Article Text

Abstract
Visceral hypersensitivity is currently the most widely accepted mechanism responsible for abdominal pain. Inflammatory mediators are known to sensitise primary afferents and to recruit silent nociceptors. Recent evidence suggests that non-inflammatory mediators also have the potential to trigger visceral pain. This sequence of events may constitute part of an alerting system which prompts the central nervous system to correct gastrointestinal responses to ingestion.
- visceral hypersensitivity
- pain mediators
- inflammation
- stress
- mast cells
- ATP, adenosine triphosphate
- CGRP, calcitonin gene related peptide
- CNS, central nervous system
- IBS, irritable bowel syndrome
- IL, interleukin
- NGF, nerve growth factor
- NKA, neurokinin A
- PGE 2
- prostaglandin E2
- SP, substance P
- TNF, tumour necrosis factor
- 5-HT, serotonin
Statistics from Altmetric.com
- ATP, adenosine triphosphate
- CGRP, calcitonin gene related peptide
- CNS, central nervous system
- IBS, irritable bowel syndrome
- IL, interleukin
- NGF, nerve growth factor
- NKA, neurokinin A
- PGE 2
- prostaglandin E2
- SP, substance P
- TNF, tumour necrosis factor
- 5-HT, serotonin
SUMMARY
Visceral hypersensitivity is currently the most widely accepted mechanism responsible for abdominal pain and contributes to intestinal motor abnormalities in functional gastrointestinal disorders such as irritable bowel syndrome (IBS). Inflammatory mediators are known to sensitise primary afferents, especially C fibre polymodal receptors, and to recruit silent nociceptors. Local tissue injury results in the release of a variety of chemical substances (for example, potassium, adenosine triphosphate (ATP), bradykinin, and prostaglandin E2 (PGE2)) which directly activate nerve endings and trigger the release of algesic mediators directly from immunocytes and mast cells. At the same time, other sensory neuropeptides released by axon reflexes activate neutrophils, fibroblasts, and mast cells resulting in the so called “neurogenic phase” of inflammation. Recent evidence suggests that non-inflammatory mediators also have the potential to trigger visceral pain or to lower the threshold of neuronal responses to mechanical stimuli. Stress, for example, affects visceral sensitivity without triggering an intestinal immune response. Chemicals such as glycerol sensitise primary afferent neurones; peripherally released glutamate and trypsin also have direct actions on nociceptive fibres. This sequence of events may constitute part of an alerting system which prompts the central nervous system (CNS) to correct gastrointestinal responses to ingestion.
INFLAMMATORY MEDIATORS OF VISCERAL PERCEPTION
Origin and nature of inflammatory mediators
Local tissue injury prompts the release of chemical mediators (potassium, hydrogen ions, ATP, and bradykinin) and inflammatory mediators (for example, PGE2) from inflammatory cells. These substances directly activate nerve endings and trigger the release of algesic mediators (for example, histamine, serotonin (5-HT), nerve growth factor (NGF), and prostanoids) from other cells and afferent nerves.1–3 This sensitises the endings of afferent nerve terminals resulting in an increased response to painful stimuli.
Prostaglandins and other arachidonic acid derivatives increase the sensitivity of nerve terminals to bradykinin and other pain producing substances. A cascade of events involving substance P (SP), histamine, 5-HT, and cytokines in secondary neurones causes nearby nociceptors to become sensitised. The close proximity between mast cells and the endings of sensory neurones results in an amplifying loop in which SP released from nerve endings activates mast cell degranulation, releasing histamine. This induces further release of SP from sensory nerve endings and also triggers the release of NGF which further exacerbates the situation by promoting the development and enhancing the function of sensory neurones.3
Although mast cells play a major role in the sensitisation of primary afferents, there is increasing evidence that substances released from other local entities are of additional importance (fig 1). In the presence of inflammation, prostaglandins released from macrophages and sympathetic terminals are thought to have a direct action on receptors located on afferent endings. ATP is released from sympathetic nerves which innervate visceral smooth muscle. It acts as a cotransmitter for noradrenaline and is thought to bind to the P2X3 subtype of purine receptors located on terminal endings thereby generating pain signals.4 Eicosanoids such as 8(R),15(S)-dihydroxyeicosatetraenoic acid released from neutrophils directly affect adenyl cyclase activity in afferent neurones, and it is likely that cytokines secreted by the macrophages indirectly affect neuronal activity.2

Substances and major local pathways involved in triggering hyperalgesia to distension within the gut. Note that several mediators such as substance P (SP) may directly and indirectly influence the threshold of response of afferent fibres to a mechanical stimulus. CGRP, calcitonin gene related peptide; C5a, complement 5a; fMLP, formyl-methionyl-leucyl-phenylanine; IL, interleukin; NGF, nerve growth factor; NPY, neuropeptide Y; PGI2, prostacyclin I2; PGE 2, prostaglandin E2; TNF, tumour necrosis factor; VIP, vasoactive intestinal peptide; 5-HT, serotonin; 8(R),15(S)-diHETE, 8(R),15(S)-dihydroxyeicosatetraenoic acid. Modified from Coelho and colleagues.34
This cascade of events places the primary afferents in a state of permanent sensitisation resulting either from increased concentrations of bradykinin, 5-HT, eicosanoids, or ATP at specific terminal neurone receptors, or as a result of interleukin (IL) release from immunocytes. Activation of local adrenergic fibres may also sensitise primary afferent endings (fig 1). This activated cascade is characteristic of inflamed tissues and is thought to be responsible for persistent pain triggered by local inflammation as acute administration of these substances triggers a transient algesic state in animals.
