Article Text

Abstract
Background: Lamina propria T lymphocytes (LPL) of the intestinal mucosa are chronically activated in Crohn’s disease (CD). Defective apoptosis of activated LPL was proposed as a key pathogenic mechanism. In fact, increased expression of antiapoptotic molecules was observed in CD LPL. In the present work, we aimed to analyse the effects and underlying molecular mechanisms of 5-amino salicylic acid (5-ASA) and derivatives on apoptosis of LPL and peripheral blood lymphocytes (PBL) in patients with CD compared with ulcerative colitis (UC) and in non-inflammatory controls.
Methods: PBL and LPL were isolated by Ficoll-Hypopaque gradient centrifugation and the EGTA-collagenase method, respectively. PBL/LPL were stimulated with FasL, 5-ASA, sulphapyridine, and sulphasalazine for 24/48 hours and apoptosis was quantified by flow cytometry (annexin V- propidium iodide method) and immunofluorescence. The molecular mechanisms of drug induced apoptosis were analysed in wild-type and FADD−/− Jurkat T cells using western blots and caspase assays.
Results: While PBL displayed a normal apoptosis pattern after Fas stimulation in patients with active CD, LPL from inflammatory areas were highly resistant. Comparable resistance to apoptosis was observed in LPL of UC patients. In contrast with 5-ASA, which did not induce apoptosis in lymphocytes, sulphasalazine proved to be a potent proapoptotic agent. Sulphasalazine induced T lymphocyte apoptosis was independent of the Fas pathway but associated with marked downregulation of antiapoptotic bcl-xl and bcl2, activation of the mitochondrial apoptosis signalling pathway, and subsequent activation of caspase-9 and caspase-3.
Conclusion: The beneficial effect of sulphasalazine in treating inflammatory bowel disease is at least in part attributable to its proapoptotic effects on LPL which allows potent downregulation of lymphocyte activation.
- IBD, inflammatory bowel disease
- LPL, lamina propria T lymphocytes
- CD, Crohn’s disease
- 5-ASA, 5-amino salicylic acid
- PBL, peripheral blood lymphocytes
- UC, ulcerative colitis
- FasL, FasLigand
- FADD, Fas associated death domain protein
- DISC, death inducing signalling complex
- IL, interleukin
- NFκB, nuclear factor κB
- Crohn’s disease
- T lymphocyte
- apoptosis
- caspase
- sulphasalazine
Statistics from Altmetric.com
- IBD, inflammatory bowel disease
- LPL, lamina propria T lymphocytes
- CD, Crohn’s disease
- 5-ASA, 5-amino salicylic acid
- PBL, peripheral blood lymphocytes
- UC, ulcerative colitis
- FasL, FasLigand
- FADD, Fas associated death domain protein
- DISC, death inducing signalling complex
- IL, interleukin
- NFκB, nuclear factor κB
Intestinal immune responses are often characterised by activation of lamina propria T lymphocytes (LPL) with potent effector functions, as in inflammatory bowel diseases (IBD).1 However, the exact pathomechanisms leading to chronic activation of LPL in Crohn’s disease (CD) patients still remain unclear.1–3 As the intestinal mucosa is permanently exposed to a magnitude of antigens, the mucosal immune system has evolved several strategies to avoid an unnecessary and uncontrolled inflammatory reaction. Among these regulatory mechanisms, downregulation of activated T lymphocytes via apoptosis is a very potent and effective strategy, now considered as a key controlling mechanism.4 In patients with CD, the LPL apoptosis pathway was found to be profoundly disturbed.4–8 Compared with LPL from non-inflammatory intestinal mucosa, LPL from CD patients were significantly less susceptible to apoptosis in reponse to different proapoptotic stimuli.4–8 Based on these data, a new conceptual view of CD was suggested: a defect in the intrinsic downregulatory apparatus which controls apoptosis of activated T cells may lead to their inapprorpiate accumulation and result in disease perpetuation and chronic inflammation.
