Article Text

Abstract
Objective: Most mechanistic studies of pancreatitis in mice employ the secretagogue-induced model. The currently reported studies were designed to develop an alternative, and possibly more clinically relevant, mouse model of pancreatitis.
Design: Na-taurocholate (10–50 μl, 1–5%) in saline, or saline alone, was retrogradely infused into the mouse pancreatic duct. The animals were killed 6–24 hours later and the severity of pancreatitis in the pancreatic head and tail was examined by quantitating hyperamylasemia, pancreatic edema, acinar cell necrosis, and pancreatic inflammation. In addition, intrapancreatic activation of trypsinogen, generation of IL-6, intrapulmonary sequestration of neutrophils, and alterations in lung compliance were evaluated. The effects of Na-taurocholate on in-vitro acinar cell calcium transients, viability, and trypsinogen activation were examined.
Results: Little or no evidence of pancreatitis was observed in mice infused with saline alone or in the tail of pancreata removed from animals infused with Na-taurocholate. In the head of the pancreas, evidence of pancreatitis was observed 12–24 hours after infusion of 20–50 μl 2–5% Na-taurocholate and the earliest morphological changes involved terminal duct and acinar cells. Intrapancreatic trypsin activity was transiently elevated within 5 minutes of Na-taurocholate infusion and pancreatic IL-6 levels were elevated 24 hours later. Under in-vitro conditions, Na-taurocholate triggered pathological acinar cell calcium transients, cell death, and calcium-dependent trypsinogen activation.
Conclusion: This clinically relevant model of acute biliary pancreatitis yields reproducible results and its severity can be easily manipulated. It is ideally suited for use in mechanistic studies employing genetically modified mouse strains.
- acute pancreatitis
- animal model
- bile acids
Statistics from Altmetric.com
Recent advances in transgenic technology have led to the wide availability of modified mouse strains making that species an increasingly attractive subject for mechanistic studies dealing with a variety of diseases including pancreatitis.1 2 Ideally, the clinical relevance of studies using an animal model would be strengthened if the employed model recapitulated events known to play a pathogenetic role in the clinical disease and, in addition, further benefit would also be gained if experiments could be performed utilizing at least two dissimilar models of the disease because, in that case, similar results with both models would reduce the likelihood that model-specific responses were being monitored. Unfortunately, in the case of pancreatitis studies in mice, neither of these ideals have been met. The clinical relevance of the most commonly employed mouse model, i.e. the model elicited by supramaximal secretagogue (caerulein) stimulation, is open to question because human pancreatitis is not generally triggered by supramaximal secretagogue stimulation. Furthermore, the choice of alternative models in mice has been suboptimal. Although alternative models including a diet-induced3 and an l-arginine-induced4 model have been described, their clinical relevance is highly questionable and, because their use presents significant technical problems, they have been neither widely employed nor well characterized.
In contrast, an alternative and seemingly clinically relevant model of pancreatitis, elicited by retrogradely infusing the pancreatic duct with the bile salt Na-taurocholate, is available for studies involving rats5 6 or larger animals. We reasoned that, with a careful surgical technique, that rat model could be adapted for use in mice, and in this communication we report the results of our studies that have characterized such a model.
MATERIALS AND METHODS
All experiments were performed using protocols approved by the Animal Care and Use Committee of New England Medical Center, and wild-type C57BL/6 mice (20–26 g) of either sex, purchased from Charles River Laboratories (Wilmington, Massachusetts, USA). The animals were housed in temperature-controlled (23 ± 2°C) rooms with a 12:12-hour light/dark cycle, fed standard laboratory chow and given freely available water.
