Article Text

Abstract
Background and aims O6-Methylguanine-DNA methyltransferase (MGMT) removes methyl adducts from O6-guanine. Known as methylation tolerance, selection for mismatch repair (MMR)-deficient cells that are unable to initiate lethal processing of O6-methylguanine-induced mismatches in DNA is observed in vitro as a consequence of MGMT deficiency. It was therefore hypothesised that an MGMT field defect may constitute a preneoplastic event for the development of MMR-deficient tumours displaying microsatellite instability (MSI).
Methods MGMT expression was investigated by immunohistochemistry and the methylation status of the gene promoter by PCR in neoplastic, adjacent and distant mucosal tissues of patients with MSI or non-MSI (MSS) colorectal cancer (CRC). The cancers were familial (42 MSI, 13 MSS) or sporadic (40 MSI, 49 MSS) in origin, or arose in the context of inflammatory bowel disease (IBD; 13 MSI, 36 MSS). Colonic mucosa from patients with diverticulitis (n=20) or IBD (n=39 in 27 patients) without cancer served as controls.
Results Loss of MGMT expression was more frequent in MSI than MSS CRC (p=0.047). In comparison with MSS tumours, MSI CRC occurred more frequently adjacent to patches of mucosa that lacked MGMT expression (p=0.002). Overall, loss of MGMT expression was associated with MGMT gene promoter methylation (p=0.03).
Conclusion MGMT field defects are more frequently associated with MSI than MSS CRC. These findings indicate that methylation tolerance may be a crucial initiating step prior to MMR deficiency in the development of MSI CRC in familial, sporadic and IBD settings.
- O6-Methylguanine DNA methyltransferase (MGMT)
- risk factor
- colorectal cancer
- mismatch repair deficiency
- microsatellite instability
Statistics from Altmetric.com
- O6-Methylguanine DNA methyltransferase (MGMT)
- risk factor
- colorectal cancer
- mismatch repair deficiency
- microsatellite instability
Significance of this study
What is already known about this subject?
A field defect defined by epigenetic inactivation of the DNA repair gene O6-methylguanine DNA methyltransferase (MGMT) was recently reported in colorectal cancers (CRCs), leading to the proposal that MGMT status might be useful for early detection and risk assessment in these tumours.
At the basic and clinical level, two main subsets of CRC have been reported so far—that is, mismatch repair (MMR)-deficient or MMR-proficient CRCs that do and do not display a microsatellite instabilty phenotype (MSI or MSS tumours, respectively).
An MGMT field defect has been suggested to play a role in CRC development by favouring transition mutations in P53 and KRAS, two cancer-related genes whose alterations are associated with MSS CRC.
A functional link between MGMT deficiency and the MSI phenotype may also exist since MGMT deficiency has been reported to favour the selection of MMR-deficient cells in vitro, via a process called methylation tolerance, as well as the occurrence of MSI neoplasms in transgenic animals when treated with alkylating agents that generate O6-methylguanine in DNA.
What are the new findings?
An MGMT field defect is more often associated with MSI than MSS CRC, indicating that methylation tolerance due to MGMT deficiency may be a common initiating step prior to MMR deficiency in the development of this subset of colorectal tumours.
Methylation tolerance due to MGMT deficiency is likely to be responsible for the emergence of MSI CRCs in different clinical contexts, including familial and sporadic settings, as well as in patients suffering from inflammatory bowel disease (IBD).
An MGMT field defect is likely to constitute a triggering event for cell transformation through two alternative mechanisms in CRC—that is, KRAS mutation that allows cells displaying an MSS phenotype to become malignant, as already proposed, or more frequently via the development of MMR deficiency leading to MSI, as reported for the first time in this study.
How might it impact on clinical practice in the foreseeable future?
Our findings highlight the importance of detection and monitoring of the MGMT field defect for the prevention of MSI colon tumours.
MGMT might be particularly useful for early detection and risk assessment in patients who show an increased risk for the development of these tumours—for example, patients with Lynch syndrome or IBD.
Methylating compounds in food or drugs such as alkylating agents that increase the production of mutagenic and toxic O6-guanine adducts in DNA should be avoided in these patients.
Introduction
In principle, the molecular abnormalities responsible for field defects, also called field cancerisation,1 should be detected in malignant as well as in normal cells, since they occur early in the neoplastic process.2 Shen et al reported that normal-appearing adjacent and non-adjacent mucosa located 1 cm and 10 cm, respectively, from colorectal cancers (CRCs) with promoter methylation of O6-methylguanine DNA methyltransferase (MGMT) showed a higher degree of MGMT methylation than normal mucosa from control subjects without CRC. These results suggested the existence of a field defect defined by epigenetic inactivation of this DNA repair gene in CRC,3 leading the authors to propose that MGMT might be useful for early detection and risk assessment of CRC.
The DNA repair protein encoded by MGMT protects the human genome against mutagenesis by removing mutagenic methyl adducts from O6-guanine. If the methyl group is not removed by MGMT, O6-methylguanine is read as an adenine and mispairs with thymine during DNA replication,4 potentially resulting in G:C to A:T transition mutations in cancer-related genes such as P535 and KRAS. Mutation of these genes is frequently observed in the main subtype of colon tumours—that is, MSS CRC which remain mismatch repair (MMR) proficient and microsatellite stable but display high levels of chromosomal instability.3 6 7 If O6-methylguanine DNA adducts are not repaired by MGMT, mismatches are recognised by the MMR system, but this is unable to process such lesions correctly, giving rise to double-strand breaks during replication and apoptosis.8 Alternatively, MGMT deficiency also favours the selection of MMR-deficient cells in vitro through a process called methylation tolerance,9 as well as the development of MMR-deficient neoplasms displaying MSI (microsatellite instability) in transgenic animals treated with alkylating agents that generate O6-methylguanine in DNA.10–15 MMR-deficient CRCs that display the MSI phenotype have quite different biological and clinical characteristics compared with MMR-proficient, or MSS CRCs.16–18 Nevertheless, a positive link between MGMT silencing and the emergence of an MSI phenotype due to MMR deficiency has yet to be established in human carcinogenesis.
