Article Text

Abstract
Background/aims: Mutations of the SPINK1 gene encoding pancreatic secretory trypsin inhibitor have been identified in association with chronic pancreatitis. The vast majority of patients carry the N34S variant, whereas other genetic variants are relatively rare and their disease association is uncertain. The aim of this study was to characterise and compare the functional defects caused by the six published missense mutations that affect mature SPINK1—namely, N34S, D50E, Y54H, P55S, R65Q, and R67C.
Methods: Wild type and mutant SPINK1 were expressed in human embryonic kidney 293T cells via transient transfection. SPINK1 expression was characterised by RT-PCR, activity assays, and western blots.
Results: Mutations N34S and P55S did not alter secretion of SPINK1 from HEK 293T cells, whereas mutation R65Q decreased secretion about twofold. Remarkably, mutations D50E, Y54H, and R67C abolished or markedly diminished secretion, but all three mutants were detected in cell extracts, indicating intracellular retention and degradation.
Conclusions: The results identify intracellular folding defects as a novel mechanism of SPINK1 deficiency associated with chronic pancreatitis. The dramatic effects of the D50E and Y54H mutations indicate that the interaction between Asp50 and Tyr54 is critical for proper folding of the inhibitor. The disease-causing biochemical defect in the N34S mutant is unrelated to secretion or trypsin inhibitory activity and remains enigmatic. Finally, the patent functional defects in mutants D50E, Y54H, and R67C suggest disease association of these rare SPINK variants.
- GAPD, glyceraldehyde-3-phosphate dehydrogenase
- HEK, human embryonic kidney
- HRP, horseradish peroxidase
- K, equilibrium constant
- PCR, polymerase chain reaction
- RT-PCR, reverse transcriptase polymerase chain reaction
- SPINK, serine protease inhibitor, Kazal type 1
- pancreatitis
- protein misfolding
- protein folding disease
Statistics from Altmetric.com
- GAPD, glyceraldehyde-3-phosphate dehydrogenase
- HEK, human embryonic kidney
- HRP, horseradish peroxidase
- K, equilibrium constant
- PCR, polymerase chain reaction
- RT-PCR, reverse transcriptase polymerase chain reaction
- SPINK, serine protease inhibitor, Kazal type 1
The association between mutations in the SPINK1 (serine protease inhibitor, Kazal type 1) gene and chronic pancreatitis has been documented extensively in the past seven years. The SPINK1 gene encodes the pancreatic secretory trypsin inhibitor, a 6.2 kDa protein with strong inhibitory activity towards trypsin, which is believed to play a part in protecting the pancreas against premature trypsinogen activation.1 This concept received strong support from the identification of the N34S mutation as a relatively frequent genetic susceptibility factor in chronic pancreatitis.2 Thus, studies from Europe and the United States indicated that 12.6% of patients with chronic pancreatitis were heterozygous for N34S, and 3.6% were homozygous.2,3,4,5,6,7,8,9,10 Heterozygous carriers of the N34S variant were also found in the control populations at 1.9% frequency on average. The N34S mutation was also recognised as a major genetic risk factor in tropical pancreatitis in the Indian subcontinent11–14 and as a minor susceptibility factor in alcoholic chronic pancreatitis,15 but not for hereditary pancreatitis associated with PRSS1 gene mutations.16 The disease relevant functional effect of the N34S mutation on SPINK1 structure and function has remained obscure so far. A study comparing recombinant N34S and wild type SPINK1 found no difference in trypsin inhibitory activity or binding to trypsin.17
In addition to the relatively common N34S mutation, a number of rare SPINK1 variants have been also reported (see http://www.uni-leipzig.de/pancreasmutation). Although conclusive association studies have been hampered by the small sample size, in some cases the nature of the mutation may suggest an obligatory loss of SPINK1 expression because of mRNA splicing problems, early translation termination, loss of the initiator methionine codon, or large genomic deletions.2,18–20 More recently, we described two families with hereditary pancreatitis who carried the L14R mutation in the signal peptide of SPINK1.21 Functional analysis of L14R and the previously described L14P mutation indicated a complete loss of secretion. The results provided the first clear experimental demonstration that alterations, which markedly reduce SPINK1 expression, are associated with classic hereditary pancreatitis. Therefore, these variants should be classified as severe and regarded as disease causing rather than disease modifiers.