The molecular mechanisms and signalling pathways of apoptosis in activated lymphocytes have been extensively studied.9 Fas induced apoptosis of LPL was suggested to play a prominent role in the downregulation of local immune responses10 After binding of FasLigand (FasL) to its membrane receptor Fas, constitutively expressed on LPL, FasL-Fas trimerises and an intracellular multiprotein complex called DISC (death inducing signalling complex) is formed.9 DISC consists of the death domain of Fas, an adapter protein called Fas associated death domain protein (FADD) and procaspases-8 and -10. Association of caspases-8 and -10 to DISC results in its autocatalytical activation with subsequent activation of the intracytoplasmic caspase signalling cascade, allowing amplification and rapid propagation of the initial apoptotic signal. Ultimate activation of downstream caspase-3 results in the execution of apoptosis, attested by DNA cleavage into 180 bp fragments and the formation of apoptotic bodies.11 The caspase cascade is controled by several inhibtory molecules, such as the inhibitor proteins of the apoptosis family (IAP1,2, XIAP) or FLIP.12 Execution caspase activation on Fas stimulation can occur in a mitochondrial dependent or independent manner. The mitochondrial apoptosis pathway is controlled and regulated by the bcl superfamily. This protein family is composed of several pro- and antiapoptotic members, which can form heterodimers, neutralising each other’s effect.13 A shift in the bcl rheostat towards an antiapoptotic constellation results in abrogation of a proapoptotic stimulus. Therefore, the bcl gene family serves as an intracellular threshold for apoptosis. In fact, defective Fas mediated apoptosis in LPL from patients with CD was associated with changes in the mitochondrial apoptosis pathway: in CD patients, there was a shift in the mitochondrial bcl-rheostat with upregulation of antiapoptotic bcl-2 and bcl-xl molecules (which protected LPL from apoptosis 5–7). Recently, Atreya and colleagues8 provided experimental evidence that interleukin (IL)-6, which is abundantly secreted in CD, upregulates expression of antiapoptotic bcl molecules via the IL-6 trans-STAT 3 signalling pathway.
There is increasing evidence that anti-inflammatory drugs, commonly used in the treatment of IBD, interfere with activation of the inflammatory cascade via the nuclear factor κB (NFκB). Wahl and colleagues14 demonstrated that sulphasalazine (the conjugate of 5-amino salicylic acid (5-ASA) and sulphapyridine) directly inhibits phosphorylation of NFκB, thereby blocking this pathway. Similarly, Kaiser and colleagues15 observed that 5-ASA alone blocks activation and translocation of NFκB in intestinal epithelial cells. In several cell types, NFκB is not only involved in the generation of an inflammatory reaction but also in the regulation of apoptosis. For instance, in Hela, Hek, or Jurkat cells, expression of the antiapoptotic molecule FLIP or of the antiapoptotic bcl molecules is regulated by NFκB.16 As a result of different regulatory factors, a cell can thus respond to proinfllammatory stimuli in different ways (that is, activation of a proinflammatory, either pro- or antiapoptotic repertoire). Therfore, we hypothesise that modulation of the NFκB pathway by anti-inflammatory drugs (such as 5-ASA and its derivatives) may interfere with the susceptibilty of lymphocytes to apoptosis. In the present study, we analysed the effect of 5-ASA and derivates on peripheral blood lymphocyte (PBL) or LPL apoptosis in patients with CD, and subsequently studied the underlying molecular mechanisms.
MATERIAL AND METHODS
Patients
Ten paediatric patients with active CD (aged 5–15 years), four patients with active ulcerative colitis (UC) (10–14 years), as inflammatory controls, and four patients without any inflammatory pathology (5–35 years) were analysed in this study. Diagnostic endoscopy was performed in all patients with CD or UC. Control patients presented with intestinal symptoms suggestive of IBD and thus underwent endoscopic examination, which was macroscopically completely normal along with a normal histology. Diagnosis of CD or UC was based on typical clinical, biological, radiological, endoscopic, and histological findings, according to established criteria.17 Treatment consisted of 5-ASA in five CD patients (combined with budesonide in one patient), parenteral nutrition in three patients, and enteral nutrition combined with prednisone (0.2 mg/kg) in a further patient. One CD and one UC patient were not receiving any treatment. The other three UC patients received 5-ASA combined with prednisone (1 mg/kg) in one patient. The control patients received no drugs, except one treated by antibiotics.
