Article Text

Abstract
Background and aims: Pancreatic cancer is among the most dismal of human malignancies. Current therapeutic strategies are virtually ineffective in controlling advanced, metastatic disease. Recent evidence suggests that the Hedgehog signalling pathway is aberrantly reactivated in the majority of pancreatic cancers, and that Hedgehog blockade has the potential to prevent disease progression and metastatic spread.
Methods: Here it is shown that the Hedgehog pathway is activated in the Pdx1-Cre;LsL-KrasG12D;Ink4a/Arflox/lox transgenic mouse model of pancreatic cancer. The effect of Hedgehog pathway inhibition on survival was determined by continuous application of the small molecule cyclopamine, a smoothened antagonist. Microarray analysis was performed on non-malignant human pancreatic ductal cells overexpressing Gli1 in order to screen for downstream Hedgehog target genes likely to be involved in pancreatic cancer progression.
Results: Hedgehog inhibition with cyclopamine significantly prolonged median survival in the transgenic mouse model used here (67 vs 61 days; p = 0.026). In vitro data indicated that Hedgehog activation might at least in part be ascribed to oncogenic Kras signalling. Microarray analysis identified 26 potential Hedgehog target genes that had previously been found to be overexpressed in pancreatic cancer. Five of them, BIRC3, COL11A1, NNMT, PLAU and TGM2, had been described as upregulated in more than one global gene expression analysis before.
Conclusion: This study provides another line of evidence that Hedgehog signalling is a valid target for the development of novel therapeutics for pancreatic cancer that might be worth evaluating soon in a clinical setting.
Statistics from Altmetric.com
Ductal adenocarcinoma of the pancreas (pancreatic cancer) is a devastating and almost uniformly lethal malignancy that accounts for approximately 33 000 deaths in the USA every year, and is the fourth most common cause of cancer-related mortality.1 Despite decades of intense research efforts aimed at better understanding of underlying aetiological and pathophysiological mechanisms, this increased knowledge has not yet translated into improvements in clinical treatment strategies or in patient survival. In fact, the overall median 5-year survival rate for pancreatic cancer remains at around only 5%, and is among the worst of all human malignancies.1 2
Due to absence of early symptoms and lack of reliable diagnostic tools for early detection, the vast majority of pancreatic cancers are diagnosed at locally advanced or metastatic stages, precluding curative surgical resection. Even patients diagnosed at early stages and operated in curative intention tend to develop local recurrence or distant metastases, and finally succumb to metastatic growth.
Previous studies by our own group and by others had shown that the Hedgehog signalling pathway is aberrantly reactivated in several cancers arising from the gastrointestinal tract, including the majority of pancreatic cancers.3–5 Moreover, blockade of Hedgehog signalling using small molecules has been suggested as a promising new potential therapeutic option for pancreatic cancer. Using murine orthotopic xenograft models of human pancreatic cancers, Hedgehog pathway blockade with cyclopamine has recently been shown to be particularly potent in preventing metastatic tumour spread.6
While orthotopic xenograft models accurately mirror the full spectrum of genetic changes that characterise the human disease and allow the study of pathophysiological processes involved in metastatic spread, they also suffer from several known conceptual shortcomings—for example, lack of a fully intact host immune system and inaccurate modelling of tumour–stroma interactions in a xenogenic setting.7 8 Therefore, we sought to test further the impact of Hedgehog pathway blockade in an autochthonous mouse model of pancreatic cancer that recapitulates the multistep progression of the human disease.9 We show that Hedgehog signalling is aberrantly reactivated in this genetically engineered mouse model of pancreatic cancer, and that pharmacological Hedgehog blockage by continuous administration of low-dose cyclopamine prolongs survival in this model.
In order to screen systematically for additional downstream targets of the Hedgehog pathway which might play a role in pancreatic cancer progression, microarray analysis was performed on non-malignant hTERT-HPNE human pancreatic ductal cells10 overexpressing the Hedgehog transcription factor Gli1.
MATERIALS AND METHODS
Cell lines
Murine pancreatic cancer cell lines M44, M45 and M63 were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, California, USA) containing 10% fetal bovine serum (FBS; Invitrogen) and 1× penicillin/streptomycin (Biofluids, Camarillo, California, USA); hTERT-HPNE cells were grown in special media as described elsewhere.10
All cells were maintained in a humidified atmosphere with 5% CO2 at 37°C.