In the present study we examined the functional consequences of the six reported mutations that affect the coding region of mature SPINK1. In addition to N34S, the rare mutations D50E, Y54H, R65Q, R67C, and the P55S polymorphism were studied using transfection experiments with human embryonic kidney (HEK) 293T cells (fig 1). The findings presented here reveal that intracellular defects of SPINK1 transport, probably caused by mutation induced misfolding, is a common mechanism that reduces SPINK1 secretion.

Missense mutations within the human pancreatic secretory trypsin inhibitor (SPINK1). The amino acid sequence of the mature inhibitor and mutations are denoted by the single letter code. Also indicated are the reactive site Lys (Lys41) and the disulfide bonds.
MATERIALS AND METHODS
Materials
Human cationic trypsinogen was expressed in Escherichia coli as inclusion bodies and refolded and affinity purified as described previously.22N-CBZ-Gly-Pro-Arg-p-nitroanilide was from Sigma (St Louis, Missouri, USA). Horseradish peroxidase (HRP) conjugated anti-Glu-Glu tag polyclonal goat antibody was purchased from Abcam (Cambridge, MA, USA).
Plasmid construction
The pcDNA3.1(−)_SPINK1 expression plasmid was constructed previously.21 Missense mutants and the Glu-Glu tagged SPINK1 constructs were generated by overlap extension polymerase chain reaction (PCR) mutagenesis and ligated into pcDNA3.1(−). The Glu-Glu tag sequence (EYMPME) is derived from the polyoma virus medium T antigen.23 SPINK1 fusion constructs with the SUMO tag were generated in the pET-SUMO plasmid (Invitrogen) according to the manufacturer’s instructions using the primers SPINK1-SUMO sense 5′-GAC TCC CTG GGA AGA GAG GCC-3′ and SPINK1-SUMO antisense 5′-TGG TTC TCA GCA AGG CCC AGA-3′.
Expression and purification of SPINK1 from E coli as SUMO fusion proteins
SUMO fusion proteins were expressed in E coli BL21(DE3). Cultures of 200 ml were grown at 37°C to optical density (OD600) of 0.6 and induced with 1 mM isopropyl β-d-galactopyranoside. Cultures were grown for an additional 2 hours, cells were pelleted by centrifugation, resuspended in 6 ml native binding buffer (50 mM NaH2PO4, 250 mM NaCl), and disrupted by sonication (10×20 seconds). Cell debris was removed by centrifugation, and the clarified supernatant was loaded onto a 1 ml Ni-NTA column equilibrated with binding buffer. The column was washed first with binding buffer, then successively with binding buffer containing 10 mM, 50 mM, or 250 mM imidazole concentrations. SUMO fusion proteins were present mostly in fractions eluted with 250 mM imidazole. To remove the SUMO fusion tag, approximately 80 μg fusion protein was digested overnight at 30°C with SUMO Protease (25 U) in SUMO protease buffer without salt in 500 μl volume. The cleavage reaction was then loaded onto a 1 ml Ni-NTA column and the column was washed with binding buffer. Native SPINK1 was recovered in the flowthrough, whereas the SUMO tag remained bound to the column.
Cell culture and transfection
Human embryonic kidney (HEK) 293T cells were cultured in D-MEM supplemented with 10% fetal bovine serum, 4 mM glutamine, and 1% penicillin/streptomycin solution at 37°C in a humidified atmosphere containing 5% CO2. A day before transfection, approximately 106 cells were plated in six-well tissue culture plates in culture medium without antibiotics. Transfections were performed using 4 μg pcDNA3.1(−)_SPINK1 plasmid, 2 μg pSV-β-galactosidase control plasmid (Promega), and 10 μl Lipofectamine 2000 (Invitrogen) in 2 ml OptiMEM medium supplemented with 2 mM glutamine. After 5 hours of incubation at 37°C, 2 ml of feed medium (D-MEM with 20% fetal bovine serum, 4 mM glutamine) was added to each well and cells were incubated for an additional 24 hours. Cells were then washed with 1 ml OptiMEM, 2 mM glutamine and the transfection medium was replaced with 2 ml OptiMEM, 2 mM glutamine. Time courses of expression were measured starting from this medium change and were followed for 48 hours. At the indicated times, 200 μl conditioned medium (that is, 10% of total volume) were removed from each well for activity assays and replaced with 200 μl fresh medium. At the conclusion of the transfection experiments, conditioned media and cells were harvested for western blot analysis and β-galactosidase assays.
