Article Text

Abstract
Background: The CD40/CD40L system, a key regulator and amplifier of immune reactivity, is activated in inflammatory bowel disease (IBD) mucosa.
Aims: To determine whether plasma levels of sCD40L are elevated in Crohn’s disease (CD) and ulcerative colitis (UC) patients compared with normal controls, to investigate the cellular source of sCD40L, and to explore CD40L induction mechanisms.
Patients: CD, UC, and normal control subjects were studied.
Methods: The concentration of sCD40L in plasma and supernatants of freshly isolated platelets and autologous peripheral blood T cells (PBT) was measured by ELISA. Surface CD40L expression level was measured by flow cytometry in resting and thrombin activated platelets, and unstimulated and CD3/CD28 stimulated PBT before and after coculture with human intestinal microvascular endothelial cells (HIMEC).
Results: Compared with normal controls, plasma sCD40L levels were significantly higher in both CD and UC patients and proportional to the extent of mucosal inflammation. Platelets from IBD patients displayed a significantly higher surface CD40L expression than those from control subjects, and released greater amounts of sCD40L than autologous PBT. Contact with IL-1β activated HIMEC induced significant upregulation of CD40L surface expression and release by platelets.
Conclusions: Elevated levels of sCD40L in the circulation of IBD patients reflect enhanced surface expression and release of CD40L by platelets. This phenomenon translates to an increased platelet activation state apparently induced by passage through an inflamed mucosal microvascular bed, a conclusion supported by the positive correlation of plasma sCD40L levels with the extent of anatomical involvement by IBD. These results suggest that platelet-endothelial interactions critically contribute to activation of the CD40 pathway in IBD.
- inflammatory bowel disease
- Crohn’s disease
- ulcerative colitis
- platelets
- CD40 ligand
- CD, Crohn’s disease
- UC, ulcerative colitis
- IBD, inflammatory bowel disease
- sCD40L, soluble CD40 ligand
- HIMEC, human intestinal microvascular endothelial cells
- PBT, peripheral blood T cells
- CAM, cell adhesion molecule
- ICAM-1, intercellular CAM 1
- VCAM-1, vascular CAM 1
- HIF, human intestinal fibroblasts
- IL, interleukin
Statistics from Altmetric.com
- CD, Crohn’s disease
- UC, ulcerative colitis
- IBD, inflammatory bowel disease
- sCD40L, soluble CD40 ligand
- HIMEC, human intestinal microvascular endothelial cells
- PBT, peripheral blood T cells
- CAM, cell adhesion molecule
- ICAM-1, intercellular CAM 1
- VCAM-1, vascular CAM 1
- HIF, human intestinal fibroblasts
- IL, interleukin
Crohn’s disease (CD) and ulcerative colitis (UC) are the two major forms of inflammatory bowel disease (IBD). Although the cause and mechanisms of both conditions remain unknown, mounting evidence suggests that gut tissue injury is not only the result of an abnormal immune response but also involves multiple non-immune cellular systems.1,2 The complex interplay of immune-non-immune cell interactions that sustains inflammatory processes is mediated by multiple receptor-ligand systems, among which the CD40/CD40 ligand (L) pathway is increasingly recognised as playing an essential role in the pathogenesis of chronic diseases.3–5 This pathway has also been implicated in IBD pathogenesis, as suggested by the increased number of CD40 and CD40L bearing cells in the circulation and actively inflamed mucosa of CD and UC patients6–8 and the beneficial effect of blocking the CD40/CD40L pathway in animal models of experimental colitis.9,10
CD40L, a membrane glycoprotein belonging to the tumour necrosis factor family, is expressed mainly by activated T cells and activated platelets,3,11 being subsequently cleaved from the cell surface and released as a biologically active soluble form (sCD40L).11,12 Engagement of the cell bound or soluble form of CD40L by CD40 bearing cells, including non-immune cells such as fibroblasts and endothelial cells, triggers a potent activation profile in these cells, resulting in the production of a wide range of proinflammatory cytokines as well as upregulation of various cell adhesion molecules (CAM).3,4 We have previously shown that CD40L positive T cells and platelets induce chemokine production and intercellular CAM 1 (ICAM-1) and vascular CAM 1 (VCAM-1) upregulation by human intestinal fibroblasts (HIF) and human intestinal microvascular endothelial cells (HIMEC), implying that the CD40 pathway can elicit an inflammatory response by non-immune cells in the intestinal mucosa.13,14
In addition to its proinflammatory effects, recent evidence indicates that CD40L also displays prothrombotic properties.15 Binding of CD40L induces tissue factor upregulation and a procoagulant phenotype by endothelial cells,16 and CD40L appears to be essential for stabilisation of arterial thrombi.17 Patients with IBD frequently exhibit thromboembolic complications and a hypercoagulability state,18 and it is possible that CD40L may contribute to these events in subjects with CD or UC.