Induction of pancreatitis
Anesthesia was achieved with a ketamine/xylazine cocktail (100 mg/kg and 10 mg/kg, respectively; Fort Dodge Animal Health, Fort Dodge, Iowa, USA). With the aid of a dissecting microscope, a midline laparotomy was performed and the first loop of the duodenum was identified. That loop was rotated, axially, to expose the pancreas and posterior surface of the duodenum. Using a traction suture, the posterior surface of the duodenum was immobilized, and the papilla of Vater was identified as it lay at the duodeno-pancreatic junction on the posterior surface of the duodenum. A puncture wound was made in the antimesenteric surface of the duodenum and a 30G blunt-tipped catheter, attached to infusion tubing leading to an infusion pump, was passed through the puncture wound, and then into the common bile duct via the papilla of Vater. The tip of the infusion catheter was placed 1 mm into the bile duct, well before the entry of the pancreatic duct. The infusion catheter was then secured in that position with a 10–0 ligature and the bile duct, at the liver hilum, was occluded with a neuro-bulldog clamp. Varying volumes of 150 mM NaCl solution containing varying concentrations of Na-taurocholate (Sigma-Aldrich, St Louis, Missouri, USA) were retrogradely infused into the bile-pancreatic duct over a 10 minute period using an infusion pump (Harvard Apparatus, Natick, Massachusetts, USA). Methylene blue (Fisher, Fair Lawn, New Jersey, USA) was routinely included in the infusion solution to permit the identification and exclusion of animals in whom the infusion had extravasated from the duct, but, with practice, extravasation of methylene blue-containing infusate was observed in less than 5% of animals. In selected experiments, India ink (Richard-Allan Scientific, Kalamazoo, Michigan, USA) was added to the infusate to permit light microscopic studies aimed at defining the extent of saline or Na-taurocholate infusion. After completion of the infusion, the catheter as well as the securing ligature and the neuro-bulldog clamp were removed and the duodenotomy was closed using a purse-string 8–0 prolene suture. The laparotomy was closed in two layers and the animals were allowed to awaken. They were given free access to water and regular laboratory chow, and analgesia was achieved by administration of buprenorphine hydrochloride (Reckitt Benckiser Healthcare, Hull, UK). With experience, the entire infusion procedure from induction of anesthesia to closure of the laparotomy incision could be completed within 30 minutes, and animal survival for 24 hours following infusion exceeded 95%. Sham-operated animals underwent laparotomy and were subjected to duodeno-pancreatic manipulation with insertion of a cannula into the bile duct but no fluid was infused.
Evaluation of pancreatitis severity
The severity of pancreatitis was evaluated as reported previously.7 Unless otherwise stated, the animals were killed 24 hours after duct infusion. At the time of death, blood samples were obtained and used for the measurement of plasma amylase activity.8 The pancreas and attached duodenum and spleen were rapidly removed en bloc. The pancreatic head (defined as the portion of the pancreas located within 5 mm of the lesser duodenal curvature) was separated from the pancreatic body/tail regions. Pancreatic edema was evaluated by quantitating tissue water content as previously described9 and expressed as a percentage of pancreas wet weight. Pancreatic inflammation was quantitated in a separate sample of pancreatic head or body/tail tissue as previously described.7 That sample of pancreas was homogenized and bromide-dependent myeloperoxidase activity was measured fluorometrically. Pancreatic necrosis was evaluated using a third sample of the pancreas head or body/tail. That sample was fixed in formalin, embedded in paraffin, stained with hematoxylin and eosin, sectioned, and evaluated by an examiner unaware of the sample identity. Necrosis was quantitated by morphometrically determining the area occupied by necrotic acinar cells, and expressing that area as a percentage of the total area occupied by acinar cells. No distinction was made between necrotic and injured acinar cells because, on purely morphological grounds, necrotic and injured cells could not be reliably distinguished from each other. Pancreatic trypsin activity was quantitated spectrophotometrically as previously described.10 Pancreatic levels of IL-6 were quantitated using a commercially available ELISA (R&D Systems, Minneapolis, Minnesota, USA) according to the manufacturer’s instructions.
In-vitro studies
The effects of Na-taurocholate on intra-acinar cell calcium transients, cell viability, and activation of digestive enzyme zymogens were evaluated using dispersed mouse pancreatic acini, freshly prepared as previously described.11 12 The pancreatic acini were passed through a 40 μm filter. Na-taurocholate-induced intracellular calcium changes were examined in single acinar cells within small acini pre-loaded with fura-2 AM. Na-taurocholate-induced activation of trypsinogen was quantitated by measuring trypsin activity in the suspending medium using Boc-Glu-Ala-Arg-4-methylcoumaryl-7-amide (Peptides International, Louisville, Kentucky, USA) as the substrate.11 13 The effects of Na-taurocholate on cell viability were quantitated by measuring the percentage of lactate dehydrogenase (LDH) release with the LDH-L reagent from Diagnostic Chemicals Limited (Oxford, Connecticut, USA) on a Cobas-Fara autoanalyzer (Roche Diagnostics, Nutley, New Jersey, USA).