Some of the candidate proalgesic mediators released by local structures during inflammation appear to play a more prominent role in the genesis of intestinal hyperresponsiveness to mechanical stimuli. 5-HT is known to activate primary afferents. Studies on pseudoaffective (cardiovascular reflex) responses to intestinal distension suggest that its action is mediated through a 5-HT3 receptor subtype coupled to a sodium channel on primary afferent endings. Accordingly, intravenous administration of low doses of 5-HT3 antagonists have been shown to have potent visceral analgesic activity in response to duodenal distension in rats.5 These findings have been confirmed in numerous studies using different animal models of visceral pain. It is probable however that other 5-HT receptor subtypes, such as 5HT1A receptors, are also involved in mediation of visceral nociceptive input.6,7
Bradykinin receptors are localised on nociceptive sensory neurones. This mediator plays a major role in a variety of inflammatory processes8 and is involved in the mediation of pain and hyperalgesia caused by irritant substances in many animal models. Bradykinin receptors are subdivided into two subtypes. The B2 subtype is responsible for mediating most of the known physiological and pathophysiological actions of the kinins. There is however increasing evidence to suggest that B1 receptors, which bind with higher affinity than B2 receptors to the bradykinin metabolite des-Arg9-BK, may play an important role in the processes that follow certain types of tissue injury, as under these circumstances they are selectively upregulated.
The antinociceptive effects of bradykinin antagonists have already been demonstrated at visceral level. In animal studies NPC-567, a non-selective B1 and B2 receptor antagonist, has been shown to decrease pain induced by intraperitoneal administration of acetic acid and urate crystals.9 Suppression of carrageenan induced hyperalgesia and hyperthermia by local administration of this antagonist showed that bradykinin induced neuronal sensitisation was also involved in the carrageenan reaction.10
Adenosine is a neuromediator in many nerve structures. Receptors of the A2 subtype are coupled to sodium channels on terminal endings of visceral primary afferents but the role of adenosine in triggering nociceptive messages from visceral nerve endings remains to be explored.
Tachykinins and calcitonin gene related peptide (CGRP) may also have an important role in the transmission of nociceptive messages from the gastrointestinal tract. C afferent fibres are thought to contain “silent receptors” for neurokinins which become sensitised by inflammatory processes in peripheral tissues. A large body of experimental findings also supports the involvement of SP and its receptors in pain transmission.
LONG TERM EFFECT ON SENSORY PATHWAYS
Nerve remodelling occurring during inflammation can trigger chronic hypersensitivity in the submucosa and other intestinal structures.11 These changes are complex, time dependent, and related to the nature of inflammation. The acute phase of Nippostrongylus brasiliensis infection is associated with a 2.5-fold increase in nerve content of the tissues, chiefly as a result of axonal dilatation. During the recovery phase (14–28 days later) when mast cell hyperplasia persists, the mean cross sectional area of the nerves decreases but there is an increase in the diameter of small fibres. This observation is consistent with the idea that nerve regeneration changes sensitivity. Additional data in rats suggest that mucosal nerves, particularly B-50 immunoreactive nerves, are in a constant state of modelling.12
It has been known for many years that afferent fibres that have been injured as a result of tissue damage become more sensitive to mechanical, chemical, and probably thermal stimuli.13 More information is now available concerning the nature of the mediators involved in this response, particularly those which interact at polymodal nociceptors.