Because of its dual role in inflammation and thrombosis, the soluble form of CD40L has been measured and found to be elevated in the plasma of patients with a variety of chronic inflammatory and autoimmune conditions as well as thrombotic states, including IBD, systemic lupus erythematosus, rheumatoid arthritis, and various cardiovascular syndromes.19–27 To date, no information exists on which cell type is the major source of sCD40L in vivo, and the possible mechanisms of platelet CD40L induction in IBD. Thus the present study was designed to measure levels of sCD40L in the circulation of CD and UC patients, including some with thromboembolic complications, to determine whether platelets or T cells are the main source of sCD40L in IBD, and to test whether contact with activated HIMEC leads to expression of CD40L on platelet surface.
MATERIALS AND METHODS
Patient population
Patients with active and inactive CD and UC were studied, and healthy subjects were enrolled as normal controls. The control group consisted of 46 subjects, 22 males and 24 females, with a mean age of 42 years (19–79). An additional subgroup of 18 IBD patients with thromboembolic complications was also studied. These included 11 males and seven females, with a mean age of 44 years (21–72), 13 with deep venous thrombosis, four with stroke, and one with superficial phlebitis. Patients with thromboembolism not associated with IBD were included as controls. Ten were male and six female, aged 43 years (19–73), 13 with deep venous thrombosis and three with stroke. All patients and controls were matched for sex and age. Patients and controls were recruited from three centers: Department of Medicine, University of Milan, Milan, Italy (n = 145), Division of Gastroenterology, University Hospitals of Cleveland, Cleveland, Ohio, USA (n = 35), and Department of Internal Medicine, Catholic University of Rome, Rome, Italy (n = 31). Of the 18 IBD patients with thromboembolic events, 17 were from Milan and one was from Cleveland. All samples from patients with active thrombosis were collected within three months since the acute events. All subjects with thrombosis were negative for factor V Leiden and prothrombin gene mutations. Diagnosis of a thrombotic event was made by routine clinical and imaging evaluation. Clinical disease activity was assessed by the Harvey-Bradshaw activity index and the colitis activity index.28,29 All diagnoses were confirmed by clinical, radiological, endoscopic, and histological criteria. Anatomical disease extension was assessed by radiological and endoscopic examination. This study was approved by the institutional review board of all three participating centres.
Isolation of platelets and PBT
Platelets were isolated using an established consensus protocol that prevents stasis and activation.30 Briefly, peripheral venous blood samples were collected without tourniquet using 10% sodium citrate as anticoagulant, and transferred into polypropylene tubes. Platelet rich plasma was obtained by centrifugation at 180 g for eight minutes at room temperature. Platelets were isolated by centrifugation (1200 g for two minutes), washed, and resuspended in Tyrodes-HEPES buffer (10 mmol/l HEPES, 12 mmol/l NaHCO3, 137 mmol/l NaCl, 2.7 mmol/l KCl, and 5 mmol/l glucose, pH 7.4). The resulting platelet population was essentially free (<0.1%) of erythrocytes and peripheral mononuclear cells, and >99% pure, as assessed by flow cytometry (see below) for expression of the platelet specific antigen CD42b.31 To ensure that the isolation procedure did not artificially stimulate platelets, their activation state was assessed before and after isolation by measuring P-selectin expression levels in whole blood and purified platelets by flow cytometry. Peripheral blood T cells (PBT) were isolated as previously reported.32 Briefly, heparinised venous blood was processed using a Ficoll-Hypaque density gradient and PBT obtained by negative selection, as previously described.
Flow cytometric analysis for detection of membrane bound CD40L on platelets and PBT
Expression of molecules on the platelet surface was assessed using an established cytometric characterisation method,30 before and after activation with 0.5 U/ml thrombin (Sigma, St Louis, Missouri, USA) for five minutes at room temperature. Platelets were fixed in 1% paraformaldehyde and incubated with antibodies against CD40L (FITC conjugated anti-CD40L; BD, San Jose, California, USA), P-selectin (CD62P; PE-anti-P-selectin; BD), or CD42b (Santa Cruz Biotechnology, Santa Cruz, California, USA), for 30 minutes at room temperature. PBT were analysed before and after 24 hours of stimulation with polystyrene beads coated with murine monoclonal antibodies to human CD3 and CD28 (Dynal, Lake Success, New York, USA), and stained for CD40L, as described for platelets. Isotype matched FITC and PE conjugated antibodies (all from BD) were used to assess non-specific binding. Samples were analysed by quantitative flow cytometry using a Coulter Epics XL Flow Cytometer (Beckman Coulter, Inc., Fullerton, California, USA), and each analysis was performed on at least 10 000 events. Quantification of CD62P and CD40L expression was obtained using the Winlist software program (Verity Software House, Topsham, Maine, USA).
Measurement of sCD40L in plasma and supernatants from platelets and PBT
Whole blood was collected into EDTA containing tubes, immediately centrifuged at 1000 g at 4°C, and plasma stored at −0°C. Platelets and PBT present in 10 ml of blood were isolated and cultured for 24 hours. Freshly isolated platelets were cultured in Tyrodes-HEPES buffer, whereas PBT were cultured in serum free RPMI 1640. Supernatants were harvested by centrifugation at 800 g for five minutes at 4°C, and stored at −70°C. Plasma and supernatant samples were thawed once and analysed for sCD40L content in duplicate using a commercially available ELISA kit with an assay reproducibility of >95% (R&D Systems, Minneapolis, Minnesota, USA).