MSI CRCs can be inherited when they arise in the context of hereditary non-polyposis colorectal cancer (HNPCC), or Lynch syndrome. This condition is characterised by heterogeneous MMR defects affecting MLH1, MSH2, MSH6 or PMS2.19 20 However, the large majority of MSI CRCs are sporadic tumours that arise following methylation-induced transcriptional silencing of the MLH1 gene.21 Contradictory results have been reported concerning the putative impact of MGMT inactivation on the emergence of CRCs showing a low level of microsatellite instability (MSI-low phenotype) which is not due to MMR deficiency.7 22–26 Since the production of O6-methylguanine DNA adducts occurs physiologically and increases within cells during inflammation,10 we hypothesised that a field defect defined by loss of MGMT expression in the colonic mucosa of patients with CRC might favour the emergence of MMR-deficient clones and could be a crucial initiating step in the development of MSI colon tumours. We therefore investigated the expression of MGMT and its methylation status in the tumour, and normal-appearing adjacent and non-adjacent mucosal tissues of patients with familial or sporadic MSI or MSS CRC. These results were compared with those obtained in a patient control group without CRC. We recently reported the existence of an MSI phenotype in patients with inflammatory bowel disease (IBD) who develop CRC due to heterogeneous MMR defects affecting MLH1, MSH2, MSH6 or PMS2 and low frequency of MLH1 promoter methylation.27 Therefore, in the current study we also screened a series of patients with IBD with MSI or MSS CRC, as well as IBD controls without CRC.
Material and methods
Patients and specimens
The following clinicopathological data were collected for each patient: sex, age at surgery, anatomical distribution of the tumour (right colon (proximal to and including the splenic flexure), left colon (sigmoid and descending colon) and rectum) and tumour stage (table 1 and supplementary table 1 online). All CRC samples were selected from the Pathology Department of Saint-Antoine Hospital, AP-HP, Paris, France, on the basis of availability. Ethics approval was obtained from the Human Research Ethics Committee of the hospital. All CRCs were tested for MSI using a single fluorescent multiplex system comprising five quasi-monomorphic mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24 and NR-27), as previously described.28 A total of 89 sporadic CRCs (40 MSI, 49 MSS) and 55 inherited CRCs (42 MSI occurring in the context of HNPCC and 13 MSS occurring in the context of familial adenomatous polyposis (FAP)) were included in this study. We also included 13 MSI and 36 MSS IBD-associated CRCs that were previously characterised for MSI status.27 MSI CRCs were considered as probably sporadic or HNPCC according to the presence or absence of MLH1 promoter methylation, respectively. Tumours displaying loss of MSH2, MSH6 or PMS2 expression were considered as likely HNPCC cases.
Clinicopathological features of patients
The control groups included specimens of non-inflammatory colon mucosa from 20 patients who underwent surgery for colonic diverticulitis and colonic samples (n=39) from 27 patients with a history of IBD (15 patients with Crohn's disease and 12 patients with ulcerative colitis) but absence of neoplastic colonic lesions. MMR protein expression (MLH1, MSH2, MSH6 and PMS2) of MSI CRCs was determined by immunohistochemistry as described previously.27
DNA was extracted from formalin-fixed, paraffin-embedded neoplastic and non-neoplastic tissues as described previously.29 Selected areas of neoplastic and non-neoplastic tissues were macrodissected under guidance from the H&E-stained section.
Immunohistochemical analysis of MGMT protein expression
MGMT protein expression was examined by immunohistochemistry in the neoplastic tissues (Neo), non-neoplastic adjacent mucosa (A) and non-neoplastic non-adjacent mucosa (D for distant) in all patients and controls. Mucosa was defined as adjacent to a carcinoma if detected on the same histological slide as the cancer (<1 cm). Non-adjacent mucosa was located at least 10 cm away from the cancer. The same paraffin block was used for both DNA extraction and immunohistochemical analysis. Sections of 4 μm were incubated with an MGMT mouse monoclonal antibody for 60 min (clone MT3:1, Neomarkers, Fremont, California, USA; dilution 1:50). Sections were scored for the intensity of staining (0, no staining; 1, weak; 2, moderate; and 3, strong) (figure 1). Cycling cells of the lower crypt of normal epithelium, stromal cells within the lamina propria and lymphocytes in each section provided positive internal controls. MGMT expression in nuclei was evaluated as present (2/3 labelling) or lost (0/1 labelling). When samples displayed heterogeneous staining for MGMT, the expression was classified as ‘loss’ if at least 50% of nuclei were unstained (0/1 labelling). Two authors (MS and JFF) reviewed the slides without knowledge of the MSI status of the tumours.

Evaluation of O6-methylguanine-DNA methyltransferase (MGMT) expression by immunohistochemistry in tumour cells. (A) No staining (labelling 0). Inflammatory cells serve as an internal positive control (arrow). (B) Weak nuclear staining (labelling 1). (C) Moderate nuclear staining (labelling 2). (D) Strong nuclear staining (labelling 3) (×400 magnification).
Determination of MGMT and MLH1 promoter methylation by methylation-specific PCR (MSP)
The DNA methylation patterns for the MGMT and MLH1 promoter regions were determined by MSP of bisulfite-treated DNA (1 μg). The following oligonucleotide primers were used: 5′-GTAGGTTGTTTGTATGTTTGT-3′ (forward primer) and 5′-AACCAATACAAACCAAACA-3′ (reverse primer) for amplification of unmethylated DNA (PCR product size 121 bp) and 5′-GGTCGTTTGTACGTTCGC-3′ (forward primer) and 5′-GACCGATACAAACCGAACG-3′ (reverse primer) for amplification of methylated DNA (PCR product size 118 bp).3
The DNA methylation pattern for the MLH1 promoter region was determined by MSP on bisulfite-treated DNA using the primer sequences reported by Park et al.30
Laser capture microdissection
Six serial 5 μm paraffin-embedded sections from four cases were cut and then mounted onto membrane slides (PALM Membranes Slides, Bernreid, Germany). The slides were stained by H&E after deparaffinisation. Neoplastic cells and non-neoplastic epithelial cells from adjacent normal mucosa were dissected from the laser capture slides and catapulted into the adhesive caps of tubes using the PALM Laser Microdisection system (PALM Microlaser Technologies, Bernreid, Germany).