Lymphocyte isolation and cell lines
PBL were isolated from heparinised blood on a Ficoll gradient centrifugation according to a standard procedure. LPL were isolated from colonic biopsies using either the EGTA-collagenase method, as previously described,18 or according to the method of Kugathasan and colleagues.19 To avoid bacterial or fungal contamination, we enriched the medium with antibiotics (cifloxacilline 100 μg/ml, fungizone 25 μg/ml). To characterise the cell populations after isolation, T cells were phenotyped by flow cytometry using anti-CD2, CD3, CD4, and CD8 surface markers (all BD, Pont de Claix, France). Only cell populations containing >90% CD3 positive cells were included in the present study. The number of cells isolated from two grouped biopsies from the same section varied from 6×105 to 4×106 cells. The degree of colonic inflammation was documented using an established endoscopic score (taking into account the presence of erythema, and superficial or profound ulcerations20). Biopsies from inflammatory areas were always taken close to ulcerations. In addition, a histological score21 was applied to quantify the degree of inflammation on biopsies adjacent to the tissue studied. PBL or LPL as well as Jurkat A3 cells (wild-type) (ATCC) and Jurkat I2-1 cells (FADD−/− cells22) were resuspended in RPMI 1640 with 10% human serum AB and cultured for variable time in 24 multiwell plates (Falcon) in a humidified incubator (5% CO2) at a concentration of 1×106 cells/ml.
Apoptosis assays
For analysis of apoptosis, the different (freshly isolated or IL-2 activated) PBL, LPL, or Jurkat cell lines were stimulated with a bioactive FasL supernatant obtained from Neuro 2a-FasL cells (dilutions 1:32 to 1:4) known to potently induce apoptosis via Fas.23 In separate experiments, apoptosis was induced by IL-2 withdrawal in PBL or LPL after a seven day stimulation period with IL-2 (50 IU/ml). In parallel experiments, cells were stimulated for 24–48 hours with sulphasalazine, 4-ASA, 5-ASA, sulphapyridine, and N-acetyl-cysteine, alone or in combinations (all Sigma-Aldrich, Taufkirchen, Germany) at concentrations from 0.01 to 5 mM freshly dissolved and directly diluted in RPMI 1640. In a different experimental setting, the broad range caspase inhibitor zVAD-fmk or the caspase-3 specific inhibitor zDEVD-fmk (100 μmol, both Alexis, Grunberg, Germany) was added before the above described stimulation of PBL, LPL, or different Jurkat cells. Apoptosis was monitored using the annexin V assay with propidium iodide counterstaining (Becton Dickenson, Pont de Claix, France) allowing quantification by flow cytometry (BDLSR; Becton Dickinson). In parallel, typical morphological characteristics of apoptosis were confirmed by immunofluorescence microscopy (Axioplan II; Zeiss, Jena Germany) and photodocumented after staining with the DNA-dye HOECHST 33342 (1 μg/ml) using a cooled CCD camera (Spot 2; Diagnostic Instruments, Serling Heights, Michigan, USA).
In parallel experiments, Jurkat cells stimulated as indicated above were lysed in ice cold lysis buffer (50 mM Tris, 150 mM NaCl, 10 mM EDTA, 1% Triton) supplemented with a mixture of protease inhibitors (Boehringer, Mannheim, Germany). Equivalent protein samples (20 μg) were resolved on 8–14% sodium dodecyl sulphate-polyacrylamide gels and transferred onto nitrocellulose membranes (Bio-Rad, Marnes-la-Coquette, France). For immunodetection, membranes were incubated overnight with anti-caspase-3 (0.5 μg/ml; Clinisciences, Paris, France), anti-caspase-9 (1:800 dilution; BD), anti-FLIP (1 μg/ml; Alexis-Coger, Paris, France), or anti-cIAP1 (1.0 μg/ml) and anti-cIAP2 antibody (1.5 μg/ml; both R+D Systems, Wiesbaden, Germany) in Tris buffered saline containing Tween 20–5% milk powder, followed by incubation with the corresponding horseradish peroxidase conjugated antibody (antimouse IgG 1:5000, antirabbit IgG 1:6000; Biosource, Hamburg, Germany). In parallel, expression and drug induced modulation of various bcl proteins were analysed in the same manner using highly specific rabbit-anti-bak antibody (1:800 dilution), rabbit-anti-bax antibody (1:700 dilution), rabbit anti-bcl-2 antibody (1:700; all three antibodies from BD Pharmingen, Heidelberg, Germany), and an anti-bcl-xl antibody (1 μg/ml; R+D Systems). Labelled bands were read by enhanced chemiluminescence (ECL-kit; Amersham, Freiburg, Germany) and photodocumented.
Statistical analysis
Results of apoptosis analyses are reported as mean (SEM) of triplicate samples. Significance was established at 95%, and determined by the Wilcoxon test (SPSS for Windows).