Western blot analysis for platelet associated CD40L
Platelets from normal and IBD subjects were isolated as described above and lysed in ice cold 2× immunoprecipitation buffer (2×TBS, 2% TX-100, 2 mM EDTA, plus 1:100 phosphatase and 1:100 protease inhibitors) for protein extraction. Western blotting was performed as previously reported,33 using a CD40L specific polyclonal goat antibody (Santa Cruz Biotechnology). Protein bands were scanned on a Bio-Rad Gel Dock 1000 (Bio-Rad, Hercules, California, USA) and densitometry relative to the glyceraldehyde-3-phosphate dehydrogenase content of the same bands was performed with Multi-Analyst 1.0.2 software (Bio-Rad).
Platelet-HIMEC cocultures
Isolation and culture of HIMEC were performed as previously reported.34 HIMEC were plated on fibronectin coated wells of a 24 well cluster plate at a density of 5×104/ml/well. HIMEC were left untreated or exposed to interleukin (IL)-1β (100 U/ml) for 16 hours, and induction of a state of activation confirmed by upregulation of VCAM-1 expression by flow cytometry. After extensive washing to remove IL-1β, each HIMEC monolayer was overlaid with 100×106 normal platelets. After a 30 minute coculture, non-adhering platelets were removed by gentle washing, and the monolayer was rinsed with Tyrode buffer. The remaining adherent platelets were stained in situ for CD40L, CD62P, and CD42b as detailed above, and detached by gentle pipetting into a single cell suspension for flow cytometric analysis. In some experiments, the supernatants of the platelet-HIMEC cocultures were collected, centrifuged, and their sCD40L content measured by ELISA, as previously described.
Statistical analysis
Data were analysed using Graphpad software (San Diego, California, USA). The Student’s t test was used for comparison of platelet counts between active and inactive IBD patients. The Kruskal-Wallis test followed by Dunns’ post hoc test was used for analysis of sCD40L or CD40L surface expression among multiple clinical groups. Repeated measures ANOVA, followed by the Bonferroni’s test, were used for analysis of CD40L expression and sCD40L release by platelets after coculture with HIMEC. For correlation between sCD40L and platelet number, the Spearman’s test was performed. Data are expressed as mean (SEM), and statistical significance was set at p<0.05.
RESULTS
Elevated plasma sCD40L levels in UC and CD patients
A total of 68 UC, 81 CD, and 46 healthy control subjects were enrolled, and their clinical characteristics are shown in table 1. Among the IBD patients, 13 UC and five CD subjects had evidence of active thromboembolic phenomena at the time of the study. A total of 16 subjects with thromboembolic events not associated with IBD were also studied, and were matched for sex and age with the 18 IBD patients with thrombotic complications.