Gli-responsive reporter assays
ASPC1 cells were seeded into 24-well plates at a density of 60 000 cells per well and co-transfected on the next day with 1 μg of Gli-responsive 8xGli and 10 ng of SV40-Renilla plasmids with FuGene6 transfection reagent (Roche, Indianapolis, Indiana, USA). Medium was changed after 6 h and cells were incubated for another 48 h in culture medium containing 0.5% FBS, supplemented with the MEK (mitogen-activated protein kinase kinase) inhibitor U0126 (Calbiochem, San Diego, CA, USA) at a concentration of c = 10 μM or solvent (dimethylsulfoxide (DMSO)) as control, respectively. Next, cells were lysed with passive lysis buffer, and luminescence determined on a luminometer (Wallac Victor2 1420, PerkinElmer, Boston, Massachusetts, USA) using the Dual-Luciferase Reporter Assay System (Promega, Madison, Wisconsin, USA). All experiments were performed in triplicate, and means and standard errors were calculated.
Cell growth assays
In vitro cell growth was determined by means of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assays (Promega) as previously described.6 11
Proliferation assays
Cell proliferation was assessed by flow cytometry using the CellTrace CFSE Cell Proliferation Kit (Invitrogen). M45 cells were labelled following the “alternative method to label adherent cells” provided by the manufacturer. Next, cells were incubated in low-serum medium (1% FBS) in the presence of cyclopamine (6 μM) or DMSO for 4 days. Fluorescence was measured on a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, New Jersey, USA).
Apoptosis assays
M45 cells were incubated with cyclopamine (6 μM) or solvent (DMSO) in low-serum medium for 12 h. Apoptosis rates were determined by measuring annexin V–fluorescein isothiocyanate (FITC) binding using the ApoDETECTAnnexin V-FITC Kit (Invitrogen).
Immunohistochemistry
SHH (Sonic Hedgehog ligand) staining of formalin-fixed, paraffin-embedded tissue sections was performed as previously described.11
Generation of stably transfected cell lines
hTERT-HPNE cells were seeded into 24-well plates at 60 000 cells per well and transfected with 1 μg of pCEFL-3xHA-hGLI1 plasmid5 using FuGene6 transfection reagent (Roche). Cells carrying the plasmid were selected for by adding 400 μg/ml of geneticin (Invitrogen) to the cell culture media.
Subcloning of stably transfected cell lines
Stably transfected cells were diluted to a concentration of 5 cells/ml in full medium. Next, 200 μl of this suspension were added to each well of a 96-well plate and cells were given time to adhere overnight. On the next morning, wells with one single, living cell were marked and further observed, until cells became confluent. Subclones derived from single cells were expanded, attributed specific numbers to identify individual subclones, and tested for Gli1 overexpression by quantitative real-time reverse transcription–PCR (RT–PCR).
RNA extraction and real-time RT–PCR
RNA extraction and cDNA synthesis were performed as previously described.6 For amplification of human or murine PGK1, Gli1, Ptch and SHH, Assays-on-Demand (Applied Biosystems, Foster City, California, USA) were used. Primer sequences for other targets (listed in table 1) were either derived from Primer Bank12 or designed using primer3 online primer design software and run with the QuantitectSYBR Green PCR kit (Qiagen) on a 7300 Real Time PCR System (Applied Biosystems). Specificity was confirmed by melting curve analysis. Glyceraldehyde phosphate dehydrogenase (GAPDH) primers have been described previously.13 Relative fold mRNA expression levels were determined using the 2(–ΔΔCt) method.14 All reactions were done in triplicate, and the results are presented as means (SE).
Generation of LsL-KrasG12D;Ink4a/Arflox/lox;Pdx1Cre transgenic mouse cohorts
All animal experiments conformed to the guidelines of the Animal Care and Use Committee of Johns Hopkins University, and animals were maintained in accordance with the guidelines of the American Association of Laboratory Animal Care.