Increased sensitivity at the level of the sensory neurones, presumably at their peripheral endings, is not the only way by which mediators can enhance sensitivity. Other mechanisms which promote hypersensitivity include direct receptor induced opening of calcium and sodium channels, upregulation or downregulation of receptors on nerve endings (triggered by changes in the number and proximity of resident immunocytes), and changes in the size or distribution of sensory neuronal endings.2
Peripheral injury of primary afferent sensory neurones and permanent activation from directly or indirectly released local algesic mediators are both known to increase excitability of the dorsal horn. The role of peripheral injury in maintaining a hyperexcitable state is important as it facilitates amplification of nociceptive inputs (the so called “wind up” phenomenon) which may persist for hours or days after the peripheral noxious stimulus has disappeared. This form of hyperexcitability may play an important role in the pathogenesis of hyperalgesia.
It has also being postulated that injury evokes action potentials that are conducted antidromically along branches of nociceptor axons, or other terminal endings, where they cause release of pain provoking chemical mediators which sensitise other nociceptors in the same proximity. Until now, evidence of cross sensitisation between visceral and somatic afferents has been lacking, even though both are known to project spinally on the same interneurones.14 There is now evidence that visceral inflammation affects sensory inputs from the inflamed viscera as well as from noxious stimuli originating on the surface of the skin. In a cat model of urinary bladder inflammation, sensitisation at the level of the spinal cord was shown to selectively increase the intensity and number of ascending postsynaptic messages.15
Interestingly, in Crohn's disease only 40% of patients exhibit rectal hypersensitivity which suggests that the “wind up” phenomenon is not constant for visceral pain or that it is counterbalanced by activation of descending bulbospinal inhibitory pathways, a possibility that is supported by the fact that somatic hypoalgesia frequently accompanies the condition.16
EXPERIMENTAL MODELS OF INFLAMMATION INDUCED HYPERSENSITIVITY
Intraperitoneal injection of acetic acid to rats triggers abdominal contractions as a manifestation of pain. Tachykinin antagonists, such as SR 48968, which act on the neurokinin NK2 receptor subtype, selectively reduce abdominal cramps while antagonists at the NK1 receptor are inactive.17 Abdominal ileus following surgery is also reduced by NK2 but not by NK1 antagonists. These observations suggest that nociceptive messages from the inflamed peritoneum involve neurokinin A (NKA) rather than SP as a mediator.
Systemic infusion of selective NK1 (GR 73632) or NK2 (GR 64349) receptor agonists have been shown to have different effects during graded rectal distension in non-inflamed rectum. NK1 stimulation selectively enhances the rectocolonic inhibitory reflex while NK2 stimulation selectively increases the frequency of abdominal cramps.18 The same selectivity of effect was observed with antagonists directed to NK1 (CP 96345, RP 67580) and NK2 (SR 48968, MEN 10376) receptors. Thus NK1 antagonism reversed colonic inhibition in response to rectal distension without affecting the abdominal response whereas NK2 antagonism selectively reduced visceral pain and reduced the frequency of abdominal contractions.19 The involvement of SP in visceral hyperalgesia related to intestinal inflammation has been demonstrated by the positive correlation between colonic inflammation with abdominal pain and increased concentrations of SP in chemically induced colitis.20
CGRP is present in a large number of splanchnic afferents. Its immunoreactivity largely disappears from the gastrointestinal tract after splanchnic section or treatment with the sensory neurotoxin capsaicin.21 Approximately 50% of CGRP immunoreactive afferent neurones also show SP or NKA immunoreactivity. CGRP released from peripheral terminals of primary afferents is thought to play an important role in the development of visceral hyperalgesia. It is also possible that sensory inputs are modified by other peripherally released peptides which cause changes in blood flow, smooth muscle contractions, immune reactivity, and/or mast cell degranulation.