Patient characteristics
Plasma sCD40L levels were significantly (p<0.05) higher in both UC and CD patients with inactive disease compared with those of healthy controls (fig 1). When UC and CD patients with clinically active disease were studied, they displayed significantly higher sCD40L levels not only in comparison with controls (p<0.01) but also with IBD patients with inactive disease (p<0.05). No significant differences were found between inactive IBD patients with and without thrombosis, or between active IBD patients with and without thrombosis. Plasma sCD40L levels of patients with thromboembolic events not associated with IBD were comparable with those of normal controls. No statistically significant difference existed among normal controls, active, or inactive IBD patients with thromboembolic events.
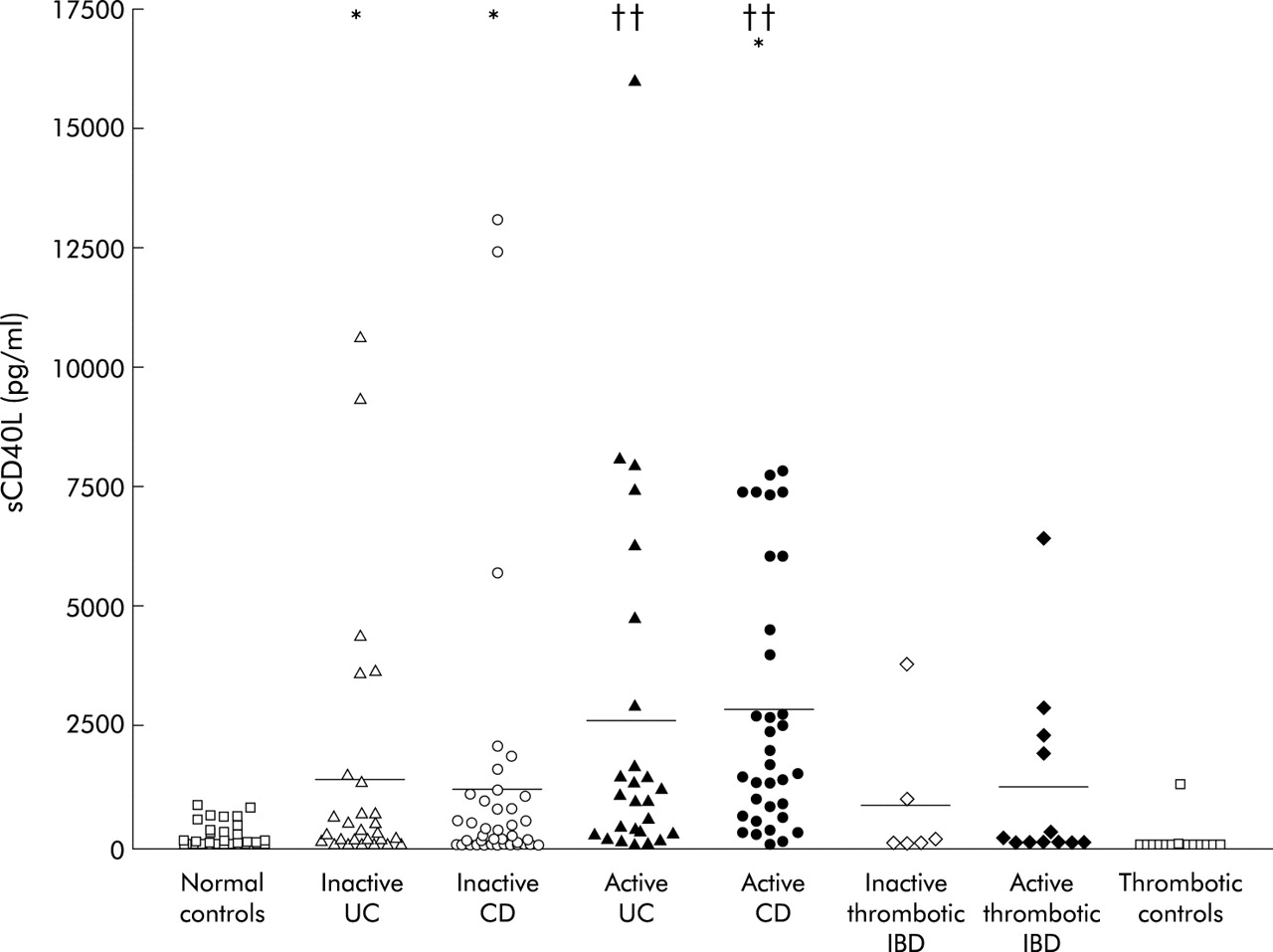
Levels of soluble CD40 ligand (sCD40L) in plasma of inflammatory bowel disease (IBD) patients. Each symbol represents an individual patient and bars represent the mean values of each group. *p<0.05 for inactive UC and CD patients compared with normal controls, and active compared with inactive CD patients; ††p<0.01 for active UC and CD patients compared with normal controls. CD, Crohn’s disease; UC, ulcerative colitis.
In addition, to investigate a link with IBD clinical activity, sCD40L levels were also evaluated with regard to other clinical parameters. Disease duration, sex, disease type, and treatment had no influence on sCD40L plasma levels, which were comparable among patients from the three recruitment centres. However, IBD patients with reactive thrombocytosis (platelet count >450×103) had significantly higher sCD40L levels than IBD patients without reactive thrombocytosis (2975 (708) v 1803 (304) pg/ml, respectively; p<0.01). Only a weak correlation (r = 0.387) existed between platelet counts and sCD40L levels. When disease extension in active and inactive IBD was taken into account, UC patients with pancolitis had higher sCD40L levels than those with left sided or distal colitis (fig 2). Among CD patients, those with ileocolonic or colonic disease had higher levels of sCD40L than those with pure ileal or rectal involvement (fig 2).
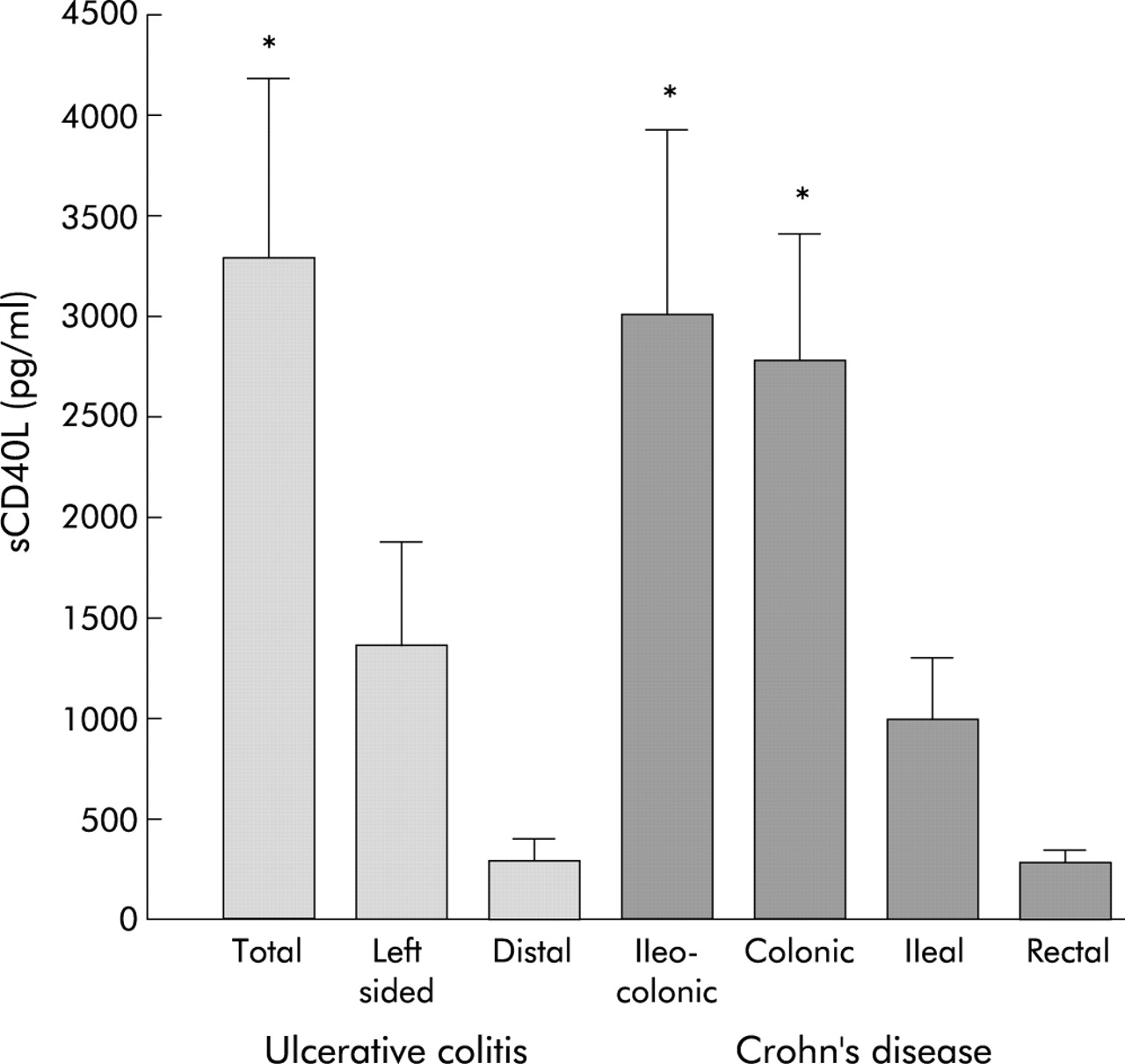
Levels of soluble CD40 ligand (sCD40L) in the plasma of inflammatory bowel disease patients according to anatomical disease extension. *p<0.05 for total compared with distal disease, and for ileocolonic and colonic compared with rectal disease in ulcerative colitis (UC) and Crohn’s disease (CD), respectively. UC: total, n = 22; left sided, n = 23; distal, n = 10. CD: ileocolonic, n = 21; colonic, n = 19; ileal, n = 25; rectal, n = 11.
