repeated administration of NS-398 and indomethacin on the healing of gastric ulcers (fig.5). On day 0, gastric ulcers had clearly developed. There were round and well-defined ulcers in all animals, the ulcerated area of 46.9 ± 3.8 mm2. Thereafter, the ulcer size spontaneously decreased. NS-398 and indomethacin were administered at daily intervals from day 0. On day 3, ulcer healing was not affected by NS-398 or indomethacin. NS-398 at 3 and 6 mg/kg had no effect to day 7, but significant impairment of ulcer healing was noted on day 10. On day 14, NS-398 at 3 mg/kg continued to delay the healing, although the inhibitory effect of the drug at 6 mg/kg was still significant. At 10 mg/kg, NS-398 significantly prevented ulcer healing from day 7 to 14. The preventive rates with NS-398 were 15.0, 21.9 and 32.6% with 3, 6 and 10 mg/kg on day 10, and 12.1 and 29.3% with 6 and 10 mg/kg on day 14, respectively.
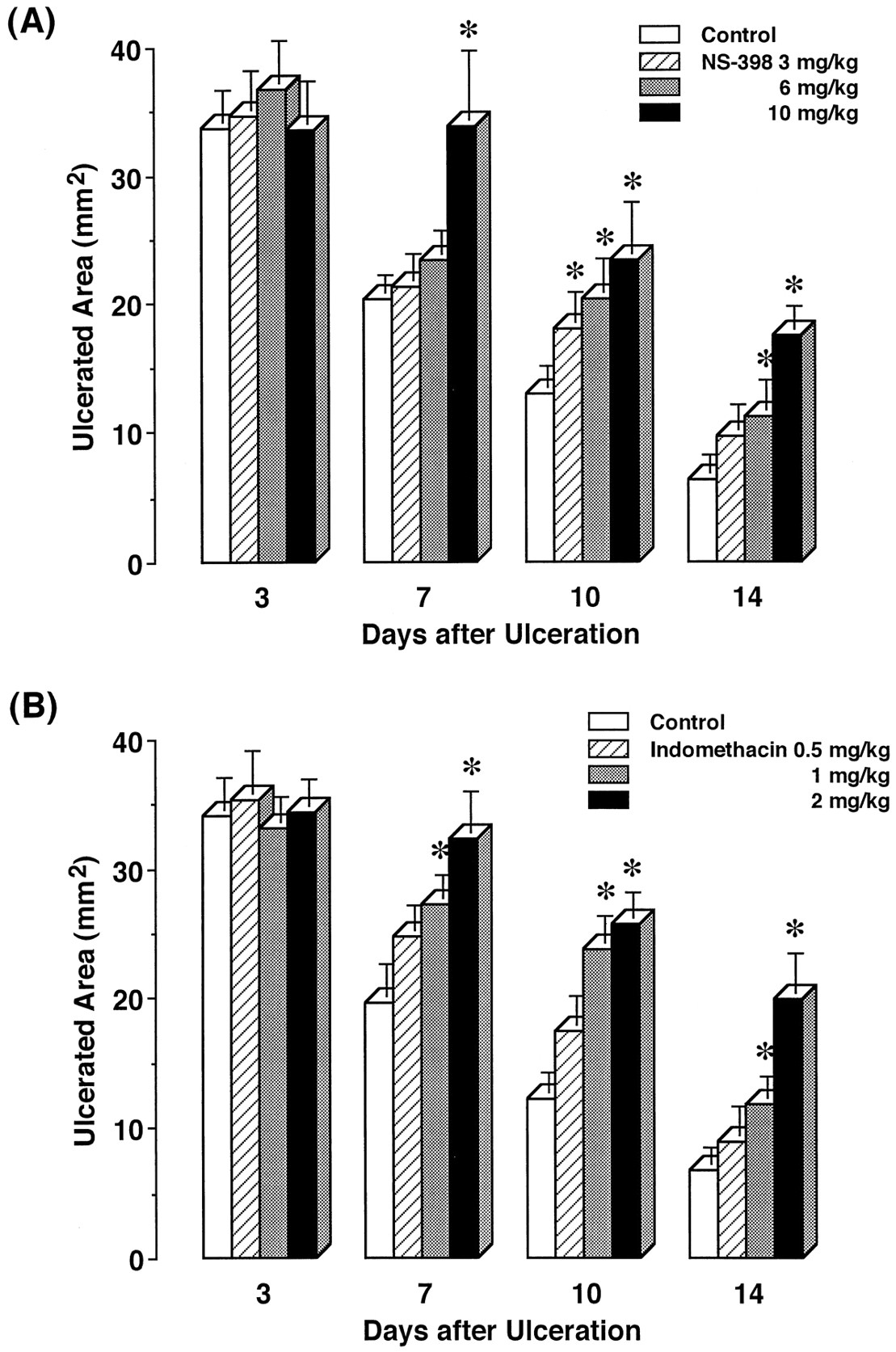
Effects of repeated administration of NS-398 and indomethacin on the healing of gastric ulcers in rats. The vehicle (control), NS-398 or indomethacin was repeatedly administered starting from day 0, and then the ulcerated area was determined on the indicated days. The data are presented as means ± S.E. (n = 6–8). * Significantly different from the corresponding control.
Indomethacin at 0.5 to 2 mg/kg dose-dependently impaired the healing from day 7 to 14. This effect was significant with doses of 1 and 2 mg/kg on these days. The preventive rates of 1 and 2 mg/kg indomethacin were 27.6 and 46.3% on day 7, 33.2 and 38.7% on day 10, and 12.5 and 32.7% on day 14, respectively. The delay by 10 mg/kg NS-398 was similar to that by 2 mg/kg indomethacin.
The gross appearances of acetic acid ulcers after treatment with 6 mg/kg NS-398 and 2 mg/kg indomethacin for 10 days are shown in figure6.