Intracerebroventricular injection of CGRP in mice inhibits the nociceptive response in the writhing test.22 Intravenous administration of the CGRP1 receptor antagonist (h-CGRP8–37) has been shown to suppress abdominal cramps induced by intraperitoneal administration of acetic acid in conscious rats.23 Unlike the selective NK2 antagonists, CGRP antagonism also blocks inhibition of gastric emptying in animals with peritonitis. Intravenous administration of CGRP itself mimics the influence of acetic acid on gastric and abdominal responses. It has also been demonstrated that CGRP is involved in the mediation of pain in response to lower intestinal distension. In accordance with these findings, the CGRP antagonist h-CGRP8–37 has been shown to reverse the sensitising effects (allodynia) of acetic acid on nociceptive response to colorectal distension following intracolonic administration of acetic acid to rats.23
Mast cell degranulation induced by administration of BrX 537 induces rectal allodynia in rats 4–6 hours after administration. Its mechanism of action involves 5-HT, but not histamine, mainly via an action on 5-HT1A receptors.24 Intraperitoneal administration of endotoxin (lipopolysaccharide from Escherichia coli) delays biphasic rectal allodynia which accompanies mast cell degranulation and cytokine (IL-1β and tumour necrosis factor α (TNF-α)) release from the CNS. A process of degranulation is implicated because allodynia is suppressed by doxantrazole (a mast cell stabiliser) and also by intracerebroventricular administration of an IL-1 receptor antagonist, a selective inhibitor of IL-1 converting enzyme, or a soluble form of the TNF-α receptor (fig 2). Vagal fibres also play an important role in limiting centrally mediated hyperalgesia as the lipopolysaccharide induced hyperalgesic state is enhanced after vagus nerve treatment with capsaicin.25

{kind=link}
{kind=link}
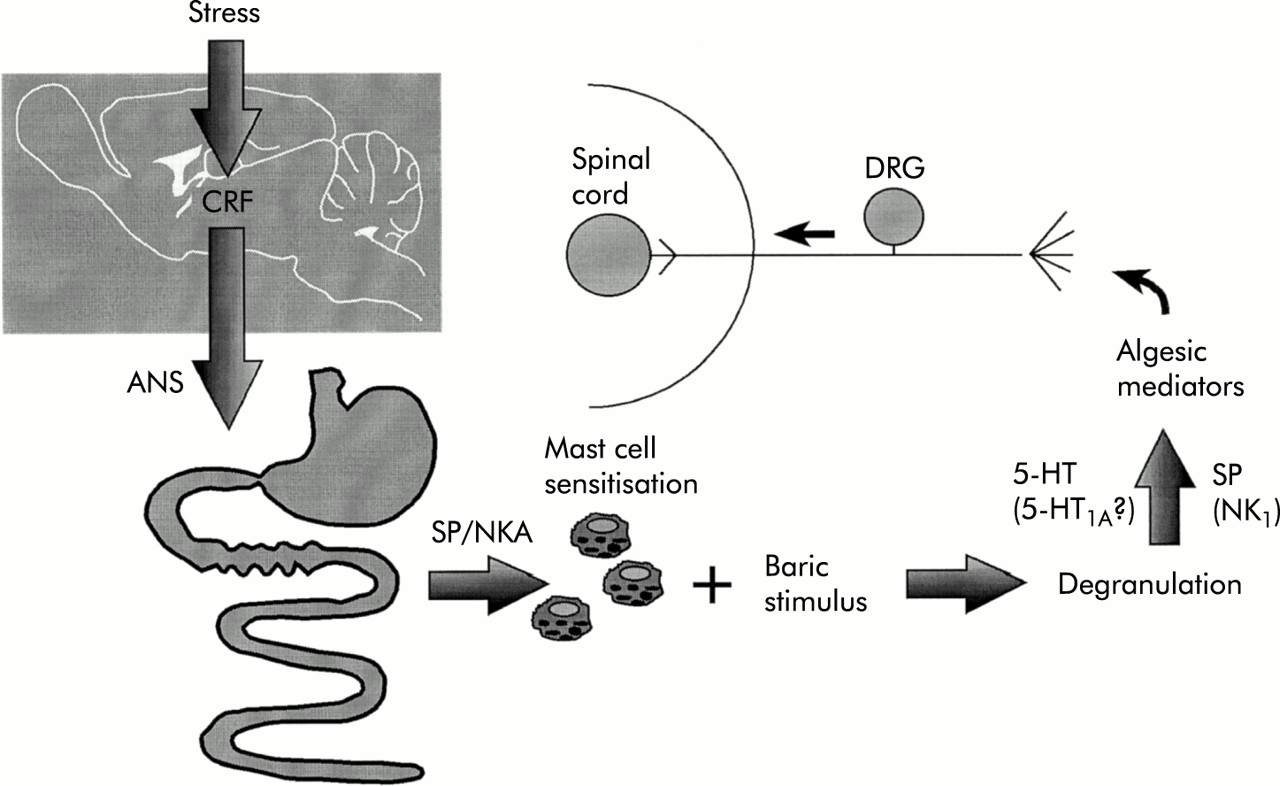
Pathways, structures, and mediators involved in stress induced hyperalgesia to visceral (rectal) mechanical stimulus. ANS, autonomic nervous system; CRF, corticotrophin releasing factor; DRG, dorsal root ganglia; NKA, neurokinin A; SP, substance P; 5-HT, serotonin.