Elevated surface CD40L expression by IBD platelets
Because sCD40L in the circulation can only derive from CD40L shed from the surface of platelets or T cells3,11 we next investigated its expression level on the surface of these two cell types isolated from active IBD patients. Unstimulated UC and CD platelets displayed a significantly increased expression of CD40L compared with that of control subjects (p<0.001 and p<0.01, respectively) (fig 3). After thrombin activation, CD40L expression increased in all groups, but proportionally and significantly more in UC and CD (both p<0.05) than control platelets. When CD40L expression was evaluated in freshly isolated and anti-CD3/CD28 stimulated PBT, no significant differences were detected between control and IBD T cells, both before and after activation (not shown).

Platelet surface CD40 ligand (CD40L) expression. CD40L mean fluorescent intensity (MFI) was measured by flow cytometric analysis in unstimulated and thrombin activated platelets. Each symbol represents an individual experiment and bars represent the mean values of each group. *p<0.05 for CD compared with normal control platelets; ††p<0.01 for UC compared with normal controls. CD, Crohn’s disease; UC, ulcerative colitis.
Differential release of sCD40L by platelets and PBT
Having demonstrated that platelets and PBT can both express CD40L, we next compared the relative capacity of each cell type to release sCD40L per unit of blood to establish which of the two is the main source of sCD40L in the circulation. Platelets and PBT present in a fixed volume of blood were isolated and cultured alone. Platelets isolated from IBD patients spontaneously released significantly (p<0.05) greater amounts of sCD40L than platelets from normal subjects (fig 4). When levels of sCD40L were measured in supernatants from normal and IBD PBT, they were undetectable and significantly lower (p<0.05), respectively, than those of autologous resting platelets.

Relative contribution of platelets and peripheral blood T cells (PBT) to soluble CD40 ligand (sCD40L) levels. Autologous platelets and PBT from normal and inflammatory bowel disease (IBD) subjects were cultured alone for 24 hours. Data are representative of four normal controls and seven IBD patients. *p<0.05 for normal and IBD platelets compared with normal and IBD PBT, respectively.
Because IBD patients have increased but widely variable numbers of platelets in the circulation35 (table 1), we adjusted sCD40L levels according to individual subject platelet counts. As shown in fig 5, resting and thrombin activated IBD platelets released significantly higher (both at p<0.05) amounts of sCD40L than control platelets, even after adjustment to a fixed platelet number.