Gross appearance of gastric ulcers after repeated administration of NS-398 and indomethacin to rats (day 10). The ulcerated area was larger in the drug groups than in the controls. Indomethacin more strongly impaired ulcer healing than NS-398 did. A, Control; B, 6 mg/kg NS-398 and C, 2 mg/kg indomethacin.
When 6 mg/kg NS-398 and 2 mg/kg indomethacin were repeatedly administered for 14 days starting from day 10, ulcer healing was not inhibited. The ulcerated area was around 1 mm2 in all groups. The administration of 6 mg/kg NS-398 or 2 mg/kg indomethacin for 14 days to normal rats did not cause any lesions or ulcers in their stomachs.
Histological studies also revealed impairment of ulcer healing in NS-398-treated and indomethacin-treated animals (fig.7). Regeneration of the mucosa on the ulcer base was apparently inhibited by NS-398 and indomethacin, compared with the control. Table 1presents the length of the regenerated mucosa, thickness of the ulcer base and density of microvessels in the base after repeated administration of NS-398 and indomethacin for 14 days. In the animals treated with NS-398 at 6 mg/kg and indomethacin at 2 mg/kg, the length of the regenerated mucosa was significantly shorter than that in the controls. There was also a significant difference between the NS-398-treated and indomethacin-treated groups. The ulcer base also significantly thickened in response to NS-398 and indomethacin. The microvessel density in the base was significantly lower in the NS-398-treated group that in the controls. Indomethacin also decreased the density of microvessels, this inhibitory effect on angiogenesis being significantly more potent than that of NS-398.

Microscopic appearance of the gastric ulcer margin after repeated administration of NS-398 and indomethacin to rats (day 14). Regeneration of the mucosa on the ulcer base was apparently prevented by NS-398 and indomethacin. The prevention by indomethacin was more pronounced than that by NS-398. A, Control; B, 6 mg/kg NS-398 and C, 2 mg/kg indomethacin.