LsL-KrasG12D;Ink4a/Arflox/lox transgenic mice were interbred with Pdx1-Cre;Ink4a/Arflox/lox mice. For genotyping, genomic DNA was extracted from tail cuttings using the REDExtract-N-Amp Tissue PCR kit (Sigma-Aldrich, Saint Louis, Missouri, USA). Two PCRs were carried out for each animal, to test for the presence of oncogenic Kras (using LoxP primers) and Pdx1-Cre transgene constructs (using Cre-specific primers along with Gabra (γ-aminobutyric acid (GABA-A) receptor subunit α1) as positive control), respectively (fig 4A). The presence of the transgenic Ink4a/Arf locus, carried by all breeders, was only occasionally confirmed by PCR (not shown). Primer sequences are listed in table 2.9

Expression of SHH was also examined in another, previously described, transgenic mouse model of pancreatic cancer exploiting oncogenic Kras expression under the control of Pdx1-Cre.15 Murine pancreatic intraepithelial neoplasias (designated mPanINs in the following)16 and invasive cancers were evaluated by immunohistochemistry and real-time RT–PCR, respectively.
Drug treatment
Drug treatment was initiated at the age of 5 weeks. Pdx1-Cre;LsL-KrasG12D;Ink4a/Arflox/lox transgenic mice were randomly assigned to receive either mock treatment or cyclopamine. In cases where littermates were available for drug treatment, only the first mouse was randomly assigned to one of the two given treatment groups; the second littermate was then assigned to the ‘matched’ control arm, and so forth. This scheme was chosen in order to obtain the highest possible degree of consistency and to avoid randomisation bias as far as possible.
Cyclopamine was dissolved in a 0.1 M sodium citrate/phosphate buffer (pH 3.0) containing 30% (w/w) 2-hydroxypropyl-beta-cyclodextrin (Sigma-Aldrich) at 30 g/l.
Cyclopamine solution or solvent, respectively, was loaded into 7D-Alzet osmotic pumps (Alzet, Cupertino, California, USA). Mice were anaesthetised using volatile gas anaesthesia with isofluorane (Vedco, St Joseph, Missouri, USA), and two pumps containing cyclopamine (Group B) or solvent (Group A) were subcutaneously implanted at the flanks or at the back of treated animals under sterile conditions. Pumps in all treatment groups were replaced weekly.
For determination of target gene downregulation, Pdx1-Cre;LsL-KrasG12D;Ink4a/Arflox/lox mice with palpable intrapancreatic tumours were treated with cyclopamine (n = 4) or solvent (n = 3) for 5 days by subcutaneous implantation of osmotic pumps. Next, mice were euthanised and tumours harvested for RNA extraction.
Statistical analysis
Mann–Whitney U test and two-tailed t test were performed using GraphPad Prism version 5.01 for Windows; p<0.05 was regarded to be statistically significant.
Microarray analysis
In order to screen for Hedgehog target genes in human pancreatic ductal cells, the stable subclone HPNE-Gli1(IC3B10) was picked, which showed 57-fold overexpression of Gli1 mRNA as compared with mock-transfected cells.
Global gene expression analysis by means of cDNA microarrays was performed using established guidelines of the Johns Hopkins Cancer Center Microarray Core Facility. A detailed description of the experimental procedure is provided in the Supplementary material.
Analysis of microarray data
Microarrays were scanned using an Agilent Scanner controlled by Agilent Scan Control 7.0 software. Data were extracted with Agilent Feature Extraction 9.1 software. Differentially expressed targets were identified by using processed signal intensities and p value log ratios as described elsewhere.17
The list of genes found to be upregulated or downregulated more than twofold was compared with previous global gene expression studies of differentially expressed genes in pancreatic cancer.18–30
RESULTS
The Hedgehog pathway is active in a genetically engineered murine pancreatic cancer model
Pdx1-Cre;LsL-KrasG12D;Ink4a/Arflox/lox mice develop pancreatic cancer with short latency; hereafter, these mice will be referred to as Kras Ink4a/Arf animals. The histologies resembled ductal adenocarcinomas of the pancreas observed in humans (fig 1A).

We harvested intrapancreatic tumours from these mice and stained them for SHH expression using immunohistochemistry. As illustrated in fig 1B, pancreatic cancer cells showed positive staining for SHH, while no staining was observed in surrounding stroma and non-neoplastic epithelial cells. In addition, strong SHH staining was observed in preinvasive mPanINs, the precursors to pancreatic cancer (data not shown). These observations demonstrate that Hedgehog signalling is activated throughout the course of pancreatic carcinogenesis, from the earliest disease stages to advanced invasive disease.