Release of soluble CD40 ligand (sCD40L) per platelet. Levels of sCD40L in supernatants of unstimulated and thrombin activated platelets were normalised to a fixed number of platelets (109). *p<0.05 for unstimulated and activated inflammatory bowel disease (IBD) platelets compared with unstimulated and activated normal platelets, respectively.
To demonstrate whether, in addition to increased release of its soluble form, IBD platelets also have an increased content of CD40L protein, western blot analysis was performed. The amount of each of the two CD40L isoforms was higher in CD and UC than in normal platelets (fig 6), and this difference was statistically significant (p<0.05) when measured by densitometric analysis between IBD and normal platelet proteins.

CD40 ligand (CD40L) protein levels in normal and inflammatory bowel disease platelets. Western blot analysis of three normal, two UC, and two CD platelet extracts. The 33 and 28 kDa protein bands represent the two isoforms of CD40L. NL, normal; CD, Crohn’s disease; UC, ulcerative colitis.
Upregulation of platelet CD40L by activated HIMEC
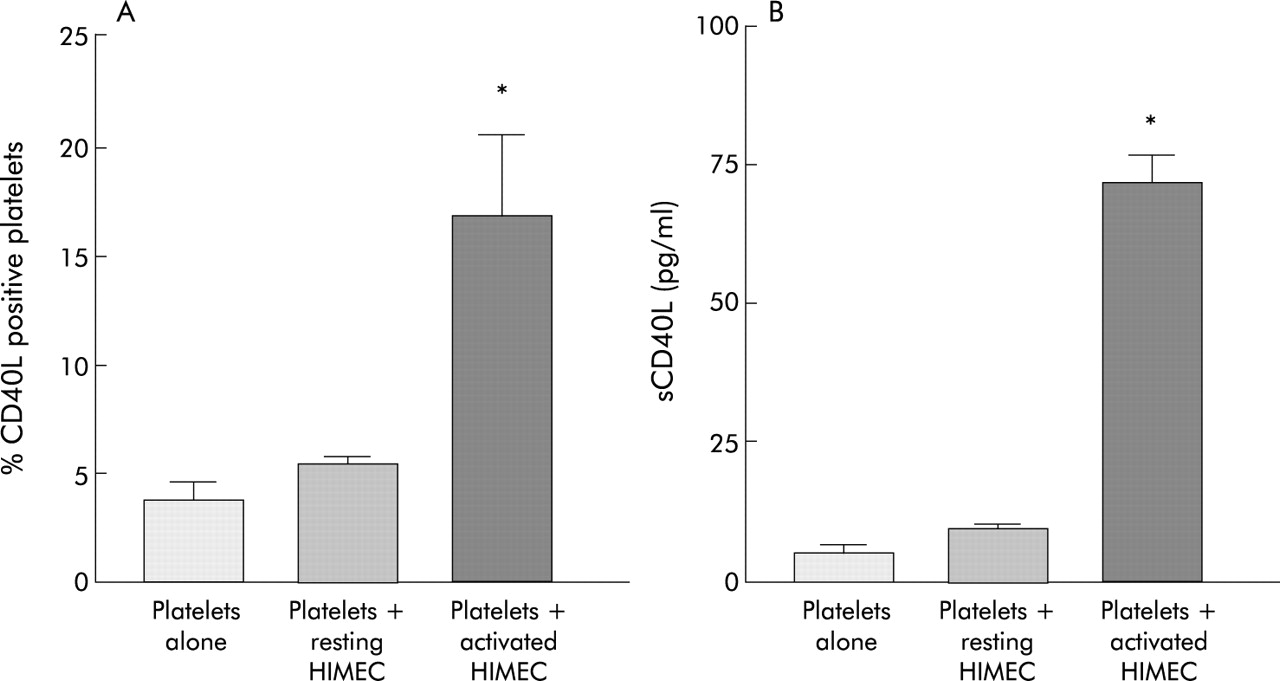
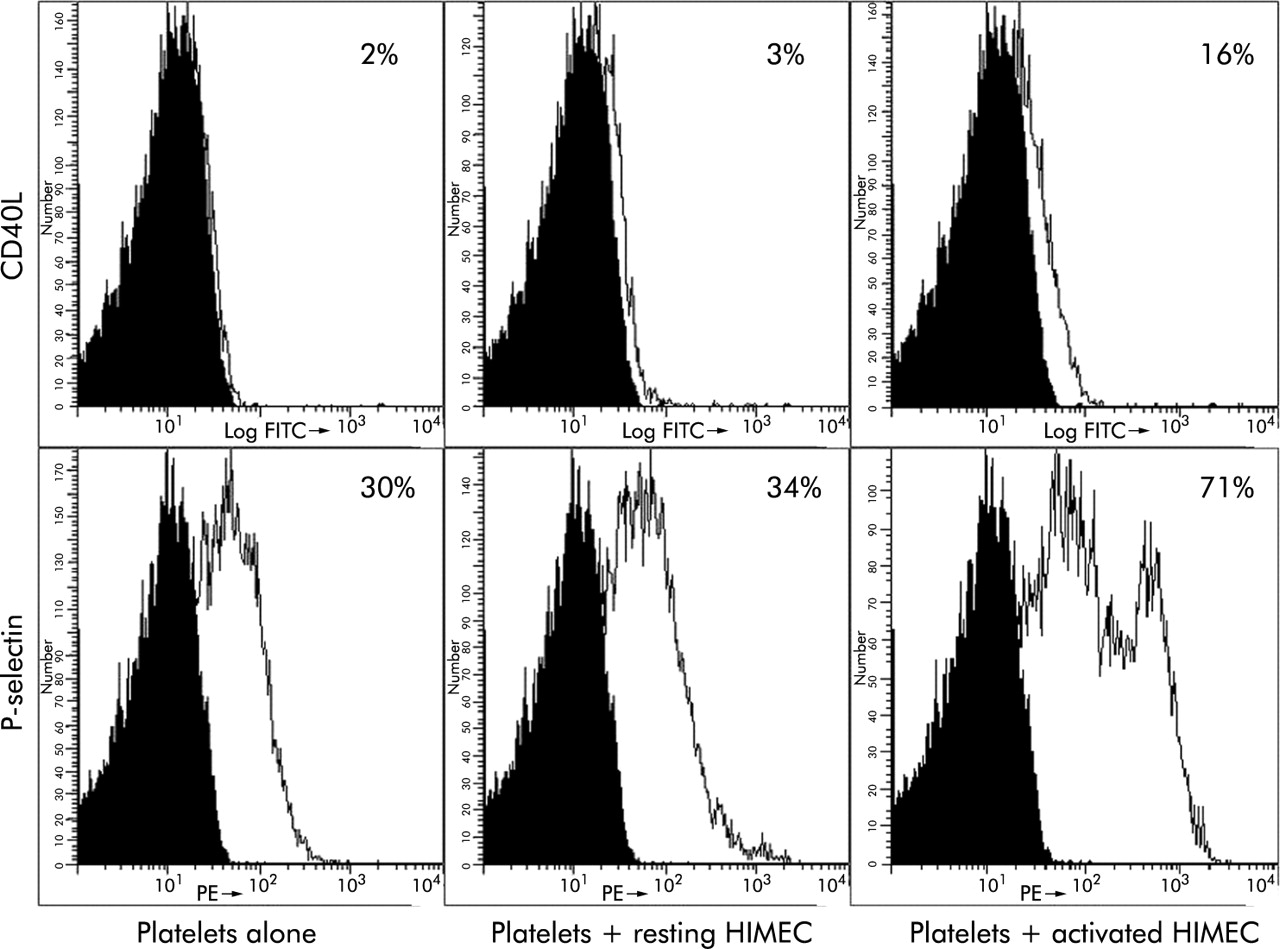
The observation that sCD40L levels are higher in IBD patients with more extensive involvement (fig 2) and platelets are the major source of sCD40L (fig 4), prompted us to investigate whether HIMEC exposed to an inflammatory milieu could trigger platelet activation resulting in CD40L upregulation. Normal platelets were analysed for CD40L and P-selectin expression before and after coculture with resting HIMEC and