Trypsin inhibitory activity determinations
Conditioned media from transfected cells were centrifuged for 5 minutes and 40 μl of the supernatant was mixed with 4 mg/ml bovine serum albumin and 80 nM recombinant human cationic trypsin (final concentrations) in a final volume of 50 μl and incubated for 1 minute at room temperature. The chromogenic substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide (150 μl) was then added to 0.14 mM concentration and 1 minute rates of p-nitroaniline release were measured at 405 nm. Residual trypsin activities were converted to inhibitor concentrations using a calibration curve obtained by assaying serial dilutions of purified recombinant SPINK1.
β-Galactosidase activity assays to determine transfection efficiency
Cells were co-transfected with 2 μg pSV-β-galactosidase plasmid. Forty-eight hours after transfection, the growth medium was removed and the cells were washed twice with phosphate buffered saline. Reporter lysis buffer (Promega; 200 μl) was added to each well and the plate was incubated for 15 minutes at room temperature. Cells were then scraped from the wells; the lysates were briefly vortexed and centrifuged for 5 minutes at 4°C. The protein concentration of the supernatants was measured with the Lowry method (Sigma) and the supernatants were assayed for β-galactosidase activity by measuring rates of o-nitrophenyl β-d-galactopyranoside hydrolysis at 420 nm, using the β-galactosidase Enzyme Assay System (Promega). The resulting specific β-galactosidase activity rates (expressed in mOD/min/mg protein) were used to normalise SPINK1 expression to variations in transfection efficiency.
Total RNA isolation and RT-PCR
RNA was isolated from cells transfected with wild type and mutant SPINK1 constructs using the RNAqueous kit (Ambion) and 0.75 μg RNA was reverse-transcribed with the SMART PCR cDNA Synthesis Kit (Clontech). SPINK1 cDNA was measured by semi-quantitative PCR using a gene specific primer pair described previously.21 As a reference control, the cDNA of the glyceraldehyde-3-phosphate dehydrogenase (GAPD) gene was also amplified using pseudogene-free amplification conditions.24
Western blotting
Samples were run on a 21% Tris-glycine gel under reducing conditions and transferred onto an Immobilon-P membrane (Millipore) at 300 mA for 3 hours. The membrane was blocked with 5% milk powder solution overnight and incubated with an HRP conjugated goat polyclonal antibody raised against the Glu-Glu tag at a concentration of 0.1 μg/ml for 1 hour at room temperature. HRP was detected using the SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Kinetic analysis of trypsin inhibition by SPINK1
The concentrations of purified SPINK1 preparations were determined by titration against human cationic trypsin. Titrations were performed with 40 nM trypsin concentration, which is about two orders of magnitude above the Ki value for SPINK1 inhibition. Under these conditions dissociation of the inhibitor is negligible in the calculations. The rate constants for SPINK1 inhibition of human cationic and anionic trypsin were determined with progress curve analysis. Cationic and anionic trypsin were incubated at 10 nM concentration with 20 nM SPINK1 and 140 μM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween-20. The increase in absorbance at 405 nm was monitored continuously for 10 minutes at room temperature. The progress curves generated were used to determine the SPINK1 association (k3) and dissociation (k-3) rate constants using the KinTekSim (KinTek Corporation) program, and the following equations.

where E = enzyme, S = substrate, P = product, and I = inhibitor.
The k1, k-1, and k2 rate constants were fixed values, which were determined from separate progress curves generated in the absence of inhibitor. The equilibrium dissociation constant (KD) for SPINK1 binding, which corresponds to the Ki, was calculated by dividing k-3 by k3.
RESULTS
Inhibitory activity of SPINK1 mutants expressed in E coli
To examine the effect of the reported mutations on the inhibitory activity of SPINK1, we expressed wild type, N34S, D50E, Y54H, P55S, R65Q, and R67C SPINK1 in E coli as His tagged SUMO fusion proteins and purified the inhibitor using metal chelating chromatography. After removal of the fusion tag, the concentration of the inhibitor was determined by titration against human cationic trypsin. The equilibrium dissociation constant (KD) for SPINK1 binding to cationic trypsin was then calculated from the association and dissociation rate constants, which were determined using progress curve analysis as described in Materials and methods. In this type of assay, trypsin is incubated with a chromogenic trypsin substrate and substrate cleavage is monitored continuously until all substrate is depleted. Addition of a trypsin inhibitor results in slower substrate consumption and the rate constants for SPINK1 binding can be calculated by non-linear regression analysis of the progress curves. Surprisingly, the mutations had only minor effects on inhibitor binding and all KD values were below nanomolar (reviewed but not shown).