Determination of MGMT and MLH1 promoter methylation by pyrosequencing
The analysis of MGMT and MLH1 promoter methylation was performed by pyrosequencing as previously described.31 Primers for the pyrosequencing assay were designed to amplify CpG dinucleotides but not CpGs within the primer sequence (kit PyroMark Q24 CpG MGMT and kit PyroMark Q24 CpG MLH1, Qiagen, Courtaboeuf, France). Methylation of target CpGs was assessed by determining the ratio of cytosine to thymine incorporated during pyrosequencing; cytosine incorporation indicated a methylated CpG, while thymine incorporation indicated an unmethylated CpG. Thus, the ratio of incorporated cytosine to thymine was proportional to the degree of methylation at a particular CpG site in the template DNA. Analysis of non-CpG cytosines provided an internal control for the completeness of bisulfite treatment. For data analysis, the percentage methylation obtained for each CpG was averaged across the CpGs analysed in duplicate PCRs (average methylation per sample). In each case, DNA methylation patterns for the MGMT and MLH1 promoter regions were analysed before and after laser capture microdissection of samples in order to evaluate the frequency of methylated cells in epithelial neoplastic and non-neoplastic adjacent mucosa.
PCR amplification and sequencing of the coding regions of MLH1 and MSH2
All MLH1 and MSH2 exons were amplified by PCR and the products sequenced on an ABI 3730 DNA sequencer (Applied Biosystems, Foster City, California, USA). The resulting sequence data were analysed and compared with the reference sequences of the MLH1 and MSH2 genes (accession numbers NM_000249.1 and NM_000251, respectively).
BRAF and KRAS mutation screening in MSI CRC
Mutational analysis of BRAF was performed by single-stranded conformation polymorphism and heteroduplex analysis, as reported previously.32 Mutational analysis of KRAS was performed by denaturing gradient gel electrophoresis.
Statistical analysis
Two-by-two table contingency analyses were performed using a χ2 test with Yate correction when some numerical values were <5. A p value of ≤0.05 was considered to be significant.
Results
MGMT expression in the distant normal-appearing colorectal mucosa of patients with CRC
Results for immunohistochemical MGMT expression in neoplastic (Neo), adjacent (A) and non-adjacent (D) mucosa of patients with MSI or MSS CRC are summarised in table 2.
MGMT expression in cohorts of patients
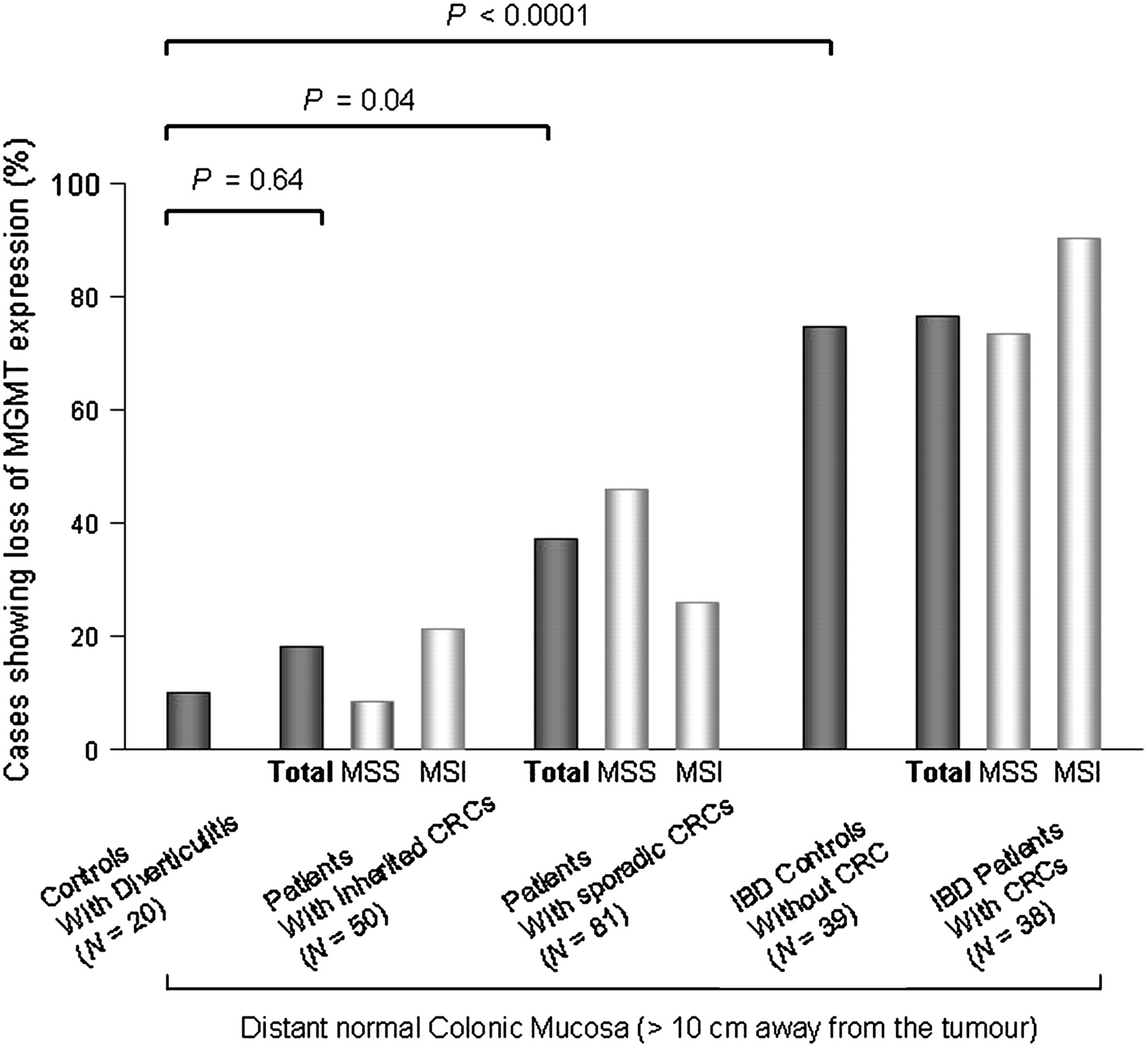
In colonic mucosa from 20 controls with diverticulitis, expression of MGMT was lost in 2/20 (10%) cases. Compared with these controls, MGMT expression was lost in 9/50 (18%) samples of D mucosa from patients with inherited CRC (8/38 HNPCC and 1/12 FAP, p=0.64) and in 30/81 (37%) samples of D mucosa from patients with sporadic CRC (9/35 MSI and 21/46 MSS, p=0.04). These results suggest the presence of an MGMT field defect in sporadic CRC (figure 2).