RESULTS
LPL but not PBL of CD patients are resistant to FAS induced apoptosis
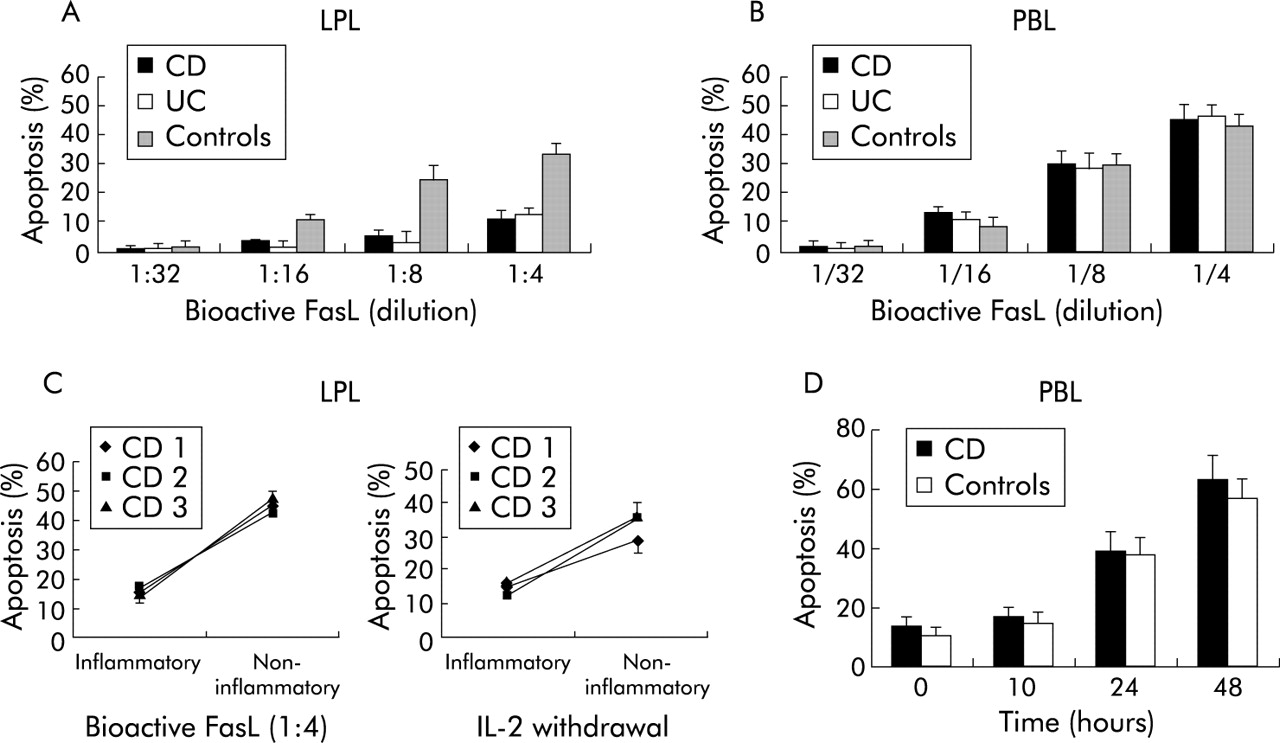
To address the question of a general T lymphocyte apoptosis defect in CD, we analysed and compared Fas induced apoptosis of autologous LPL of highly inflammatory colonic mucosa (histological score >5) and PBL (from the same patient). As shown in fig 1 (A, B), Fas induced apoptosis (at all FasL concentrations tested) was markedly lower in LPL (mean 11 (4%), median 9.2% at FasL 1:4) compared with activated PBL (mean 43 (7)%, median 46.3%) (p<0.005). In keeping with the findings of Boirivant and colleagues,6 LPL derived from the inflamed tissue were markedly more resistant to apoptosis not only in CD but also in UC patients than LPL isolated from histologically normal colon in controls (fig 1A). To analyse the role of the inflammatory reaction in LPL apoptosis, we compared LPL from highly inflammatory areas (histological score >5) to uninvolved tissue (histological score 0) in three CD patients (fig 1C). Susceptibility to Fas induced apoptosis was markedly lower in LPL derived from inflammatory areas than in LPL from non-inflammatory sections. Similarly, withdrawal of IL-2 in IL-2 pre-stimulated LPL induced almost no apoptosis in cells from inflammatory (median 15.2%) compared with non-inflammatory (median 35.3%) areas. In contrast, Fas or IL-2 withdrawal induced apoptosis of IL-2 activated PBL was comparable in CD patients and controls (fig 1D).