Expression of SPINK1 mutants in HEK 293T cells via transient transfection
The cDNA constructs encoding wild type, N34S, D50E, Y54H, P55S, R65Q, and R67C variants of SPINK1 (fig 1) were cloned into the pcDNA3.1(−) plasmid and expressed in HEK 293T cells using transient transfection. To measure transfection efficiency, cells were routinely co-transfected with a plasmid encoding β-galactosidase. With the exception of the R65Q mutant, transcription from the wild type and the mutant SPINK1 constructs were comparable, as judged by semi-quantitative RT-PCR analysis of cDNA preparations from transfected cells (reviewed but not shown). The R65Q mutant exhibited slightly reduced mRNA levels by RT-PCR analysis.
Trypsin inhibitory activity of conditioned media
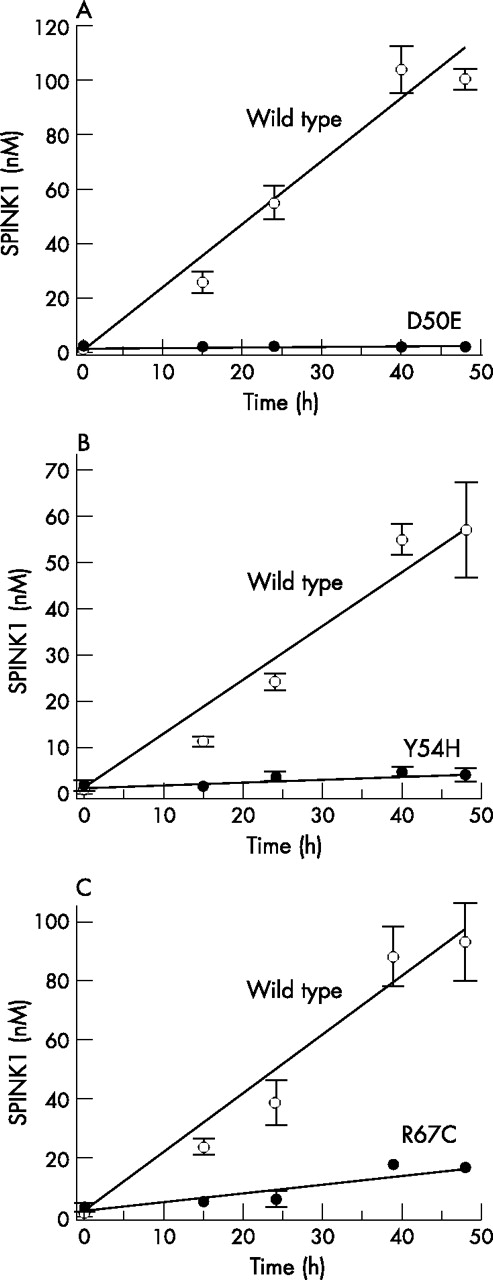
Secretion of SPINK1 protein by transfected cells was followed by measuring the trypsin inhibitory activity of conditioned media. The inhibitory activity was then converted to SPINK1 concentrations using a calibration curve prepared with purified wild type SPINK1 and human cationic trypsin. The SPINK1 concentrations obtained were then normalised to β-galactosidase expression. Because the mutations do not alter the inhibitory capacity of SPINK1, this assay should report on SPINK1 secretion. The results indicated that secretion of wild type or mutants N34S and P55S were comparable during the 48 hours post-transfection time course, whereas secretion of mutant R65Q was reduced approximately twofold (fig 2). In contrast, mutant D50E was not secreted to a detectable extent, and mutants Y54H and R67C exhibited 10-fold and fivefold reductions in secretion, respectively, relative to wild type SPINK1 (fig 3).

Effect of mutations (A) N34S, (B) P55S, and (C) R65Q on the secretion of SPINK1 from HEK 293T cells. At the designated times post-transfection, SPINK1 inhibitory activity of the conditioned media was determined against human cationic trypsin, as described in Materials and methods. Inhibitory activities were converted to SPINK1 concentrations using a calibration curve prepared with purified, recombinant SPINK1 and human cationic trypsin. Data represent the mean of two experiments with the deviation from the mean shown by the error bars. Open circles, wild type SPINK1; solid circles, mutant SPINK1.