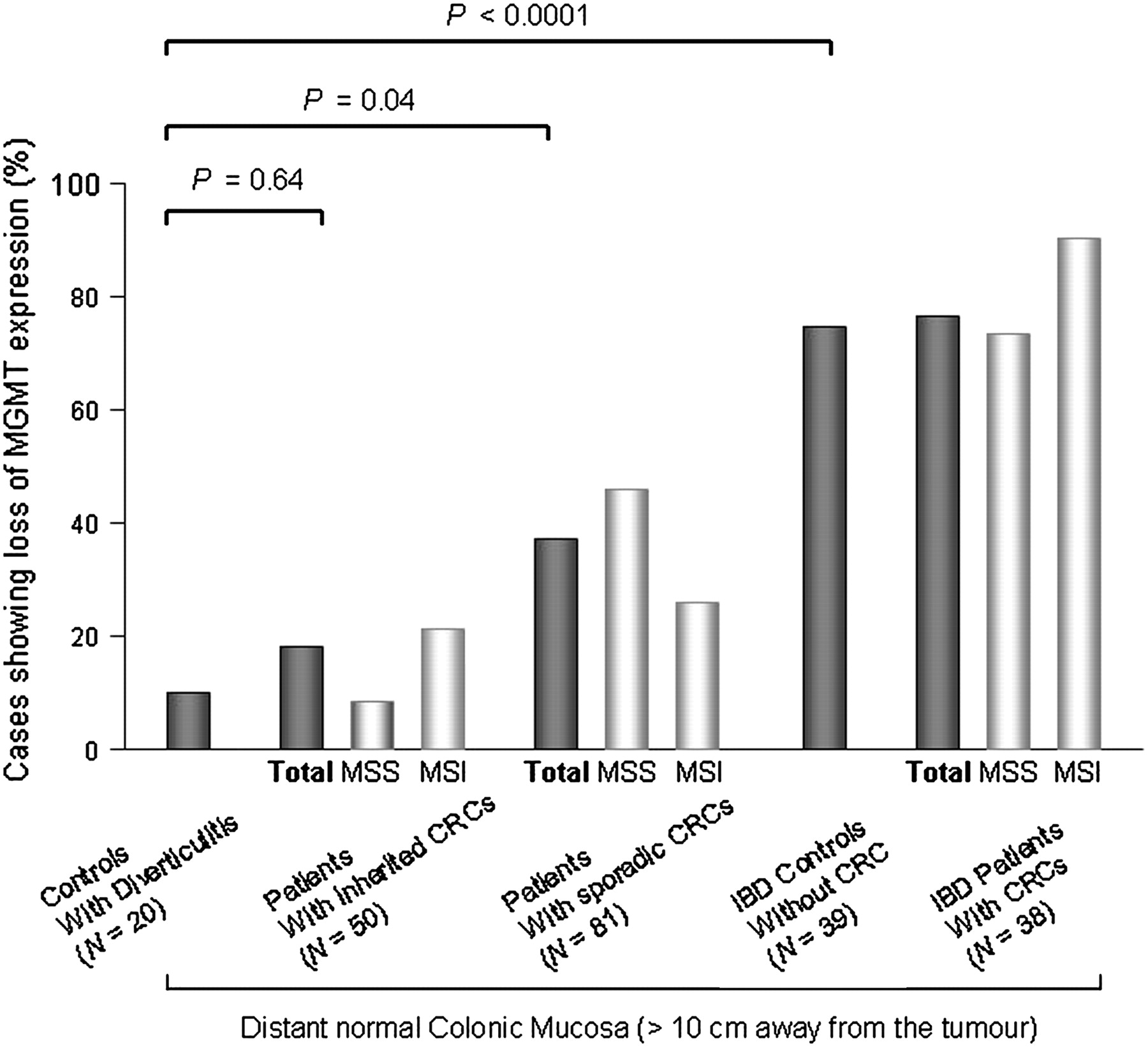
O6-Methylguanine-DNA methyltransferase (MGMT) expression in the distant normal colonic mucosa of patients with microsatellite-unstable (MSI) or microsatellite-stable (MSS) colorectal canver (CRC) arising in different clinical contexts as compared with control subjects without CRC. IBD, inflammatory bowel disease.
In patients with IBD, a frequent loss of MGMT expression was observed in the D mucosa of patients with CRC (29/38, 76.3%, comprising 9/10 MSI and 20/28 MSS) and in non-cancer controls (29/39, 74.4%). This suggests the existence of an MGMT field defect in the clinical context of IBD that is independent of tumour development. The frequency of loss of MGMT expression in the D mucosa was very different between patients with and without IBD (29/39 vs 2/20, p<0.0001, figure 2).