Response to Fas stimulation (FasLigand (FasL)) in lamina propria T lymphocytes (LPL) and peripheral blood lymphocytes (PBL). (A) LPL derived from inflammatory lesions of Crohn’s disease (CD) and ulcerative colitis (UC) patients but not LPL from non-inflammatory controls showed a marked resistance to Fas induced apoptosis. In contrast (B), PBL from CD as well as from UC displayed a high degree of apoptosis after Fas stimulation comparable with controls. (C) Comparison of LPL derived from inflammatory and non-inflammatory sections in three CD patients: LPL of inflammatory areas were resistant to Fas induced apoptosis. Similarly, after pre-stimulation with interleukin 2 (IL-2 50 IU/ml for seven days), cytokine withdrawal induced only apoptosis in LPL from non-inflammatory sections whereas LPL from inflamed tissue were resistant to IL-2 withdrawal induced apoptosis. (D) Time-kinetic studies in Fas stimulated PBL revealed an identical apoptosis pattern for inflammatory CD and control patients.
Sulphasalazine but not sulphapyridine or 5-ASA induces apoptosis in T lymphocytes of CD patients
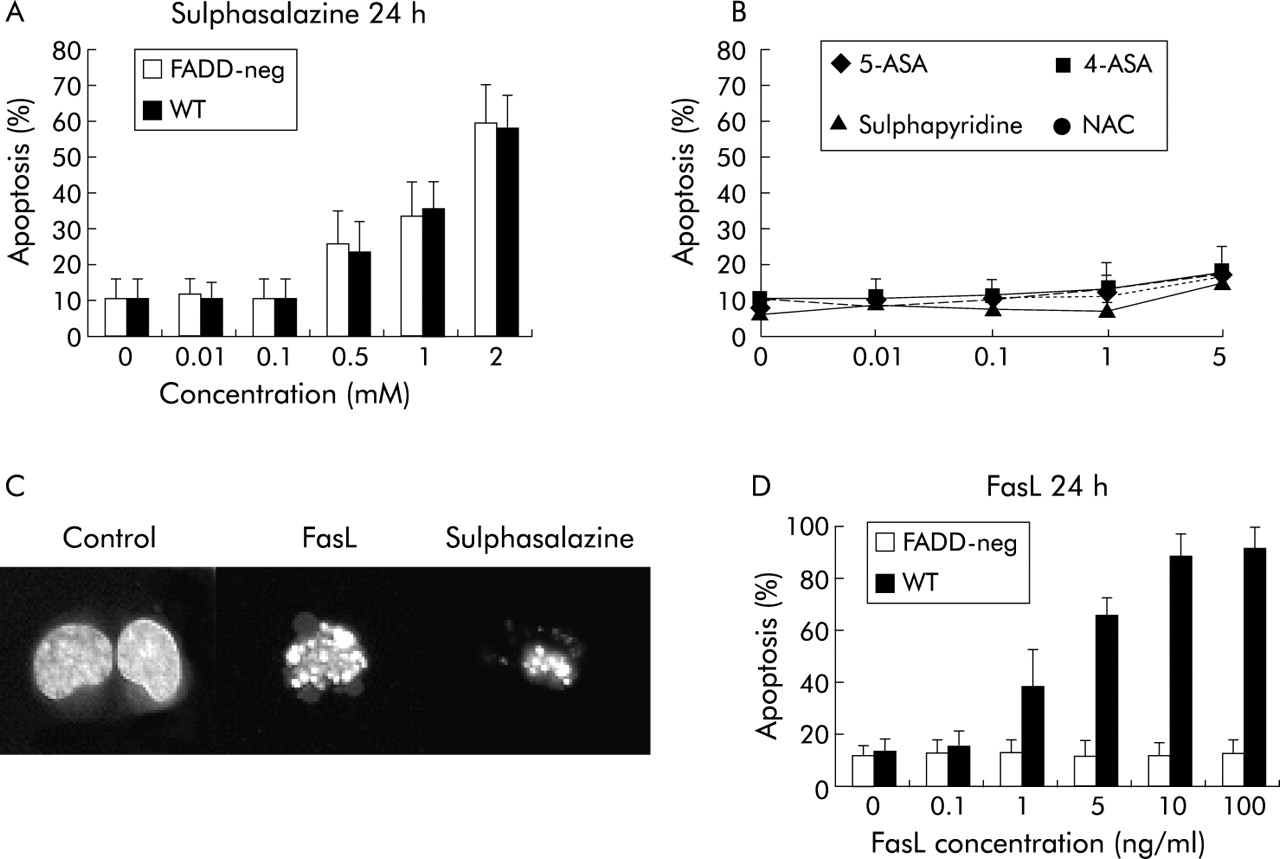
In the next step, we analysed the effects of 5-ASA and derivates on PBL and LPL apoptosis. As shown in fig 2, sulphasalazine was a potent inducer of lymphocyte apoptosis, both in PBL and LPL of CD patients. A maximal effect was observed at a 2.0 mM concentration, the proportion of apoptotic cells reaching 78 (3)% and 83 (5)% (p<0.001 versus control) for PBL and LPL, respectively. In contrast, no apoptotic response was noted in PBL or LPL of CD patients after stimulation with 5-ASA or sulphasalazine alone or in combination. These findings indicate that only the intact molecule sulphasalazine (5-ASA linked to sulphapyridine) has a specific proapoptotic effect. As additional controls, 4-ASA and N-acetyl cysteine were used, which were both without proapoptotic effects (data not shown). No significant difference in sulphasalazine induced apoptosis in LPL or PBL was observed between CD or UC or between CD and non-inflammatory controls. The apoptotic nature of cell death in sulphasalazine treated PBL and LPL was confirmed by immunofluorescence which showed condensation and fragmentation of the nuclei and formation of apoptotic bodies. In addition, annexin V staining of sulphasalazine treated PBL or LPL allowed identification of the flip of phosphatidyl serine to the outer plasma membrane in early apoptotic cells (fig 2C). Induction of apoptosis by sulphasalazine was independent of the activation status of lymphocytes as the response to sulphasalazine was comparable in freshly isolated PBL and IL-2 activated PBL (data not shown).