HIMEC preactivated by IL-1β to mimic the IBD mucosa conditions. Coculture with resting HIMEC failed to upregulate platelet bound or soluble CD40L (fig 7; fig 8A, 8B). On the other hand, contact with IL-1β pretreated HIMEC elicited an activated profile from platelets, demonstrated by a significant increase in the expression of both CD40L and P-selectin (fig 7, fig 8A), as well as a significantly increased release of sCD40L by platelets (fig 8B).

Induction of human intestinal microvascular endothelial cell (HIMEC) contact dependent CD40 ligand (CD40L) on platelets. Normal platelets were left untreated, or cocultured with unstimulated or interleukin 1β (IL-1β) activated HIMEC, stained with CD40L and P-selectin antibodies, and submitted to flow cytometric analysis. The histogram shows the percentage of positive platelets, and is representative of one of three separate experiments.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Induction of human intestinal microvascular endothelial cell (HIMEC) contact dependent CD40 ligand (CD40L) on platelets. Normal platelets were left untreated, or cocultured with unstimulated or interleukin 1β (IL-1β) activated HIMEC, stained with CD40L and submitted to flow cytometric analysis. The coculture supernatants were collected and the sCD40L content measured by ELISA. (A) Percentage of CD40L positive platelets. (B) Levels of sCD40L. Data represent the values of three and four individual experiments for (A) and (B), respectively. *p<0.05 for platelets cocultured with activated HIMEC compared with platelets alone or cocultured with resting HIMEC.