Histological evaluation of gastric ulcer healing after repeated administration of NS-398 and indomethacin to rats
PGE2 production in ulcerated tissue was measured after repeated administration of NS-398 and indomethacin (fig.8). In the control animals, PGE2 production was significantly elevated from day 3 to 10, as compared with in the normal mucosa (see fig. 4). This increased PGE2 production gradually decreased from day 10. NS-398 at 3 to 10 mg/kg inhibited the production in a dose-related manner. The effect of NS-398 at 3 mg/kg was only significant on day 7. NS-398 at 6 mg/kg reduced the stimulated PGE2 production to near normal levels throughout the experiment, although a significant effect was observed from day 3 to day 10. In contrast, NS-398 at 10 mg/kg potently inhibited the production throughout the experiment to a level significantly lower than the normal production level.
Indomethacin at 0.5 to 2 mg/kg inhibited PGE2 production in a dose-dependent manner throughout the experiment. Significant inhibition by indomethacin was observed with doses of 1 and 2 mg/kg at all observation times. At 2 mg/kg, indomethacin potently reduced PGE2 production to below normal levels, the inhibition remaining more than 80% at all points.
Discussion
Our results indicate that COX-2 expression is induced by gastric ulceration in rats and that the level of COX-2 mRNA decreases with ulcer healing. In contrast, COX-1 mRNA was expressed in both intact and ulcerated tissues, the levels remaining constant during ulcer healing. These findings are consistent with those of Mizuno et al.(1997) concerning mice. Furthermore, we found that COX-2 is only localized in ulcerated tissue. Immunohistochemical staining showed that the COX-2 protein is expressed in fibroblasts, macrophages/monocytes and granulocytes in the upper portion of the ulcer base. Severalin vitro studies have revealed the expression of COX-2 in these cell types in response to various stimuli (Mitchell et al., 1995; Herschman, 1996). In ulcerated tissue, PGE2production was significantly elevated when compared with that in normal tissue, and this increased production returned to the normal level in parallel with ulcer healing. This change in PGE2 production was well correlated with COX-2 mRNA expression. The increased PGE2 production in the ulcerated tissue was dose-dependently inhibited by NS-398, but the production in other tissues was unaffected. These results indicate that COX-2 contributes to the elevation of PGE2 production in the ulcerated tissue. In addition, NS-398 administered at 6 mg/kg reduced the increased PGE2 production to near the normal level, without affecting PGE2 production in the intact tissue throughout the experiment. This finding also suggests that the increased PGE2 in the ulcerated tissue might be predominantly due to COX-2.
We clearly demonstrate that COX-2 plays a crucial role in the healing of gastric ulcers. The inhibition of only COX-2 activity by NS-398 at 3 and 6 mg/kg caused a significant delay in the ulcer healing in rats. The healing was also impaired by indomethacin and 10 mg/kg NS-398, which inhibited both COX-1 and COX-2 activities. Furthermore, we histologically confirmed that regeneration of the mucosa is prevented by 6 mg/kg NS-398 and 2 mg/kg indomethacin. The delay was well associated with the inhibition of PGE2 production in the ulcerated tissue. But, neither NS-398 nor indomethacin affected ulcer healing on day 3. It is probable that the duration from day 0 to day 6 is a lag period in which the delay in ulcer healing had not yet become manifest. The persistent impairment of healing responses caused by the inhibition of PG production leads to visible healing impairment after day 6. In contrast, when both NS-398 and indomethacin were administered starting from day 10, no delay in ulcer healing was observed. These results suggest that the persistent inhibition of increased PG production in the early phase of the healing process leads to impairment of ulcer healing.