Evaluation of pancreatitis-associated lung injury
The severity of lung injury was examined 24 hours after retrogradely infusing the pancreatic duct with 50 μl saline or saline containing 2% Na-taurocholate. Lung inflammation and microvascular permeability were evaluated by quantitating lung myeloperoxidase activity and leakage, into the bronchoaveolar space, of intravenously administered FD-4, respectively, as described.14 Lung compliance was quantitated using the Flexivent system.15
Analysis of data
Results are reported as means ± SEM obtained from three or more independent experiments. In all figures, vertical bars denote SEM values. Statistical evaluation of the data was accomplished by Student’s t-test, and p < 0.05 was considered to indicate significant differences.
RESULTS
Definition of a “standard protocol” for duct infusion-induced pancreatitis in mice
Infusion of 50 μl 2% Na-taurocholate into the mouse pancreatic duct induced acute necrotizing pancreatitis in the pancreatic head. As shown in fig 1, 24 hours after completion of this infusion there was a significant elevation in plasma amylase activity, and edema of the pancreatic head was detectable when the latter parameter of pancreatitis was quantitated by measuring tissue water content. In addition, 24 hours after Na-taurocholate infusion (50 μl, 2%), myeloperoxidase activity in the pancreatic head was significantly elevated indicating that neutrophils had been sequestered within that region of the pancreas and approximately 25% of acinar cells in the pancreatic head appeared to be either injured or necrotic. In contrast, only mild pancreatic edema and no increase in myeloperoxidase activity or evidence of acinar cell injury/necrosis were observed when samples of the pancreatic tail were examined after infusion of Na-taurocholate. Furthermore, infusion of saline (50 μl), alone, caused only mild hyperamylasemia but no significant pancreatic edema, acinar cell injury/necrosis, or intrapancreatic neutrophil sequestration even in the pancreatic head. Representative photomicrographs reflecting the light microscopic appearance of pancreas head and tail samples obtained 24 hours after Na-taurocholate (50 μl, 2%) or saline infusion are shown in fig 2. Pancreatic edema, inflammation, and necrosis were observed in pancreatic head samples taken from Na-taurocholate-infused mice but not in samples obtained from saline-infused or sham-operated animals or in pancreatic tail samples prepared from Na-taurocholate-infused animals.