Comparative analysis of MGMT expression in neoplastic, adjacent and non-adjacent mucosa of patients with MSI or MSS CRC
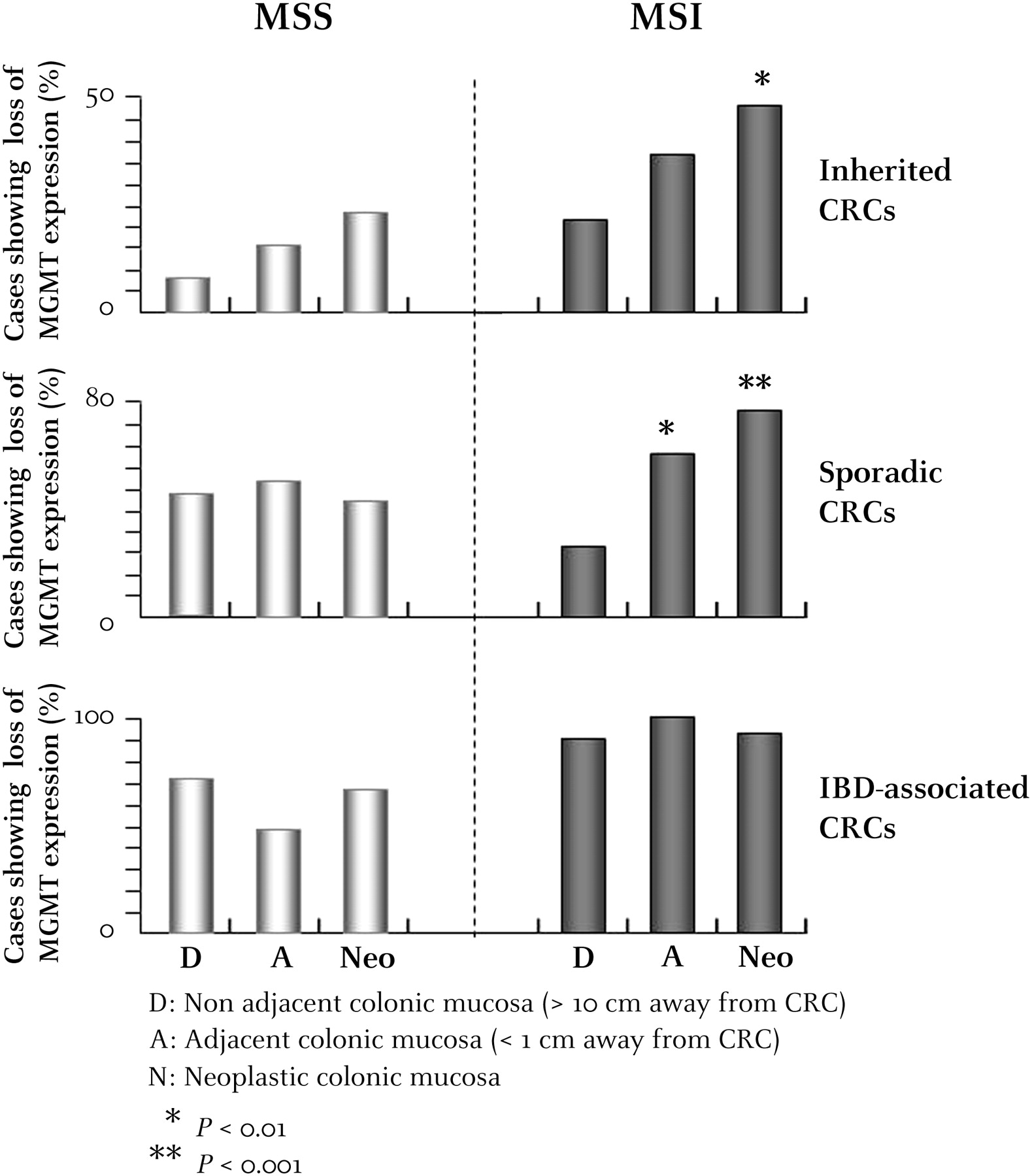
Overall, the loss of MGMT expression was more frequent in Neo tissues from MSI compared with MSS CRC (60/95 (63.2%) vs 48/98 (49%), p=0.047, all patients included). This association was significant for the sporadic CRC subgroup (28/40 (70%) MSI vs 21/49 (42.9%) MSS; p=0.01) but not for inherited CRC (20/42 (47.6%) HNPCC MSI vs 3/13 (23.1%) FAP MSS; p=0.2) or IBD-associated CRC (12/13 (92.3%) MSI vs 24/36 (66.7%) MSS; p=0.15).
In patients without IBD with MSI CRC, the loss of MGMT expression was more frequent in A and Neo mucosa compared with D mucosa (sporadic MSI, pAdjacent/Distant=0.005 (18/30 vs 9/35) and pNeo/Distant <0.0001 (28/40 vs 9/35); inherited MSI, pAdjacent/Distant=0.13 (15/41 vs 8/38) and pNeo/Distant=0.01 (20/42 vs 8/38)). This difference was not significant for patients without IBD with MSS CRC (sporadic MSS, pAdjacent/Distant=0.68 (22/44 vs 21/46) and pNeo/Distant=0.78 (21/49 vs 21/46); inherited MSS, pAdjacent/Distant=0.94 (2/13 vs 1/12) and pNeo/Distant=0.65 (3/13 vs 1/12)) (figure 3).