In vitro experiments establish murine pancreatic cancer cell lines derived from Kras Ink4a/Arf mice as Hedgehog dependent
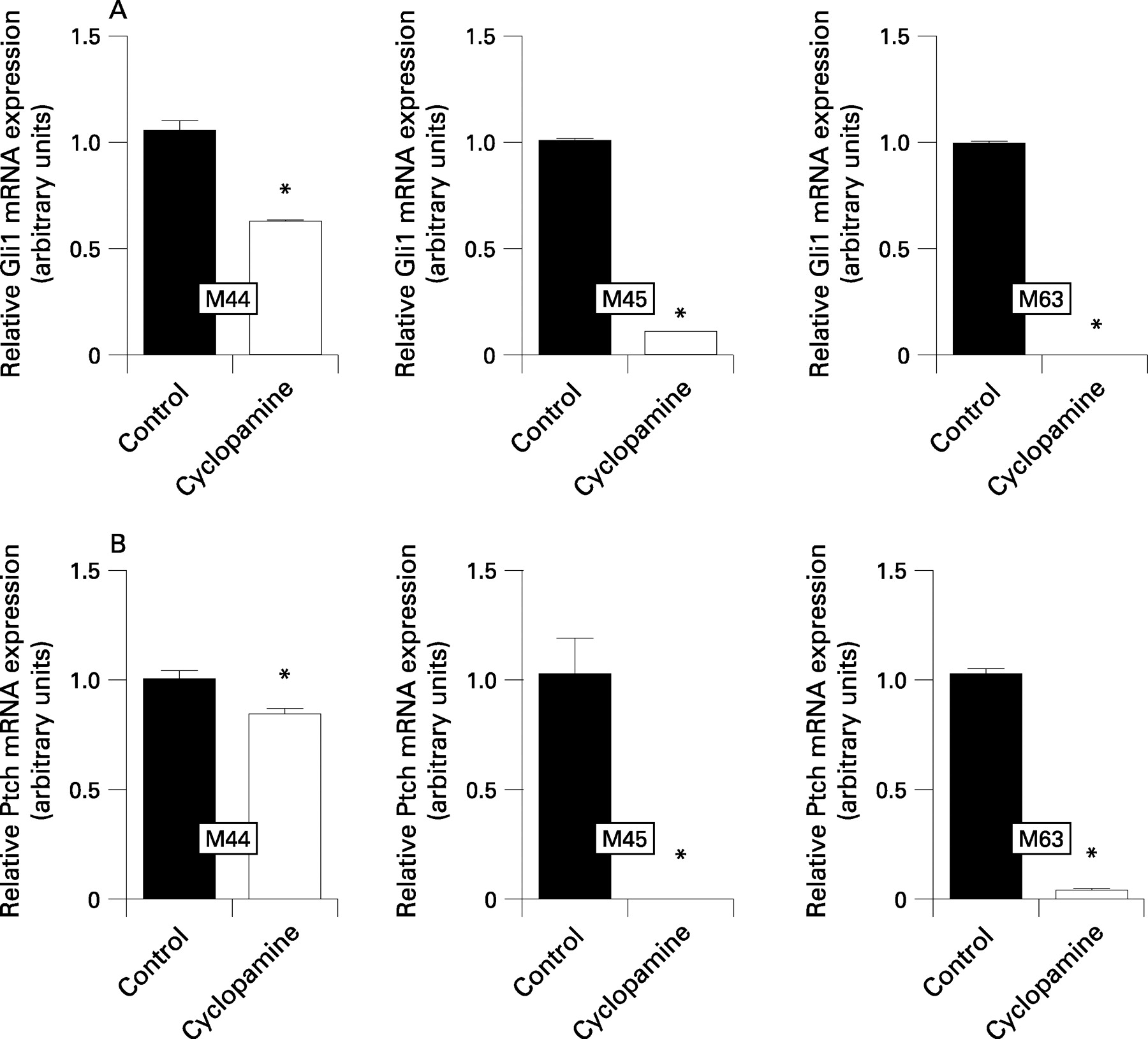
Hedgehog inhibition with cyclopamine (6 μM) in low serum conditions led to marked growth inhibition of >50% after 96 h in all three murine pancreatic cancer cell lines studied in vitro as determined using MTT assays (fig 2A). Cyclopamine-treatment (6 μM) of M45 cells caused prolonged retention of carboxyfluorescein diacetate succinimidyl ester (CFSE) staining as well as annexin V binding, as observed using flow cytometry, in line with inhibition of cell proliferation and induction of apoptosis, respectively (fig 2B,C). Moreover, cyclopamine caused significant downregulation of the Hedgehog target genes Gli1 (fig 3A) and Ptch (fig 3B) at the mRNA level as observed using real-time quantitative RT–PCR, in line with robust pathway inhibition.