Effect of mutations (A) D50E, (B) Y54H, and (C) R67C on the secretion of SPINK1 from HEK 293T cells. Experimental conditions are given in figure 2. Data represent the mean of two experiments with the deviation from the mean shown by the error bars. Open circles, wild type SPINK1; solid circles, mutant SPINK1.
Kinetic analysis of SPINK1 mutants N34S and R65Q expressed in HEK 293T cells
We have purified wild type SPINK1 and mutants N34S and R65Q from the conditioned media on a bovine trypsin column. Kinetic parameters of inhibition were then determined against human cationic trypsin and human anionic trypsin using progress curve analysis. The calculated KD values were all below nanomolar, confirming that mutants N34S and R65Q do not exert their pathogenic effect through impairment of the trypsin inhibitory activity of SPINK1 (fig 4).

Kinetic analysis of trypsin inhibition by wild type SPINK1 and mutants N34S and R65Q. Inhibition of human cationic trypsin (A), anionic trypsin (B), and mesotrypsin (C) by wild type SPINK1 and mutants N34S and R65Q was measured using progress curve analysis as described in Materials and methods. The trypsin concentration was 10 nM, the SPINK1 concentration was 20 nM, and the nominal trypsin substrate concentration was 140 μM. Representative progress curves are shown. The indicated KD values were calculated from two or three progress curves. Control progress curves were generated in the absence of SPINK1. Mesotrypsin is not inhibited by SPINK1 under these conditions.
Western blotting of conditioned media and cell lysates
To demonstrate by an alternative technique the reduced secretion of SPINK1 mutants western blotting was utilised. Previously we have successfully used a rabbit polyclonal antibody raised against full length human SPINK1 to follow SPINK1 secretion from mammalian cells.21 To validate the utility of this antibody against the mutants, first we performed immunoblots on SPINK1 mutants expressed in E coli. Unexpectedly, mutants D50E, Y54H, P55S, and R67C yielded minimal or no immunoreactive signal, whereas wild type SPINK1 and mutants N34S and R65Q were readily detected (reviewed but not shown). These results indicate that some of the SPINK1 mutations affect important immunological epitopes and the rabbit polyclonal antibody is an unsuitable reagent to compare secreted SPINK1 levels in conditioned media.