DISCUSSION
The results of this study show that plasma sCD40L levels are significantly elevated in CD and UC patients compared with those found in normal individuals, and that platelets display enhanced expression of surface CD40L in both forms of IBD. Elevation of circulating sCD40L is not a phenomenon unique to IBD, being found in a variety of inflammatory, autoimmune, and thrombotic disorders.20–26 However, combined with increased expression of CD40 in affected tissues, it provides important clues to disease pathogenesis, as shown in other chronic inflammatory conditions such as atherosclerosis and rheumatoid arthritis.4,5 Similarly, overexpression of CD40 in actively inflamed IBD mucosa7,8 demonstrates the presence of resident cells which, upon binding of CD40L, can become activated and produce proinflammatory mediators. We have previously reported that this is indeed the case, as T cell bound, platelet bound, or soluble CD40L induce chemokine production, CAM upregulation, and increased leucocyte adhesion by HIF and HIMEC.13,14
Because CD40L also displays prothrombotic properties, and high concentrations of sCD40L represent a risk factor for vascular events,36 in view of the increased incidence of thromboembolic phenomena in IBD patients, we specifically measured plasma levels of sCD40L in a subgroup of CD and UC patients with active thromboembolic complications. Levels in this clinical subgroup were comparable with or lower than those found in IBD patients with both active and quiescent disease, a finding perhaps explained by the utilisation of sCD40L in the inflamed mucosa. In fact, sCD40L clusters inside platelet-endothelial aggregates during thrombus formation,14 and is sequestered inside thrombi where it is essential for clot stabilisation,17 both of these events resulting in sCD40L consumption.
Because sCD40L derives primarily from activated platelets or activated T cells,11 establishing which cell type is the main source of the abundant sCD40L circulating in IBD patients is clearly relevant to disease pathogenesis. In spite of its obvious importance, this key issue has never been addressed by the multiple reports evaluating plasma sCD40L in other disease states.20–25 When we first assessed its expression on the platelet surface, both freshly isolated and thrombin activated IBD platelets expressed significantly higher CD40L levels compared with those of healthy controls, a finding in agreement with the enhanced state of platelet activation in both CD and UC.37 In contrast, no difference existed between CD40L expression by PBT of IBD and normal subjects, regardless of whether or not they were activated. Together, these results point to platelets as the major source of plasma sCD40L in IBD. To corroborate this conclusion, we isolated and cultured total platelets and PBT from a fixed volume of blood from normal and IBD subjects, and quantified the relative levels of spontaneously released sCD40L in cultures of each cell type. Platelets but not PBT from normal subjects released sCD40L in the supernatants, and platelets from IBD patients released vastly greater amounts of sCD40L than autologous PBT, confirming that platelets are indeed the almost exclusive source of sCD40L in the circulation of IBD patients.
Under the above experimental conditions, both unstimulated and activated IBD platelets released greater quantities of sCD40L than normal platelets. Nevertheless, these results could be deceiving because the number of platelets/volume of blood tends to be higher in IBD patients and vary from sample to sample.38,39 To control for these variables, levels of sCD40L in the supernatants were normalised to the concentration of platelets in each blood sample. Even after this adjustment, platelet supernatants from IBD patients contained more sCD40L than those from normal subjects. In addition, as demonstrated by immunoblotting, IBD platelets themselves contain greater amounts of CD40L protein than normal platelets. These observations confirm that platelets actually release increased quantities of sCD40L in both CD and UC, a finding in agreement with elevated expression of surface CD40L secondary to their increased state of activation. In fact, the increased state of activation rather than the platelet number appears to be the critical factor leading to enhanced sCD40L release, as only a weak correlation was found between platelet number and sCD40L levels.
Further supporting increased release of sCD40L by IBD platelets is a recent report showing that the high plasma levels of sCD40L in patients with inflammatory disease associated reactive thrombocytosis are not solely due to increased platelet numbers but also to an increased content of CD40L in the platelet cytoplasm.40 Overall, the results of our study support the conclusion that, in vivo, activated platelets are the dominant source of plasma CD40L in IBD.
Finally, we explored a possible mechanism that could induce elevated expression of surface CD40L and explain enhanced levels of its soluble form in IBD. Platelet activation can be triggered by a wide variety of substances and cells,41 but in IBD an obvious possibility is activation in the vast intestinal microvascular bed, where they encounter and cross talk with inflamed endothelial cells.35,42–45 This is the reason why we adopted a platelet-HIMEC coculture system as it allows platelet contact with and stimulation by activated intestinal mucosa microvascular cells, mimicking the in vivo IBD conditions. IL-1β stimulated HIMEC elicited platelet activation, proven not only by the upregulation of P-selectin expression but, more importantly, the increase in membrane bound as well as released CD40L. This mechanism is strongly corroborated by the observation that levels of sCD40L in IBD were proportional to the anatomical extent of mucosal inflammation which reflects the size of the endothelial cell surface area encountered by platelets.
In summary, this study reports the presence of elevated levels of sCD40L in the circulation of CD and UC patients, and demonstrates that sCD40L originates primarily from platelets likely activated in the IBD microvascular bed. The importance of these observations resides with the potent and far reaching biological activities triggered by activation of the CD40 pathway in a multitude of immune and non-immune cells types.3,11 In view of the demonstration that recombinant or platelet derived sCD40L can effectively trigger the CD40 pathway in intestinal cells,13,14 the presence of elevated levels of sCD40L in both CD and UC patients provides a novel platelet dependent mechanism that, by amplifying and sustaining inflammation, contributes to IBD pathogenesis.
Acknowledgments
This work was supported by grants from the Crohn’s and Colitis Foundation of America, Inc., the Italian Society of Gastroenterology (SIGE), and the Sirio Lentini Memorial Fund to SD, and the National Institutes of Health (DK30399 and DK50984) to CF.
REFERENCES
Linked Articles
- Digest