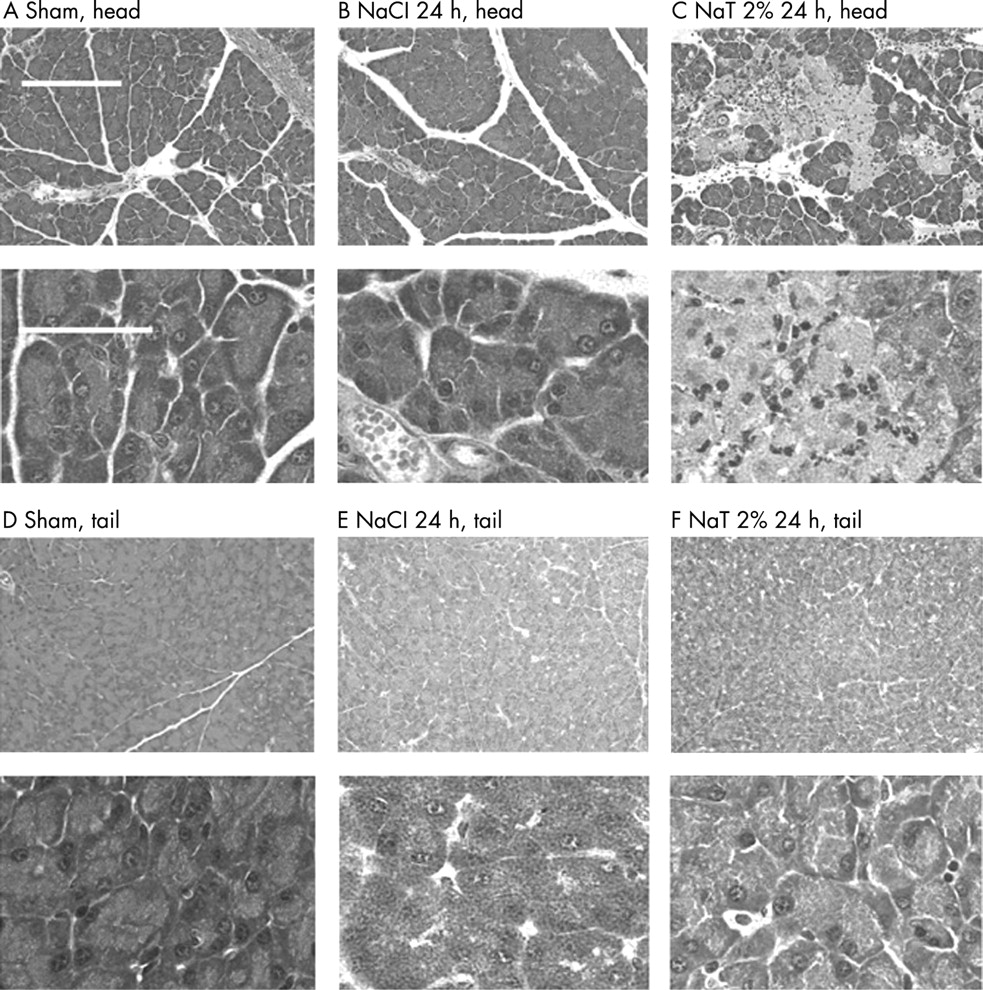

Effect of time on duct infusion-induced pancreatitis
The effects of time on the changes associated with duct infusion-induced pancreatitis are shown in fig 3. When compared with saline-infused animals, serum amylase activity first became significantly elevated 12 hours after duct infusion, and remained significantly elevated 24 hours after the completion of duct infusion. Significant pancreatic edema and elevated pancreatic myeloperoxidase activity were first observed 24 hours after duct infusion, whereas pancreatic injury/necrosis was first significantly increased when compared with saline-infused animals 12 hours after duct infusion. Over the subsequent 12 hours, the extent of injury/necrosis further increased but this increase was transient and, by 72 hours after duct infusion, the fraction of pancreatic tissue that appeared injured or necrotic had markedly decreased.
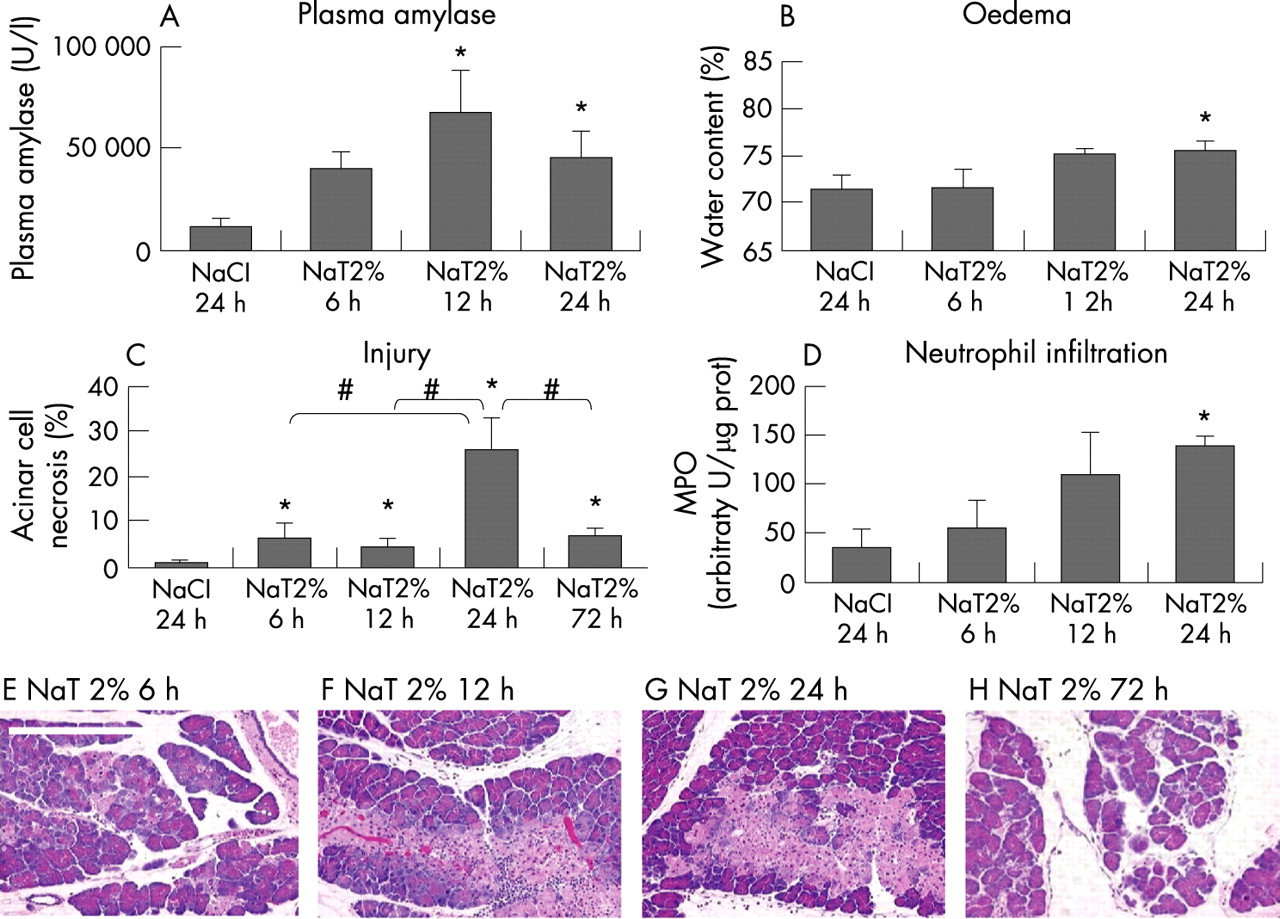
Effect of Na-taurocholate concentration on duct infusion-induced pancreatitis
Mice were infused with 50 μl saline containing varying concentrations of Na-taurocholate and killed 24 hours after duct infusion. As shown in fig 4, serum amylase activity, pancreatic water content, pancreatic myeloperoxidase activity, and the extent of pancreatic injury/necrosis were all significantly increased in animals receiving either 2 or 5% Na-taurocholate but no significant increase, when compared with saline-infused animals, was observed when 1% Na-taurocholate was infused.

Effect of infusate volume on duct infusion-induced pancreatitis
Mice were infused with 50 μl saline or 10–50 μl 2% Na-taurocholate and the effects of varying infusate volume on serum amylase activity, pancreatic myeloperoxidase activity, and pancreatic injury/necrosis were examined 24 hours later. As shown in fig 5, a significant rise in all three manifestations of pancreatitis was observed when 50 μl Na-taurocholate was infused. In contrast, a significant increase in serum amylase activity but not in either tissue myeloperoxidase activity or pancreatic injury/necrosis was observed when only 20 μl Na-taurocholate had been infused.