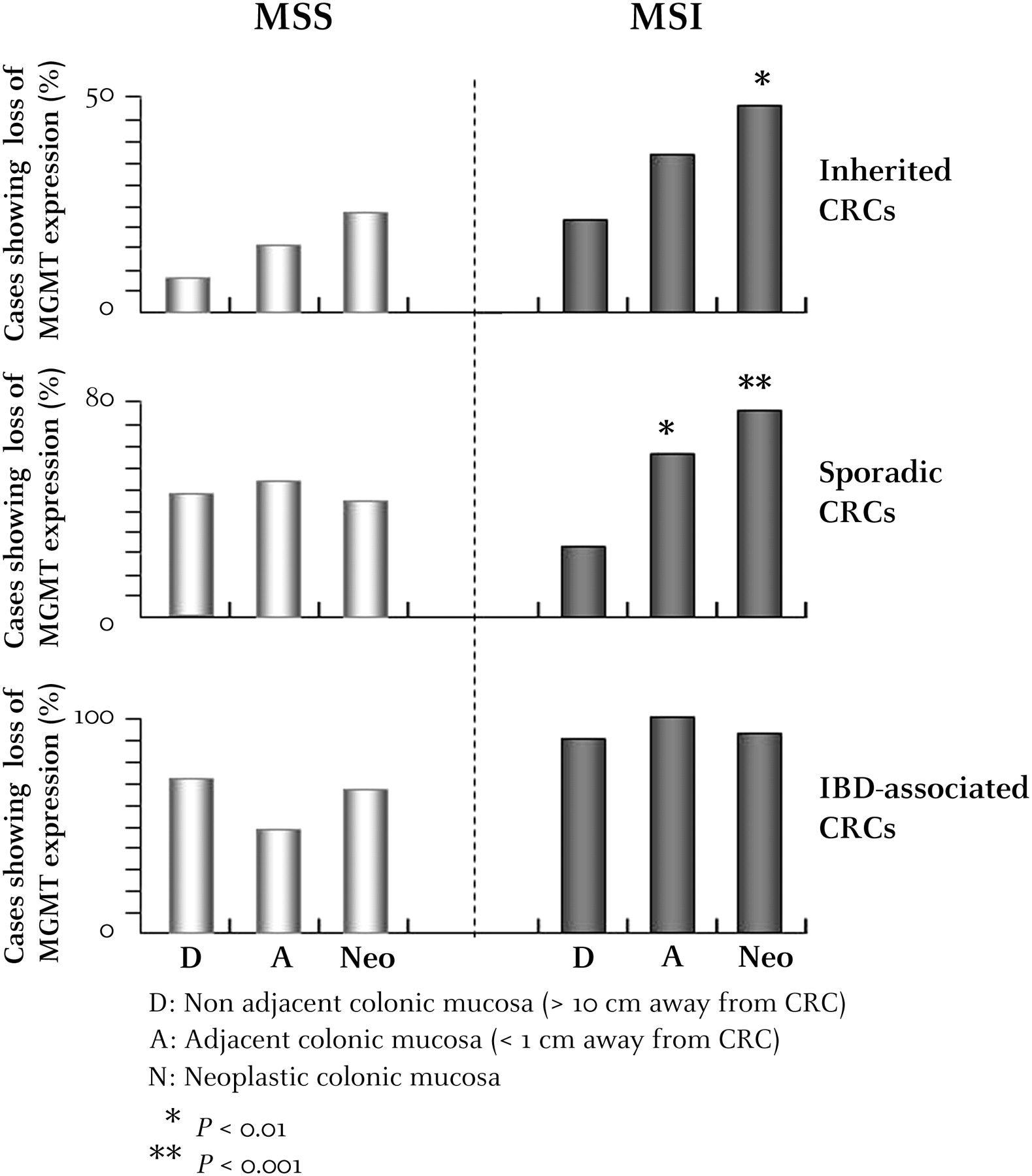
Comparative analysis of O6-methylguanine-DNA methyltransferase (MGMT) expression in neoplastic tissues (Neo), non-neoplastic adjacent mucosa (A) and non-neoplastic distal mucosa (D) from patients with inherited, sporadic or inflammatory bowel disease-associated colorectal cancer (CRC). p Values showing significantly different MGMT expression in A and Neo mucosa compared with D mucosa for patients with microsatellite-unstable (MSI) or microsatellite-stable (MSS) CRC are indicated by an asterisk (*p<0.1; **p<0.01;***p<0.001).
Since MGMT expression was lost in the D mucosa of almost all patients with IBD, there was no evidence for positive selection, regardless of the MSI status of the tumours (figure 3).
Comparative paired analysis of MGMT expression status within neoplastic and adjacent mucosa of patients with MSI or MSS CRC
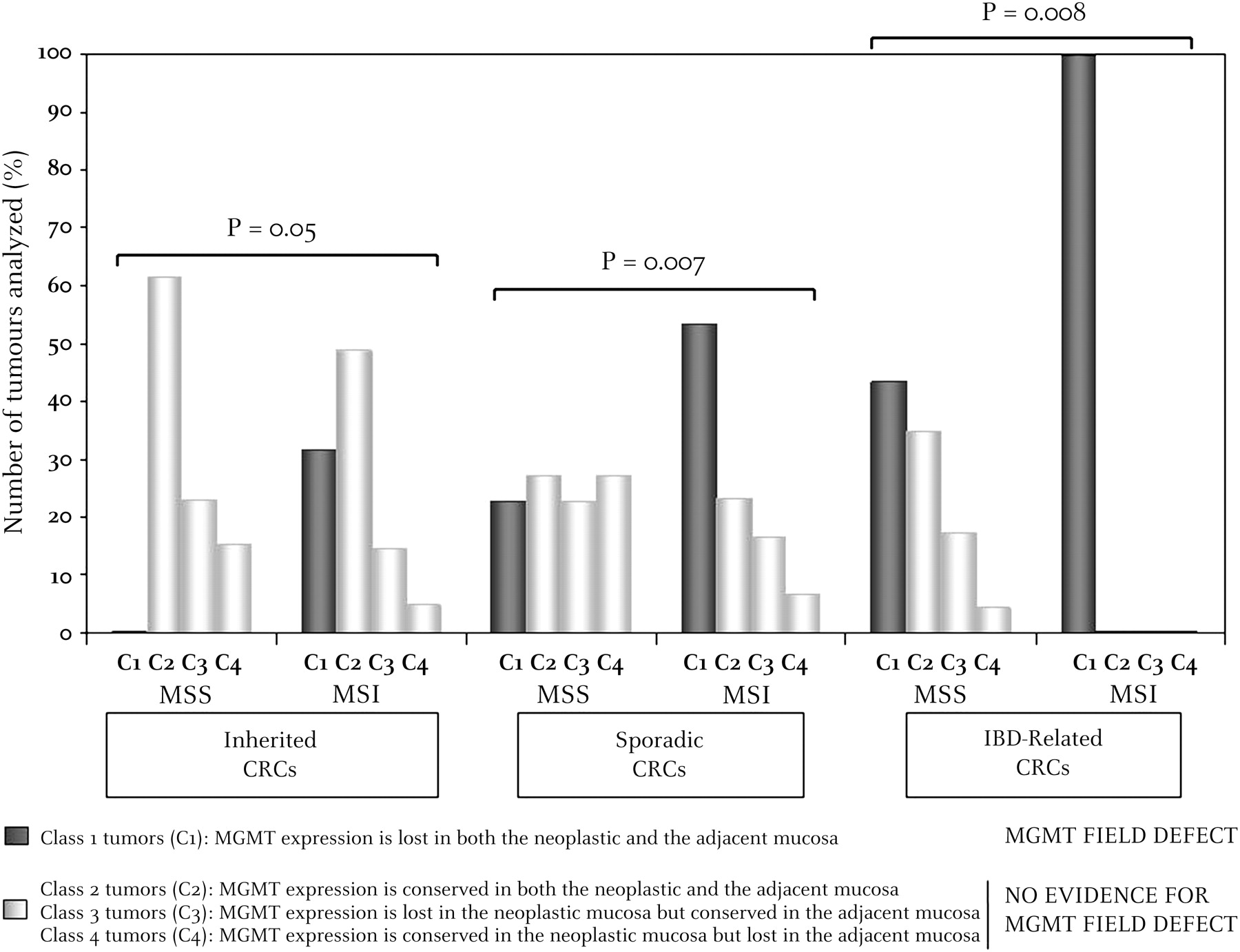
Patients were classified into four classes: (1) class 1, patients for whom MGMT expression was lost in both Neo and A tissues (figure 4A); (2) class 2, patients for whom MGMT expression was conserved in both Neo and A tissues (figure 4B); (3) class 3 (figure 4C) and (4) class 4 (figure 4D), patients for whom MGMT expression status was either lost in Neo but conserved in A tissues, or conserved in Neo but lost in A tissues, respectively.

Immunohistochemical staining of O6-methylguanine-DNA methyltransferase (MGMT) in neoplastic tissues and non-neoplastic adjacent mucosa (A). (A) Class 1 tumour, MGMT expression was lost in both the tumour and adjacent mucosa (MSI sporadic