Oncogenic Kras signalling might contribute to Hedgehog pathway activation in pancreatic cancer
The basis for Hedgehog pathway activation early in pancreatic cancer development is not defined. Since Kras drives PanIN development in the mouse and in the human disease, we sought to determine whether Kras could directly promote Hedgehog pathway activity. Expression of oncogenic KrasG12D in non-malignant hTERT-HPNE human pancreatic ductal cells lead to significant overexpression of SHH as observed using real-time RT–PCR. Likewise, the MEK inhibitor U0126 inhibited Gli-responsive reporter activity in the Kras mutant pancreatic cancer cell line ASPC131 (fig 4A,B). Of note, marked overexpression of SHH was also detected in mPanIN lesions (fig 4C) and invasive cancers (fig 4D) derived from another mouse model of pancreatic cancer,15 expressing oncogenic Kras under the control of Pdx1-Cre in the absence of additional Ink4a/Arf knockout. These findings suggest that SHH pathway activation may be part of the Kras-induced pancreatic cancer transformation programme.
Hedgehog inhibition prolongs survival of Kras Ink4a/Arf mice
Kras Ink4a/Arf mice were identified by PCR amplification of the transgenic Kras and Pdx1-Cre constructs in DNA isolated from tail cuttings (fig 5A). Overall, ∼50% of the offspring from LsL-KrasG12D;Ink4a/Arflox/lox×Pdx1-Cre; Ink4a/Arflox/lox crosses showed the desired Pdx1-Cre;LsL-KrasG12D;Ink4a/Arflox/lox genotype, in line with the percentage predicted by the classical Mendelian rules of inheritance.

Based on the strong activation of the Hedgehog pathway in the murine pancreatic cancers and on the sensitivity of derivative cell lines to cyclopamine we assessed the impact of cyclopamine administration on tumour development in our mouse model. Hedgehog inhibition by continuous subcutaneous application of cyclopamine (0.72 mg/day) using implantable osmotic Alzet pumps prolonged overall median survival of Kras Ink4a/Arf mice (n = 11) by 6 days as compared with mock-treated (n = 10) animals (median survival 67 vs 61 days; p = 0.026; fig 5B). We did not observe any weight loss or behavioural abnormalities in cyclopamine-treated mice.
Cyclopamine treatment led to significant downregulation of intratumoural steady-state Gli1 and Ptch mRNA levels, in line with robust pathway inhibition (fig 5C).
Necropsies could not be performed on animals included in this study, since animals were usually found dead in the cage on the next morning, precluding reliable assessment of metastases due to often already considerable amounts of autolysis.
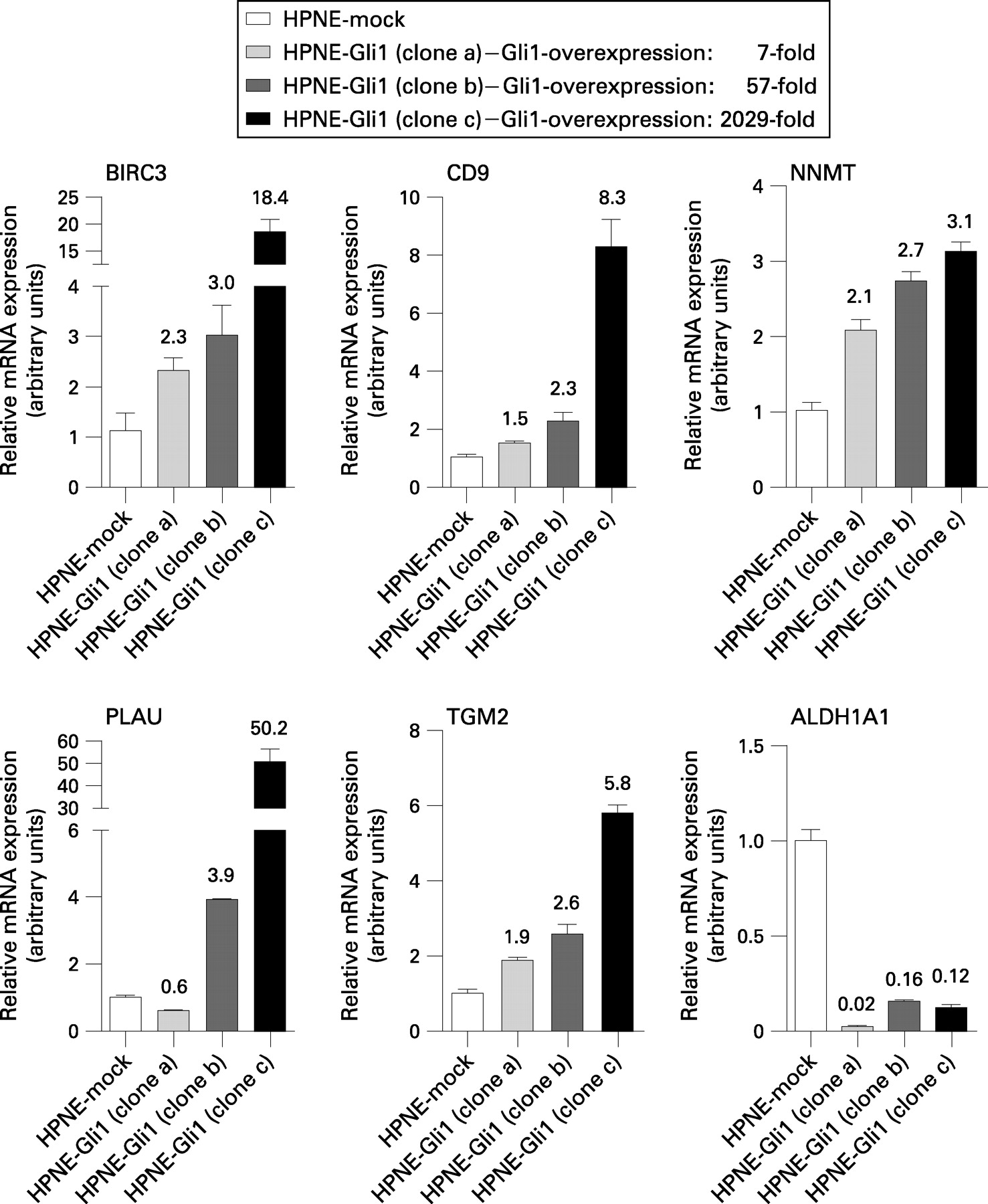
Microarray analysis of HPNE-Gli1 identifies genes differentially expressed in pancreatic cancer as potential downstream targets of aberrant Hedgehog signalling