Effect of Na-taurocholate on distribution of infusate and evolution of injury
To examine these issues, India ink particles were added to the duct infusate (50 μl) and the animals were killed 10 minutes, 3 hours, or 6 hours after duct infusion. As shown in fig 6, light microscopic examination of samples prepared from either the pancreatic head or the pancreatic tail revealed collections of India ink particles in small pancreatic ducts and within acinar lumena. Small areas of extravasation were observed at the duct/acinus junction. Those collections were first noted 10 minutes after completion of the infusion and they persisted for at least 6 hours. Neither the frequency nor the size of the collections was altered by the inclusion of Na-taurocholate in the infusate. The India ink collections were frequently noted in the head of the pancreas but were only rarely seen in the tail of the gland, although the size and appearance of the individual India ink collections was similar regardless of their location.

Evidence of cell injury could already be detected 10 minutes after infusion of saline or saline containing Na-taurocholate and that initial injury was confined to cells immediately surrounding the terminal segments of small ducts, i.e. near the duct-acinus junction (fig 6). Ten minutes after the infusion of saline, the area of injury corresponded to the area noted to be labeled with India ink and, for the most part, that injury appeared to have resolved 6 hours after completion of the saline infusion. The area of initial cell injury noted after the infusion of Na-taurocholate also corresponded to the area labeled with India ink but, with time after Na-taurocholate infusion, that area expanded and, by 6 hours, the area of injury was noted to include entire lobules. In spite of this expansion, however, the injury noted 6 hours after Na-taurocholate infusion was confined entirely to acinar cells and, perhaps, terminal duct cells. Little or no evidence of injury to vascular structures or larger pancreatic ducts was observed at the light microscope level of resolution, and the injury remained multifocal with cells and acini that appeared to be completely healthy lying in apparent close proximity to cells that appeared to be severely injured or even necrotic.