case); (B) class 2 tumour, MGMT expression was conserved in both the tumour and adjacent mucosa (MSS sporadic case); (C) class 3 tumour, MGMT expression was lost in the tumour but conserved in the adjacent mucosa (hereditary non-polyposis colorectal cancer case; MSI); (D) class 4 tumour, MGMT expression was conserved in the tumour but lost in the adjacent mucosa (MSS sporadic case) (×200 magnification). Inserts in A–D represent serial H&E sections in each case. N, normal adjacent mucosa; T, tumour. MSI, microsatellite unstable; MSS, microsatellite stable.
In inherited CRC (HNPCC and FAP), we found: (1) 13 HNPCC and 0 FAP in class 1; (2) 20 HNPCC and 8 FAP in class 2; (3) 6 HNPCC and 3 FAP in class 3; and (4) 2 HNPCC and 2 FAP in class 4. In sporadic and IBD-associated CRC, we found, respectively: (1) 26 (16 MSI and 10 MSS) and 20 (10 MSI and 10 MSS) in class 1; (2) 19 (7 MSI and 12 MSS) and 8 (all MSS) in class 2; (3) 11 (5 MSI and 10 MSS) and 4 (all MSS) in class 3; and (4) 14 (2 MSI and 12 MSS) and 1 (MSS) in class 4. The respective proportions of MSI and MSS CRC found to belong to these four different subclasses are displayed in figure 5. Overall, these results demonstrate a significant excess of class 1 tumours (MGMT field defect) compared with all other classes (no MGMT field defect) in patients with MSI CRC compared with those with MSS CRC (39/81 MSI vs 20/80 MSS, p=0.002). This remained highly significant for the sporadic (16/30 vs 10/44, p=0.007) and IBD (10/10 vs 10/23, p=0.008) subgroups, and just reached significance for the familial cancer subgroup (13/41 vs 0/13, p=0.05).