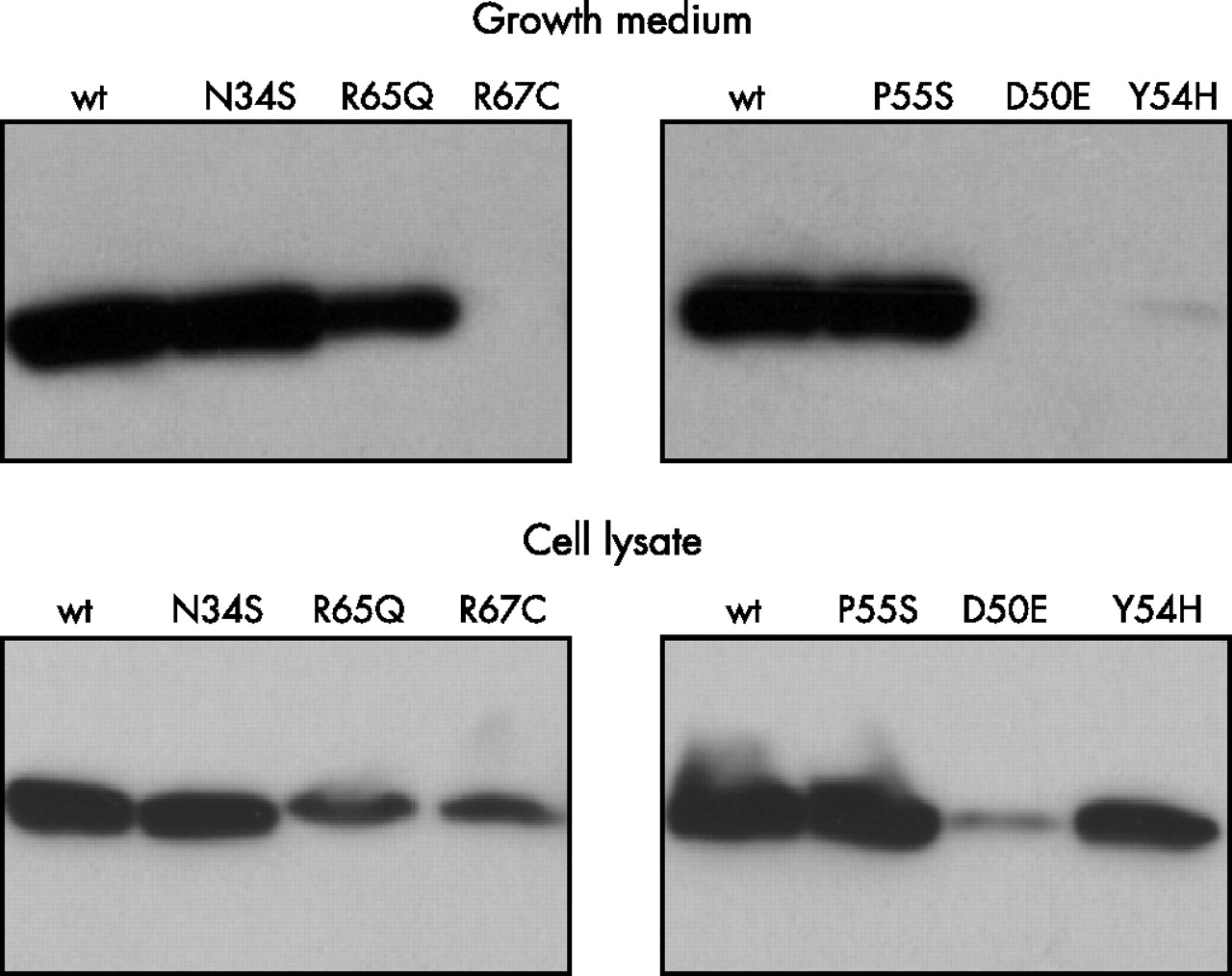
To circumvent this problem, a Glu-Glu epitope tag (EYMPME) was engineered to the C terminus of each SPINK1 construct. Epitope tagging had no effect on the secretion rate or the trypsin inhibitory activity of wild type SPINK1 and all mutants exhibited the same secretion defect as their non-tagged counterparts, as judged by activity assays. Western blots of conditioned media expressing epitope tagged SPINK1 constructs confirmed that mutants D50E, Y54H, and R67C were present in undetectable amounts and mutant R65Q showed approximately twofold reduced levels. On the other hand, conditioned media with mutants N34S and P55S exhibited immunoreactivity comparable to that of wild type SPINK1 (fig 5).

Western blot analysis of SPINK1 expression in conditioned media and cell lysates of transfected HEK 293T cells. Aliquots of conditioned media (5 μl) and cell lysates (10 μl; ∼3.5 mg/ml protein concentration) harvested 48 hours post-transfection were subjected to polyacrylamide gel electrophoresis, transferred to Immobilon P membranes and detected with a commercial anti-Glu-Glu tag antibody (Abcam).
Analysis of cell lysates revealed an interesting pattern. Compared to wild type SPINK1, intracellular levels of N34S and P55S mutants was essentially identical, whereas mutant R65Q was reduced about threefold. Remarkably, mutants D50E, Y54H, and R67C were all readily detectable in cell lysates at varying levels. Mutant Y54H gave the most prominent immunoreactive signal, which was as high as that of wild type SPINK1 (fig 5). Taken together the lack of secretion and the presence of immunoreactive SPINK1 protein in cell lysates, the observations indicate that SPINK1 mutants D50E, Y54H, and R67C are retained and degraded at varying rates inside the cell.