Effect of sulphasalazine on peripheral blood lymphocyte (PBL)/lamina propria T lymphocyte (LPL) apoptosis. Stimulation with sulphasalazine induced dose dependent apoptosis in (A) PBL or (B) LPL from inflammatory lesions in Crohn’s disease (CD) patients. In contrast, sulphapyridine, 5-amino salicylic acid (5-ASA) or the combination of both drugs had no proapoptotic effect at any concentration tested. (C) Flow cytometric analysis of PBL from an inflammatory CD patient allowed quantification of early and late apoptotic events after sulphasalazine stimulation using the annexin V-propidium iodide assay (see material and methods). ***p<0.001.
Molecular mechanisms of sulphasalazine induced T lymphocyte apoptosis
Using the Jurkat T cell line, a detailed analysis of the molecular mechanisms of sulphasalazine induced apoptosis was performed. Similar to PBL or LPL, Jurkat cells rapidly underwent apoptosis after stimulation with sulphasalazine whereas neither 5-ASA, sulphapyridine, alone or in combination, nor 4-ASA or NAC had any proapoptotic effects (fig 3). However, Jurket cells were slightly less sensitive to sulphasalazine induced apoptosis compared with PBL or LPL.

Role of Fas/Fas associated death domain protein (FADD) in sulphasalazine induced apoptosis. (A) Jurkat T cells responded with apoptosis after sulphasalazine stimulation in a way similar to peripheral blood lymphocytes (PBL)/lamina propria T lymphocytes (LPL). Wild-type (WT) and FADD-neg Jurkat showed the same apoptotic response to sulphasalazine. In contrast, 5-amino salicylic acid (5-ASA), sulphapyridine, 4-ASA, or NAC failed to induce Jurkat apoptosis (B). (C) Morphological analysis of sulphasalazine induced apoptosis (immunofluorescence after HOECHST33342 staining) revealed nuclear condensation and fragmentation of treated cells comparable with Fas induced apoptosis. (D) FADD-neg Jurkat cells were resistant to Fas induced apoptosis confirming that sulphasalazine induced apoptosis was independent of a functional death inducing signalling complex.