Previous studies suggested that Hedgehog activation might contribute to pancreatic cancer progression through several different mechanisms. Hedgehog signalling promotes epithelial–mesenchymal transition and increases motility and invasiveness of pancreatic cancer cells. Moreover, recent evidence suggested that Hedgehog signalling might play a role in maintaining subpopulations of cancer cells with increased tumourigenic potential (ie, tumour-initiating cells).6 32 Here, we sought to identify Hedgehog target genes that could contribute to the oncogenic programme.
cDNA microarray analysis of the stably transfected subclone HPNE-Gli1(IC3B10) revealed a list of 438 targets that were overexpressed at least twofold, and another 292 targets that were downregulated by at least 50% as compared with mock-transfected hTERT-HPNE cells (Supplementary table 1). Out of these, 23 upregulated targets and 3 downregulated targets were picked for validation by quantitative real-time RT–PCR. As shown in fig 6 and Supplementary fig 1, all of the 23 targets found to be overexpressed by microarray analysis were also seen to be overexpressed using RT–PCR, and the three downregulated targets were also downregulated as determined by RT–PCR. However, the observed fold changes differed considerably in some cases with high overexpression (Supplementaty fig 1), while RT–PCR and microarray results were strikingly consistent for targets that were only moderately overexpressed. This observation might be due to the overall higher dynamic range of PCR-based assays.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
We then compared these targets with previous studies carried out at our own institution18–22 or elsewhere23–30 using global gene expression techniques (ie, either transcriptomic or proteomic) to screen for differentially expressed genes in pancreatic cancer. Using this approach, we identified 26 genes that were found to be upregulated upon Gli1 overexpression in our screen, and that had previously also been found to be overexpressed in pancreatic cancer by at least one of the mentioned studies (table 3). Vice versa, 14 of the downregulated genes identified here had previously been described as underexpressed in pancreatic cancer (table 4). Of note, five genes, BIRC3, COL11A1, NNMT, PLAU and TGM2, had been found as upregulated in pancreatic cancer in at least two independent global gene expression studies. Likewise, ALDH1A1 and SERPING1 had been identified by two of the mentioned studies as downregulated in pancreatic cancer, probably conferring a somewhat higher overall degree of evidence that these candidates are in fact differentially expressed in the setting of pancreatic cancer, possibly as downstream targets of an aberrantly reactivated Hedgehog signalling pathway.