Effect of Na-taurocholate on intrapancreatic activation of trypsinogen
Mice were infused with 50 μl saline or saline containing 2% Na-taurocholate and killed 1, 5, 10, 30, 180, or 360 minutes and 24 hours after completion of the 10 minute infusion. As shown in fig 7A, a small but statistically insignificant increase in trypsin activity in the pancreatic head was observed 1 minute after completion of the infusion of either saline or Na-taurocholate. In the saline-infused animals, this was followed first by a further small increase over the subsequent 9 minutes and then by a small decrease so that, 180 minutes after the end of the infusion, pancreatic trypsin activity was approximately 2-fold elevated in the animals undergoing saline infusion. In contrast to the results noted after saline infusion, infusion of Na-taurocholate led to a profound (> 6-fold) increase in trypsin activity in the pancreatic head that was maximal 5 minutes after completion of the infusion. That rise in trypsin activity was transient and, 30 minutes after the infusion, it had diminished to the level observed in the saline-infused animals. Over the subsequent 330 minutes of observation, no further increase in trypsin activity was observed. In the tail region of the pancreas, trypsin activity was not altered by either saline or Na-taurocholate infusion and the measured levels were similar to those observed in the naive animals (fig 7A).

Effect of Na-taurocholate on intrapancreatic generation of IL-6
Pancreas samples were harvested 24 hours after infusion of saline or 2% Na-taurocholate (50 μl) and pancreatic levels of IL-6 were quantitated by ELISA. Infusion of saline alone triggered a modest increase in IL-6 (fig 7B) whereas infusion of Na-taurocholate caused a further increase in IL-6 levels.
Effects of Na-taurocholate on in-vitro acinar cell calcium transients, cell viability, and zymogen activation
We observed the following three patterns of response in fluorescence studies using Fura 2-loaded cells (fig 8A): (a) calcium oscillations superimposed upon a stable and flat resting free intracellular calcium concentration (referred to as “oscillations”); (b) calcium oscillations superimposed upon a peak-plateau rise in intracellular calcium concentration (referred to as “oscillations + peak plateau”); and (c) a rapid rise followed by a sudden fall in Fura 2 fluorescence to a level below the initial baseline (referred to as “other”). The first pattern is reminiscent of the “normal” or physiological oscillatory pattern that is observed when acini are exposed to physiological concentrations of secretagogues whereas the second pattern of response is reminiscent of the so-called “pathological response”, which is noted when acini are exposed to supramaximally stimulating concentrations of secretagogues.16 17 The third response pattern is compatible with changes that might accompany Na-taurocholate-induced cell lysis, i.e. a rapid rise in fluorescence, possibly triggered by the influx of calcium from the suspending medium, followed by a rapid fall in fluorescence as Fura 2 leaks out of the cell and/or Fura 2 fluorescence is quenched. As shown in fig 8A, the observed response pattern was dependent upon the Na-taurocholate concentration in the medium. In general, approximately 36% of the cells responded to 0.05% and 0.1% Na-taurocholate, in contrast to more than 95% that responded to caerulein that was added to the same acini after treatment with Na-taurocholate. Of the cells that responded to Na-taurocholate, 80% responded with “oscillations” whereas only 20% of cells manifested “oscillations + peak plateau” and no cells appeared to lyse when the concentration of Na-taurocholate was 0.05%. In contrast, 0.1% Na-taurocholate elicited “oscillations” in 33% of cells, “oscillations + peak plateau” in 44% of cells, and apparent cell lysis in 22% of cells.

The effects of Na-taurocholate on acinar cell viability and zymogen activation are shown in fig 8B and C. Incubation of acini with 0.2 and 0.3% Na-taurocholate led to slowly evolving concentration-dependent cell death (fig 8B) whereas a rapid rise in LDH leakage to maximal levels, indicative of sudden and extensive cell death, was observed after exposure to 1% N-taurocholate. A rapid and transient rise in trypsin activity was observed 1 minute after exposure to 1.0% Na-taurocholate (fig 8C), but no change in trypsin activity was observed when acini were exposed to 0.1% Na-taurocholate. Intermediate levels of trypsin activity were observed after exposure to 0.3 and 0.5% Na-taurocholate.