Given the fact that LPL of CD patients are resistant to Fas induced apoptosis, and that they respond to sulphasalazine induced apoptosis, we wished to test the hypothesis that the effect of sulphasalazine on T lymphocyte apoptosis was independent of Fas. Therefore, we used FADD-neg dominant mutant Jurkat cells, which are resistant to death receptor induced apoptosis, particularly to Fas (fig 3D). As observed with wild-type Jurkats and PBL/LPL of CD patients, sulphasalazine was a potent inducer of apoptosis in FADD-neg Jurkat cells, confirming that Fas and DISC formation with FADD was not required (fig 3A).
Based on the observation that sulphasalazine induced apoptosis was independent of the formation of a DISC, we analysed the role of the mitochondrial signalling apoptosis pathway after sulphasalazine stimulation. This pathway is under the close control of antiapoptotic bcl molecules. Analysis of the bcl-rheostate by western blots revealed marked downregulation of the antiapoptotic molecules bcl-xl and bcl-2 in response to sulphasalazine whereas levels of the proapoptotic molecules bax and bak remained unchanged (fig 4). 5-ASA, sulphapyridine, and the combination of both drugs did not alter expression of either bcl molecule. Similarly, 4-ASA and NAC had no effect on bcl molecule expression (data not shown). This sulphasalazine induced shift in the blc-rheostate was accompanied by activation of the mitochondrial apoptosis pathway, as seen in the activation of caspase-9, demonstrated by its total proteolysis (fig 4B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanisms of sulphasalazine induced apoptosis. (A) Sulphasalzine induced a shift in bcl homeostasis towards a proapoptotic constellation by reducing antiapoptotic bcl-xl and bcl 2 expression, without affecting expression of proapoptotic bax or bak, as shown by western blot analysis. In contrast, 5-amino salicylic acid (5-ASA), sulphapyridine, alone or in combination, did no alter any bcl molecule expression. (B) Activation of caspase-9 in response to sulphasalazine was demonstrated in an indirect way by disappearance of the proenzyme at 48/46 kDa; 5-ASA and sulphapyridine had no effect. (C) Inhibition of the caspase cascade using the broad range caspase inhibitor zVAD-fmk blocked sulphasalazine induced apoptosis, as did the caspase-3 specific inhibitor zDEVD-fmk. (D) Activation of caspase-3 after sulphasalazine stimulation was monitored by western blot, showing the occurrence of the active enzymes at 17 and 12 kDa with the proenzyme migrating at 32 kDA. Again, 5-ASA, sulphapyridine, and the combination of both drugs had no effect on caspase-3. One of three representative experiments is shown.
The role of the caspase signaling cascade in response to sulphasalazine was further demonstrated using the broad range caspase inhibitor zVAD-fmk which blocked apoptosis of Jurkat cells in a dose dependent manner (fig 4C). Furthermore, caspase-3 was identified as an executionary caspase, as demonstrated by the occurrence of activated caspase-3 in response to sulphasalazine stimulation, whereas 5-ASA and sulphapyridine alone or in combination had no effect (western blot analysis, fig 4D). Noticeably, sulphasalazine, 5-ASA, or sulphapyridine did not modify expression of antiapoptotic IAP molecules (cIAP1, cIAP2, XIAP, or FLIP, data not shown).
DISCUSSION
Under normal conditions, the intestinal immune system is in a physiological state of controlled inflammation. To limit a potentially harmful immune reaction, activated T lymphocytes are rapidly eliminated by apoptosis. However, in patients with IBD, a defect of T lymphocyte apoptosis was described.1,2,4 In the present study, we confirmed and extended previous findings of a markedly reduced susceptibility towards activation induced apoptosis of LPL in CD. However, our data do not suggest a primary intrinsic apoptosis defect in T lymphocytes in CD or UC. Only T cells in the inflamed intestinal mucosa were resistant to Fas or cytokine withdrawal induced apoptosis whereas autologous LPL from non-inflamed areas displayed a normal apoptotic response. In addition, PBL from all IBD patients responded normally to Fas induced apoptosis. Therefore, we hypothesise that the inflammatory reaction induces upregulation of antiapoptotic molecules in T lymphocytes. Accordingly, enhanced expression of antiapoptotic bcl molecules has previously been described in CD.4–7 This hypothesis is further supported by the observation that resistance of LPL to Fas induced apoptosis occurred under different mucosal inflammatory conditions, independent of the underlying disease (CD or UC).