Of note, quantitative real-time RT–PCR analysis confirmed diferential expression of the picked genes in the HPNE-Gli1 subclone used for microarray analysis as well as in two additional subclones. Moreover, levels of target gene overexpression correlated with the extent of Gli1 mRNA overexpression in the respective subclones (fig 6).
DISCUSSION
In the present study we show that the Hedgehog signalling pathway is aberrantly reactivated in a genetically engineered mouse model of pancreatic cancer.9 Hedgehog blockade with cyclopamine led to marked in vitro growth inhibition of murine cancer cell lines derived from this model as well as downregulation of the Hedgehog target genes Gli1 and Ptch, and significantly prolonged survival of Kras Ink4a/Arf mice in vivo. mPanIN lesions, including high-grade mPanINs (carcinoma in situ), were ubiquitous in the pancreata of these mice at 5 weeks,9 suggesting that we were treating cancer progression rather than initiation in this model. The data presented here support results from our previous work on murine xenograft models6 and suggest that pharmacological Hedgehog blockade might carry the potential to improve the prognosis of pancreatic cancer.
While the observed increase in overall survival is relatively small, we feel that this effect is nevertheless meaningful given the strong underlying “genetic force” of three mutational changes and the extremely rapid course of disease and short overall median survival of only 61 days in this particular model. It is tempting to speculate whether similar effects can be observed in other more recently described transgenic models—for example, exploiting Pdx1-Cre-driven oncogenic Kras expression with additional suppression of p53 function,33 or oncogenic Kras signalling in combination with SMAD4 or TGFBR2 depletion.34–36 While the observed histologies resembling moderately differentiated adenocarcinomas in those models might indicate that they are even more prone to respond to Hedgehog blockade,37 the long latency and longer overall survival observed in these mice would on the other hand require longer time frames for similar studies using those models.
The observations of this present study as well as of previous studies have obvious clinical implications, and it is our hope that potent pharmacological Hedgehog inhibitors will soon be available and evaluated in a clinical setting, since this will be the only way ultimately to learn whether or not this approach has therapeutic potential in the human disease.
Our in vitro data indicate that oncogenic Kras signalling might contribute at least partially to Hedgehog activation in the setting of pancreatic cancer. This observation is in line with recent reports by others.38–40 Whether or not this is the major underlying cause of Hedgehog activity and which other factors contribute to Hedgehog pathway activity in pancreatic cancer will have to be examined in separate future studies—for example, by correlating expression of Hedgehog pathway genes in primary tumour tissue samples or direct evidence for in vitro pathway activity using functional assays in cancer cell lines with the presence or absence of activating Kras mutations, or by directly screening for somatic mutations involving Hedgehog pathway-related genes in the setting of pancreatic cancer.
Previous studies found that Hedgehog inhibition seems to affect pancreatic cancer progression through several independent mechanisms, namely enhancement of apoptosis, inhibition of proliferation, invasion, migration and modulation of the tumour microenvironment.3 4 6 Moreover, Hedgehog signalling might confer maintenance of defined subsets of malignant cells with enhanced tumourigenic potential, and inhibiting Hedgehog signalling could prevent tumour initiation and formation of metastases by directly targeting these subpopulations.6 32
To begin to define potential additional mechanisms of Hedgehog-dependent tumour promotion systematically, we sought to identify downstream targets of an activated Hedgehog pathway in human pancreatic ductal epithelial cells. Using microarray analysis, we found several genes that were upregulated in hTERT-HPNE cells stably transfected to overexpress the main activating Hedgehog transcription factor Gli1 and that had previously been found to be overexpressed in pancreatic cancer.