To determine whether the trypsinogen activation and the cell injury are calcium dependent we used the intracellular calcium chelator 1,2-bis (2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetra (acetoxymethyl) ester (BAPTA-AM). Mouse acini, were isolated as described in Methods and resuspended in nominally calcium-free medium incubated with or without 50 μM BAPTA-AM for 30 minutes. They were then incubated with 0.3% Na-taurocholate and after 1 minute the trypsin activity was determined. We found that pre-incubation of mouse acini with BAPTA-AM reduced trypsinogen activation by 85% compared with the trypsinogen activation found in control acini, indicating that the Na-taurocholate-induced trypsinogen activation is calcium dependent (fig 9B). As a control we tested whether BAPTA-AM has an effect on the activity of isolated trypsin and found it not to be the case. Interestingly, when we tested whether Na-taurocholate-induced cell injury is calcium dependent we found that BAPTA-AM has no effect on cell death as judged by LDH leakage (fig 9A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
Passage of a biliary tract stone into or through the terminal biliopancreatic duct is believed to be the most frequent triggering event in acute pancreatitis,18 but the mechanisms by which that event elicits pancreatitis are uncertain. Theoretically, stone impaction in the terminal duct could trigger pancreatitis simply by obstructing pancreatic juice outflow and, thus, causing ductal hypertension or, alternatively, it could trigger pancreatitis by establishing a common upstream channel through which bile might reflux, retrogradely, into the pancreatic duct. In humans, evidence favoring both of these mechanisms has been presented19 20 whereas, in the opossum, the evidence to date suggests that pancreatitis is triggered by simple pancreatic duct obstruction.21
Claude Bernard,22 in experiments using olive oil, appears to have been the first to show that experimental pancreatitis could be elicited by retrogradely injecting substances into the pancreatic duct. Subsequently, an extensive literature has evolved based on the retrograde infusion of bile salts into the pancreatic duct as a means of eliciting pancreatitis in rats and larger experimental animals. In the current communication, we have shown that the retrograde infusion of Na-taurocholate into the pancreatic duct of anesthetized mice can also be used to elicit acute pancreatitis. That pancreatitis, which is severe and necrotizing but non-lethal, is characterized by the very early, but transient, intrapancreatic activation of trypsinogen, hyperamylasemia, pancreatic edema, sequestration of inflammatory leukocytes within the pancreas, pancreatic acinar cell injury/necrosis, and intrapancreatic generation of the pro-inflammatory cytokine IL-6. Its severity is dependent upon the concentration of Na-taurocholate infused as well as the volume of the infusate and the time that has elapsed after infusion. Infusion of saline, alone, causes little or no evidence of pancreatitis but infusion of 20–50 μl 2–5% Na-taurocholate results in pancreatitis that is mostly confined to the head region of the pancreas. When compared with infusion of saline alone, infusion of 2% Na-taurocholate (50 μl) does not alter lung myeloperoxidase activity, pulmonary microvascular permeability, or lung compliance 24 hours later. This indicates that the experimental pancreatitis elicited by the infusion of 2% Na-taurocholate (50 μl) in mice is not associated with significant lung injury. Future studies will be needed to determine if lung injury occurs when higher concentrations of Na-taurocholate are used.
Taken together, our findings indicate that retrograde infusion of Na-taurocholate into the mouse pancreatic duct elicits a highly reproducible and controllable non-lethal model of necrotizing pancreatitis. The severity of this model can be easily adjusted by altering the infusate volume or Na-taurocholate concentration. This model is based on a clinically relevant method of induction and is characterized by changes that are typical of clinical acute necrotizing pancreatitis. These observations suggest that this mouse model of pancreatitis would be ideally suited for use in mechanistic studies employing genetically modified animals.