DISCUSSION
In this study we characterised the functional effects of the six reported missense mutations (N34S, D50E, Y54H, P55S, R65Q, and R67C) that alter the coding sequence of mature SPINK1. These variants occur with different frequency and have different clinical significance as well. The N34S mutation is the most frequent and best studied SPINK1 variant, which is clearly a risk factor for chronic pancreatitis. The P55S variant was first described by Witt et al as a heterozygous polymorphic variant identified in a healthy control patient.2 Subsequently, a number of studies found this variant both in healthy controls as well as in patients with chronic pancreatitis, acute pancreatitis, alcoholic pancreatitis, or fibrocalculous pancreatic diabetes.3,14,25–29 Although some studies showed a modest enrichment in the patient population, taking all available data together, the P55S alteration should be defined as a polymorphic variant with no disease association. Mutation D50E was described by Pfützer et al in a patient with familial chronic pancreatitis, who also carried the N34S mutation.3 Mutation Y54H was identified in a patient with tropical calcific pancreatitis from Bangladesh, who was negative for the N34S mutation.14 The R65Q SPINK1 mutation compounded with an Y1092X nonsense CFTR mutation was first identified in 2001 in a 35-year-old German male patient, whose healthy sister and mother also carried these mutations.30 Finally, mutation R67C was found in two Japanese patients, one of whom was also heterozygous for N34S.31–33 Interestingly, two other healthy members of this patient’s family were also R67C/N34S compound heterozygotes, suggesting low penetrance. Because mutations D50E, Y54H, R65Q, and R67C are rare, genetic evidence alone is insufficient to establish whether these mutations are true genetic risk factors for chronic pancreatitis.
The findings presented here show that these rare missense mutations do not alter trypsin inhibitory activity, but can reduce SPINK1 secretion by causing intracellular retention and degradation of SPINK1, which occurs probably because of mutation induced misfolding. Mutations D50E, Y54H, and R67C result in complete loss or marked reduction of secretion, whereas mutation R65Q decreases SPINK1 secretion about twofold. Inspection of crystal structures of pancreatic secretory trypsin inhibitor reveals that Asp50 and Tyr54 form a conserved hydrogen bond (fig 6). Clearly, as the data presented here indicate, this interaction is required for proper intracellular folding of SPINK1. Mutation R67C introduces an unpaired Cys residue, which can interfere with the correct formation of the three native disulfide bonds in SPINK1 and thus result in misfolded protein. On the other hand, the crystal structure shows the Arg67 side chain pointing towards the Asp50-Tyr54 pair, which raises the possibility that Arg67 might interact with Asp50 and stabilise its interaction with Tyr54 (fig 6). The relatively modest effect of the R65Q mutation on secretion is difficult to explain, but it may be related to the lower mRNA levels observed with this mutant and not a secretion defect. It is noteworthy that complete loss of secretion was also observed when mutants D50E and Y54H were expressed in Saccharomyces cerevisiae (not shown), indicating that the folding defect is a property of SPINK1 and does not depend on the host cell. Consequently, the same folding and secretion defect probably occurs in the pancreatic acinar cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Interaction between Asp50 and Tyr54 in the tertiary structure of SPINK1. The ribbon diagram of human pancreatic trypsin inhibitor is based on the structure of a recombinantly made artificial variant35 (Protein Data Bank file 1HPT). The side chains of the amino acids affected by missense mutations are shown. In this model, the reactive site Lys41 was restored and is shown in red. For clarity, the disulfide bridges are omitted. The image was rendered using DeepView/Swiss-PdbViewer version 3.7 (www.expasy.org/spdbv/).36
A previous study found that inhibitory properties of wild type and N34S mutant SPINK1 expressed recombinantly in Saccharomyces cerevisiae were indistinguishable.17 Our data confirm and extend these observations by demonstrating that neither mRNA levels nor SPINK1 folding/secretion is altered by the mutation. Ironically, therefore, the functional defect caused by the most studied SPINK1 mutation remains enigmatic awaiting further investigations. As discussed by other authors, intronic mutations in linkage disequilibrium with N34S may be responsible for the clinical effect or the N34S mutation might affect so far uncharacterised SPINK1 functions unrelated to trypsin inhibition. Finally, as expected, the P55S polymorphic variant had no effect on SPINK1 secretion.
The observations presented here define a small subset of pancreatitis cases as “protein folding diseases.” Pancreatitis thus may join a number of well known disorders that are caused by protein misfolding, such as α1-antitrypsin deficiency; cystic fibrosis, and neurodegenerative diseases (for a recent review see Chaudhuri and Paul34).
Acknowledgments
We thank Walter Halangk (University of Magdeburg) and Jian-Min Chen (University of Brest) for valuable discussions and sharing unpublished data.
REFERENCES
Footnotes
-
Published Online First 24 May 2007
-
Funding: This study was supported by NIH grant DK058088 to MS-T.
-
Competing interests: None to declare.
-
Note added in proof: The P55S variant was first described as P32S in a subject with chronic pancreatitis and two healthy controls (Chen JM, Mercier B, Audrezet MP, et al. Mutational analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene in hereditary and sporadic chronic pancreatitis. J Med Genet 2000;37:67–9.)