Interestingly, one of these, WISP1, is a known downstream target of the WNT signalling pathway,41 which is also known to be re-activated in pancreatic cancers.42 This finding might support the hypothesis that canonical WNT signalling is turned on as a downstream target of an activated Hedgehog pathway, which is in line with recent data presented by Pasca di Magliano and colleagues.42
Five of the genes found to be upregulated upon Gli1 overexpression had been identified as overexpressed in pancreatic cancer by more than one global gene expression study. Upregulation of the apoptosis inhibitor BIRC3 is observed early during pancreatic carcinogenesis,43 and it has been shown that BIRC3 is upregulated in intestinal epithelial cells upon oncogenic ras signalling.44 Expression of BIRC3 has been linked to resistance to chemotherapy and poor outcome in multiple myeloma.45 It has been shown that repression of BIRC3 and Mcl1 by sorafenib dramatically sensitises Bax-deficient HCT116 cells to TRAIL- (tumour necrosis factor-alpha-related apoptosis-inducing ligand) induced apoptosis,46 possibly opening up new therapeutic avenues. Sorafenib has recently been approved for treatment of renal cancer, and its efficacy against several other tumour entities, including pancreatic cancer, is currently being evaluated in multiple clinical trials (www.clinicaltrials.gov). Interestingly, BIRC3 has recently been found to be amplified in 5/19 examined pancreatic cancer cell lines, supporting the hypothesis that it might play a role in pancreatic carcinogenesis.47
Transglutaminase-2 (TGM2) has been linked to activation of nuclear factor-κB in pancreatic cancer,48 and is thought to play a role in chemoresistance and development of a metastatic phenotype in cancer cells.49
Urokinase plasminogen activator (PLAU) has long been thought to play a role in cancer invasion and metastasis, and has been suggested as a prognostic marker in pancreatic cancer.50 51 Of note, small interfering RNA (siRNA)-mediated silencing of PLAU led to marked inhibition of tumour growth and metastasis in murine xenograft models of human prostate cancer.52
Another interesting target gene identified by our microarray analysis is BMP2. Though it was not found by any of the above-mentioned global gene expression profiling studies, it has nevertheless been previously demonstrated to be significantly overexpressed in pancreatic cancers, and BMP2 expression has been linked to tumour cell growth and postoperative patient survival.53 BMP2 expression has been linked to in vitro invasion and migration, as well as to metastasis in vivo in breast and prostate carcinoma, respectively.54 55 Moreover, BMP2 has been found to be essential in mediating epithelial–mesenchymal transition in cardiac cushion56; all of these are mechanisms that we previously suggested to be potentially involved in mediating the effects of Hedgehog signalling on metastatic spread.6 11
Taken together, our data presented here indicate that Hedgehog signalling is activated in the studied transgenic mouse model of pancreatic cancer and provide another piece of evidence that pharmacological Hedgehog blockade might be a valid novel therapeutic approach for pancreatic cancer which may be worth evaluating in a clinical setting. Our in vitro data support the hypothesis that oncogenic Kras signalling contributes to Hedgehog activation, and that likewise canonical WNT signalling might be activated as a downstream target of an active Hedgehog pathway.
Several genes were identified as potential Hedgehog downstream targets in the setting of pancreatic cancer that had previously been found to be mechanistically involved in tumour cell proliferation, invasion/migration and metastatic tumour spread, among them BIRC3, TGM2, PLAU and BMP2.
Acknowledgments
Cyclopamine was kindly donated by Infinity Pharmaceuticals (Cambridge, Massachusetts, USA). We thank Dr David Tuveson and Dr Sunil Hingorani for access to tissues from the Pdx1-Cre;LsL-KrasG12D mice.15
REFERENCES
Supplementary materials
web only appendix 57/10/1420
Files in this Data Supplement:
Footnotes
▸ Additional methods, a figure and a table are published online only at http://gut.bmj.com/content/vol57/issue10
Funding: Supported by the Sol Goldman Pancreatic Cancer Research Center, the Michael Rolfe Foundation, NIH SPORE (Specialized Programs of Research Excellence) in Gastrointestinal Cancer P50CA62924 and NIH R01CA113669 to AM. GF was supported by a fellowship grant within the postdoc-programme of the German Academic Exchange Service (DAAD). HA received generous support from the D’Amato Family.
Competing interests: None.