Sulphasalazine is composed of a 5-ASA and sulphapyridine moiety. This drug is considered as mainstay therapy for Crohn’s colitis or UC.24 In the past, sulphasalazine was believed to be a prodrug, with the 5-ASA moiety as the active substance and sulphapyridine as the carrier. However, recent work indicated that sulphasalazine is a biologically active substance with potent anti-inflammatory effects on intestinal epithelial cells or T lymphocytes. Via inhibition of the inflammatory pathway NFκB, sulphasalazine was shown to interfere with regulation of apoptosis.25,26 In the present study, we provided experimental data showing that sulphasalazine had a potent proapoptotic effect on T lymphocytes of CD patients. Interestingly, there was no marked difference in inducing apoptosis in PBL compared with LPL in response to sulphasalazine stimulation. 5-ASA as well as sulphapyridine failed to induce apoptosis in T lymphocytes; even the combined use of both drugs had no proapoptotic effect, confirming previous findings that intact sulphasalazine is the effector molecule.14,25 This observation may explain the beneficial effect of sulphasalazine in IBD patients who fail to respond to 5-ASA treatment. Sulphasalazine concentrations in the inflamed intestine and colon are unknown. However, serum levels in patients treated with sulphasalazine were measured between 0.025 and 0.05 mM, with stool concentrations of approximately 2 mM.27 Therefore, the sulphasalazine doses used in this study may be within the hypothetical therapeutic range achieved within the intestinal mucosa.
Analysing the molecular mechanisms of T lymphocyte apoptosis in response to sulphasalazine, we observed that this drug interferes with expression of antiapoptotic molecules of the bcl family, thereby activating the mitochondrial apoptosis pathway. In fact, the bcl family plays an important regulatory role in the control of the mitochondrial apoptosis pathway, with antiapoptotic bcl2 or bcl-xl blocking proapoptotic bak or bax by the formation of heterodimers.13 A shift towards a proapoptotic constellation can result either from upregulation of proapoptotic bak or bax or from downregulation of antiapoptotic bcl2 or bcl-xl, as observed here in sulphasalazine treated T cells. This proapoptotic shift results in the formation of bak-bak or bax-bax homodimers which change the configuration of the mitochondrial membrane.28 This allows the exit of intramitochondrial molecules, such as AIF or cytochrome-c, into the cytoplasmic compartment.28,29 Whereas AIF induces apoptosis in a caspase independent manner through nuclear translocation, cytoplasmic cytochrome-c binds to APAF1 and caspase-9 forming a so-called apoptosome.30 In the present work, we showed in Jurkat cells that stimulation with sulphasalazine results in activation of caspases-9 and -3. However, Liptay and colleagues26 recently suggested an additional caspase independent mechanism. Sulphasalazine induced apoptosis was clearly independent of Fas or other death receptor induced apoptosis as FADD-neg Jurkat cells underwent apoptosis in a manner identical to wild-type cells after stimulation with sulphasalazine. Costimulation of LPL, PBL, or Jurkat cells with Fas and sulphasalazine showed only an additive, but no synergistic, effect (data not shown).
There is increasing evidence that induction of apoptosis in immune competent cells of the lamina propria is a more general and perhaps a crucial effect in the treatment of CD. Theoretically, elimination of activated LPL via apoptosis should suppress a chronic inflammatory process. In fact, Tiede and colleagues31 recently proposed that the beneficial effect of azathioprine/6-mercaptopurine in IBD is based on induction of apoptosis in activated lamina propria T cells. Similarly, the sustained anti-inflammatory effect of one single infusion of the anti-tumour necrosis factor antibody infliximab is attributed to its apoptosis inducing effect in lamina propria monocytes and T lymphocytes.32,33 In the present work, we were able to show that sulphasalazine is a potent proapoptotic drug in T lymphocytes of patients with IBD. Induction of apoptosis in LPL or PBL was independent of their activation status which is in clear contrast with the proapoptotic effect of other therapies such as azathioprine/6-mercaptopurine. The optimal anti-inflammatory or immunosuppressive drug in the treatment of CD should selectively block or eliminate the main effector subgroup of activated intestinal immune competent cells. One mechanism could in fact be induction of apoptosis in these cells. Hopefully, in the near future more potent and more selective immune modulatory drugs will become available for the treatment of CD.
Acknowledgments
This study was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM to FMR, NCB), a DAAD scholarship (to JD), and a Ferring foundation scholarship (to FMR). We acknowledge the excellent technical help of T Rottmann.