To examine the pathophysiology of this model further, we performed a series of time-dependence studies using infusates labeled with India ink. Our experiments were designed to evaluate the effects of Na-taurocholate on the distribution of the infusate and on the relationship between that distribution and the evolution of injury in this model. We found that most of the infusate was localized to the head region of the pancreas and that areas of infusate extravasation could be found surrounding the small terminal ducts and the acinar spaces even when the infusate contained only saline. Within 10 minutes of the infusion, acinar cell injury was observed in these areas of extravasation but, when only saline had been infused, that injury had largely disappeared 6 hours later. Neither the distribution of the infusate nor the extent of its extravasation were altered by the addition of Na-taurocholate to the infusion solution, but the presence of Na-taurocholate had a profound effect on the subsequent acinar cell injury. As had been observed when only saline was infused, acinar cell injury was initially observed 10 minutes after the infusion but, in contrast to the events noted with saline alone, the injury noted after the infusion of Na-taurocholate was not transient. Rather, it expanded over time to involve additional acinar cells and, eventually, to encompass entire pancreatic lobules. Little or no injury to duct or vascular structures was observed and the injury remained multifocal rather than confluent. Taken together, these observations lead us to conclude that the presence of Na-taurocholate in the infusate triggers pancreatitis by converting a mild, reversible, and limited injury induced by infusion of saline alone into an expanding and irreversible severe injury. Furthermore, our findings indicate that the extent of injury is not dependent upon the pressure of the infusion but, rather, on the presence of Na-taurocholate.
We performed a series of in-vitro studies to examine the mechanism(s) by which Na-taurocholate might exert an injurious effect on acinar cells in this model of pancreatitis. Several recently reported studies, most of which have employed rat, rather than mouse, pancreatic acini and bile salts other than Na-taurocholate, have indicated that relatively low concentrations of certain bile salts can trigger pathological intra-acinar cell calcium transients, activate digestive zymogens such as trypsinogen, and trigger acinar cell injury/necrosis.23–26 For our studies, we focused on these phenomena shortly after exposing mouse pancreatic acini to varying concentrations of Na-taurocholate in vitro. We found that Na-taurocholate concentrations in the range of 0.3–1.0% could cause rapid and transient calcium-dependent zymogen activation that closely resembled the rapid and transient appearance of intrapancreatic trypsin activity that we had observed, under in-vivo conditions, after the retrograde infusion of 2% Na-taurocholate into the pancreatic duct. Presumably, the transience of trypsin activation observed in our studies simply reflects the propensity of trypsin to undergo auto-inactivation, but other mechanisms for the decline in trypsin activity can not be excluded. Interestingly, this trypsin activation appears significantly lower than that seen when cells are incubated with supramaximal concentrations of caerulein (100 nM). We also found that, under in-vitro conditions, exposure of acini to 0.2–1.0% Na-taurocholate led to acinar cell injury/death and that the extent of cell death was dependent upon the concentration of Na-taurocholate being used. Finally, we found that exposure of acini to 0.05–0.10% Na-taurocholate triggered calcium transients in acinar cells. With 0.05% Na-taurocholate, the transients in most cells had the appearance of physiological oscillations but, in a minority of cells, pathological peak plateau oscillations were observed. With 0.1% Na-taurocholate, on the other hand, the calcium response pattern shifted and most cells demonstrated pathological oscillations and/or changes suggestive of cell lysis.
Taken together, therefore, our in-vitro findings suggest that Na-taurocholate-induced pancreatitis after retrograde ductal infusion may be the result of Na-taurocholate-induced pathological intra-acinar cell calcium changes and/or activation of digestive enzyme zymogens. As the concentration of Na-taurocholate that is present at the site of infusate extravasation, i.e. the concentration to which acinar cells are exposed under in-vivo conditions, is not known, however, it is possible that Na-taurocholate also injures acinar cells by acting non-specifically as a membrane-permeabilizing detergent. Our studies using BAPTA-AM to chelate calcium suggest that Na-taurocholate triggers calcium-independent cell injury while it induces calcium-dependent zymogen activation.
We expect that future studies will expand our understanding of mechanisms that underlie the evolution of this mouse model of biliary pancreatitis and that this clinically relevant model will be extensively employed in studies using genetically modified mice to address mechanistic issues related to acute pancreatitis.
Acknowledgments
Supported by a fellowship from Sigrid Jusélius Foundation, Finland (J.L.) and by the Stichting Prof. Michaël-van Vloten Fonds in the Netherlands (G.J.D.vA) and the Netherlands Organisation for Health Research and Development (ZonMw, MD-Medical Research Trainee Grant 920-03-327) (G.J.D.vA) and by NIH grants DK031396-24 (M.L.S.) and AA015410 (G.P.).
REFERENCES
Footnotes
↵These authors contributed equally to this work
Conflict of interest: none declared.
- Abbreviations:
- BAPTA-AM
- 1,2-bis (2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetra (acetoxymethyl) ester
- LDH
- lactate dehydrogenase