N brasiliensis produces long lasting (1–3 months) visceral hyperalgesia in laboratory animals which is shown by exacerbation of the hypotensive response to intestinal distension.26 This effect is limited to intestinal segments where there is an increase in the number of mucosal mast cells which release tachykinin as the response is attenuated by selective NK1 as well as NK2 selective antagonists. Bradykinin and its major metabolite Des-Arg9-BK are also involved in this mechanism and administration of either substance selectively blocks the increase in reactivity without affecting the basal sensitivity.27
Repeated oral administration of low doses of diquat, a herbicide present as food residue, induces a mild gastrointestinal inflammation associated with an increased sensitivity to distension in rats. This long term effect is temporarily blocked by NK2 receptor antagonists suggesting that NKA is also involved in visceral hypersensitivity.28
NON-INFLAMMATORY MEDIATORS OF VISCERAL PERCEPTION
Stress has been shown to alter perception of colonic or rectal distension in humans, particularly in patients with IBS.29 In rats, partial restraint stress is associated with a transient hyperalgesia to distension, more so in female than in male animals.30 The finding that this effect is blocked by doxantrazole, a mast cell stabiliser, suggests that mast cell degranulation is involved in the mechanism. Stress is thought to increase the synthesis of mast cell products such as histamine31 and stress sensitised mast cells have a greater ability to degranulate under a visceral baric stimulus than unsensitised mast cells (unpublished data). This form of stress induced rectocolonic hyperalgesia however is not associated with a significant increase in colonic myeloperoxidase activity.
Intracolonic infusion of glycerol has been shown to induce spontaneous abdominal cramps accompanied by increased responsiveness to colonic distension.32 5-HT3 receptor antagonists and indomethacin both suppress the abdominal cramps triggered by glycerol. The 5-HT3 antagonists are more active when injected intracolonically than when administered by other routes suggesting a local site of action.
Local infusion of glutamate at doses that are inactive when injected intraperitoneally have also been shown to produce abdominal cramps. This response occurs without histological, biochemical, or immunological evidence of an inflammatory reaction, and is not induced by general muscular hypertonicity as concomitant recording of other somatic striated muscle cells is unaffected by intracolonic administration of glutamate. Thus the mechanism of action of glutamate remains unclear.
Proteinase activated type 2 receptors, expressed by primary afferent neurones, have been described at the periphery.33 These neurones are activated by tryptase derived from mast cells, as well as by trypsin. A tethered peptide (SLIGRL), cleaved as a result of tryptase or trypsin, activates the receptor. Intracolonic infusion of this activating receptor (PAR-2-AP) was shown to delay rectal allodynia, an effect that was not observed after systemic administration.34 The response was not associated with increased tissue myeloperoxidase activity suggesting that the process dose not involve intestinal inflammation.
CONCLUSIONS
Even though our knowledge of the importance and site of action of the mediators responsible for gastrointestinal hyperalgesia has improved considerably, the exact role played by these mediators in the pathophysiology of IBS in humans remains unclear. Despite this, it is likely that with continued research several candidate drugs will be developed in the years to come.35
Pain sensation in animal models appears to be triggered by rectal distension. Enhancement of the nociceptive response by immunological and non-immunological stimuli has been widely evaluated but few studies have investigated spontaneous nociception generated in pathophysiological situations.
Both inflammatory and non-inflammatory mediators participate in the genesis of visceral hyperalgesia in humans. Similar neuropeptides appear to be released by immunocytes, and intrinsic and extrinsic nerves. They exert their effect by binding to receptors located on sensory nerve endings.
Stress and an excess of certain mediators have the potential to trigger a form of visceral hyperalgesia which is not associated with the classic model of intestinal inflammation.