Furthermore, we examined the delayed ulcer healing mechanism through the selective inhibition of COX-2 activity. Schmassmann et al. (1995) reported that, in addition to a decrease in the ulcerated area, maturation of the ulcer base (reduction of the ulcer base size) and angiogenesis in the ulcer base are also important factors for efficient healing of gastric ulcers. In fact, the maturation and angiogenesis were reported to be suppressed in the indomethacin-induced delay of ulcer healing in rats (Schmassmannet al., 1995; Tsukimi et al., 1996). We describe that maturation of the ulcer base and angiogenesis are significantly prevented on inhibition of COX-2 activity by NS-398 alone. This finding also indicates the important role of COX-2 in ulcer healing. It should be noted that NS-398 at 6 mg/kg also prevented maturation of the base, and the preventive rates were similar in the 6 mg/kg NS-398-treated and 2 mg/kg indomethacin-treated groups. However, both regeneration of the mucosa and angiogenesis were more strongly inhibited by 2 mg/kg indomethacin than by 6 mg/kg NS-398. These results suggest that COX-2 might play a predominant role in maturation of the ulcer base, and that COX-1 as well as COX-2 might be involved in both regeneration of the mucosa and angiogenesis in the ulcer base.
Our results further suggest that NS-398 administered at 3 and 6 mg/kg only inhibits COX-2 activity, but the drug at 10 mg/kg is able to inhibit COX-1 activity as well as COX-2 activity in stomachs with ulcers. Namely, NS-398 at less than 6 mg/kg reduced the stimulated PGE2 production in the ulcerated tissue without inhibiting PGE2 production in the intact tissue of an ulcerated stomach. However, NS-398 at 10 mg/kg inhibited the production by both tissues in the same way that indomethacin did. In addition, NS-398 at 3 and 6 mg/kg failed to affect PGE2 production by the gastric mucosa of normal rats, but at 10 mg/kg it significantly reduced this production. It is evident that the intact tissue of stomachs with ulcers and the gastric tissue of normal rats only possess COX-1, as found in this study. Arai et al. (1993) and Masferreret al. (1994) reported that NS-398, administered orally at 10 mg/kg to rats, only slightly inhibits PGE2 production in the stomach. The PGE2 production assay was carried out at 30 min and 6 hr after drug administration by Arai et al.(1993) and Masferrer et al. (1994), respectively. In contrast, in the study by Futaki et al. (1993), when gastric PGE2 production was measured 3 hr after the administration of 10 mg/kg NS-398, it was significantly reduced (60% inhibition), compared with in the control. Therefore, we examined the time courses of the effects of these drugs on PGE2 production in ulcerated gastric tissue after their administration. We confirmed that the maximal effects of COX inhibitors were observed at 3 hr after drug administration. We speculated that the evaluation of the in vivo effect of NS-398 on gastric PGE2 production may have been insufficient in the studies by Arai’s and Masferrer’s groups. Thus, proper evaluation of the role of COX-2 in the stomach using NS-398 at high doses such as 10 mg/kg is unlikely. At present, it is unknown why there is only a slight difference in the doses of NS-398 between COX-2 selective inhibition (6 mg/kg) and dual COX inhibition (10 mg/kg), despite the fact that NS-398 is highly selective for COX-2 in in vitro experiments (Futaki et al., 1994;Panara et al., 1995; Chulada and Langenbach, 1997). To our knowledge, there have been no reports concerning the metabolism of NS-398 in rats, yet it seems possible that a metabolite of NS-398 having the ability to inhibit COX-1 activity may be produced in a large quantity with concentrations of more than 10 mg/kg. Alternatively, a considerable amount of NS-398 may accumulate in the gastric mucosa with 10 mg/kg NS-398 administration.
Overall, we conclude that COX-2 plays an important role in the healing of gastric ulcers in rats. COX-2-selective inhibitors are expected to be new NSAIDs without ulcerogenic effects, but they are likely to impair gastric ulcer healing if used in the early phase of the healing process.
Acknowledgments
The authors thank N. J. Halewood for critical reading of the manuscript, and M. Ishikawa, M. Shimose, M. Yoshida, S. Kitazawa and K. Matsuno for their technical assistance.
Footnotes
-
Send reprint requests to: Dr. Satoru Takahashi, Department of Applied Pharmacology, Kyoto Pharmaceutical University, Missasagi, Yamashina, Kyoto 607-8414, Japan.
- Abbreviations:
- COX
- cyclooxygenase
- PG
- prostaglandin
- NSAID
- nonsteroidal antiinflammatory drug
- Received April 6, 1998.
- Accepted May 21, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}