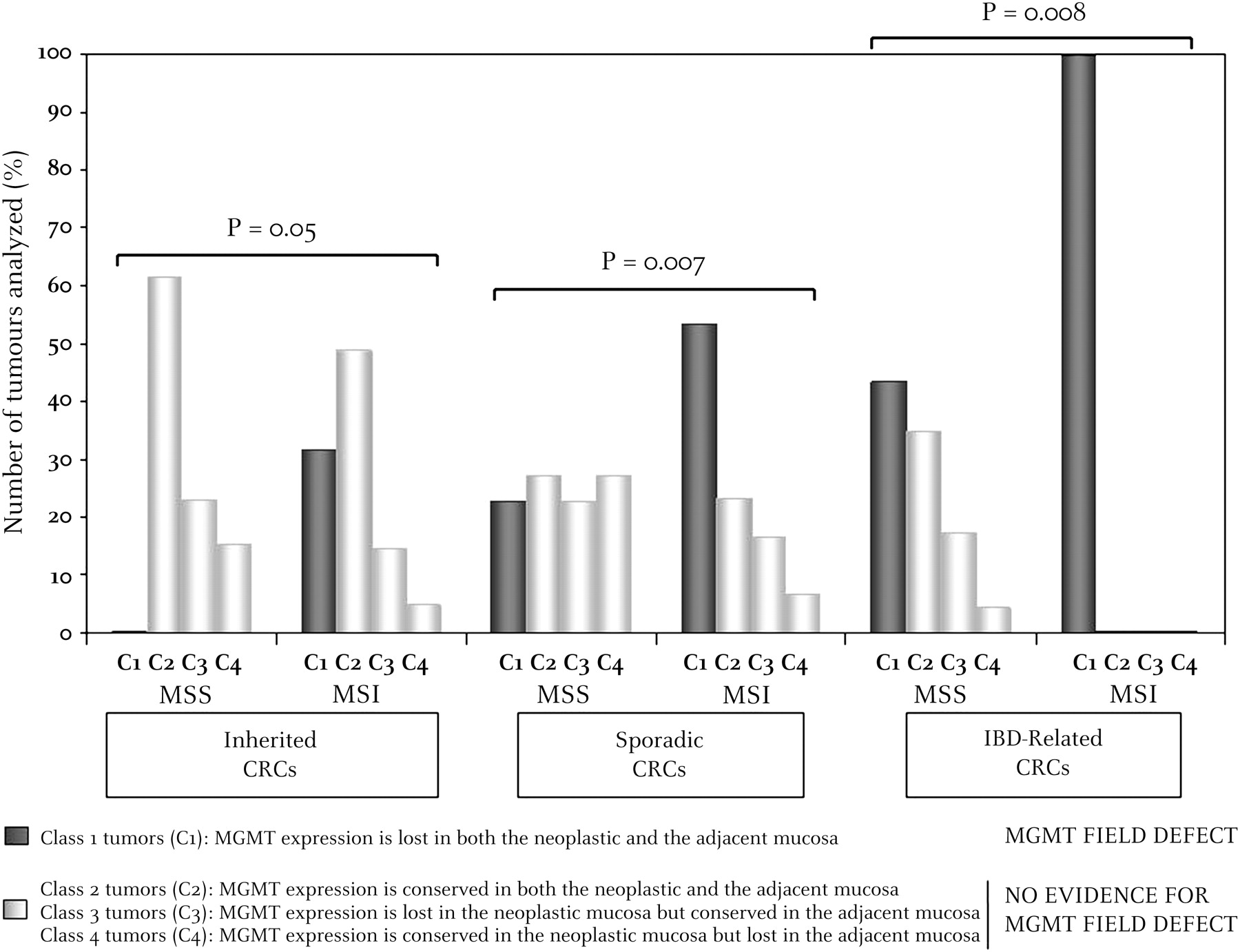
Comparative paired analysis of O6-methylguanine-DNA methyltransferase (MGMT) expression in the microenvironment (adjacent mucosa) and neoplastic tissues of microsatellite-unstable (MSI) versus microsatellite-stable (MSS) colorectal cancer (CRC). p Values (χ2 test) were calculated in each case by comparing the number of tumours belonging to class 1 (MGMT field defect) with all others (no MGMT field defect) in patients with MSI or MSS CRC.
Methylation status of the MGMT promoter by MSP and expression of MGMT in neoplastic and adjacent mucosa of patients with MSI CRC
The association between lack of MGMT expression and MGMT promoter methylation in Neo tissues was examined in a series of 69 MSI CRCs that included 21 HNPCC-related CRCs, 35 sporadic CRCs and 13 IBD-related CRCs (figure 6). Overall, MGMT promoter methylation was detected in 45/69 (65.2%) cases. Among the 45 tumours with MGMT promoter methylation, MGMT expression was lost in 34 (75.6%), whereas 12/24 (50%) cases that were unmethylated retained MGMT expression. A significant association therefore exists between methylation of MGMT and loss of protein expression in MSI CRC (p=0.03). This association did not reach significance when limited to sporadic MSI (p=0.2). Among the 24 tumours with MGMT promoter methylation, 18 (75%) showed loss of MGMT expression (scoring 0/1), whereas 5/11 (45.5%) tumours that were unmethylated retained MGMT expression (scoring 2/3) (figure 6). No association was seen in patients with IBD (p=0.66) (figure 6), even if MGMT expression was lost in almost all MSI CRCs that developed in this context (12/13). MGMT protein expression was lost in 8/9 tumours that had a methylated MGMT promoter, but also in all four tumours that had an unmethylated MGMT promoter. For inherited MSI CRC, the correlation between methylation of MGMT and protein expression was strong in unmethylated tumours, with 7/9 (78%) of unmethylated tumours retaining protein expression, but was average in methylated tumours (8/12, 67%, p=0.1) (figure 6).

Correlation between the methylation status of the O6-methylguanine-DNA methyltransferase (MGMT) promoter and loss of MGMT protein expression in microsatellite-unstable colorectal cancer (MSI CRC) (0/1 labelling).
Methylation of the MGMT promoter was sometimes detected in A tissues in 17 patients with MSI CRC. Among the 9 A mucosa samples with MGMT promoter methylation, 7 showed loss of MGMT protein expression (78%). In contrast, MGMT expression was lost in only 2/8 (25%) of A samples with an unmethylated MGMT promoter. Therefore, a trend for association also exists between methylation of MGMT and loss of protein expression in A tissues from patients with MSI CRC (p=0.09). In patients with MGMT promoter methylation in an MSI tumour, significant MGMT promoter methylation was also observed in the apparently normal adjacent mucosa. Indeed, among 16 patients for whom the methylation status of MGMT was determined in both tumour and adjacent mucosal samples, all nine cases with a methylated MGMT promoter in Neo tissues displayed MGMT promoter methylation in A tissues, while four of the seven cases with an unmethylated MGMT promoter in Neo tissues displayed an unmethylated MGMT promoter in A tissues (p=0.02; figure 7A)

(A) O6-Methylguanine-DNA methyltransferase gene (MGMT) promoter methylation in tumour and adjacent mucosa of patients with microsatellite-unstable colorectal cancer (MSI CRC) determined by methylation-specific PCR. The presence of a PCR product in lanes marked M indicates methylated MGMT; product in lanes marked U indicates unmethylated MGMT. M ctrl, methylated control (Co115 MSI CRC cell line); U ctrl, unmethylated control (TC71 MSI CRC cell line). (B) MLH1 and MGMT promoter methylation status in tumour, adjacent and distant mucosa in a series of 14 MSI CRCs. M, methylated promoter; U, unmethylated promoter. It is noteworthy that we did not observe any loss of MLH1 expression in adjacent mucosa using immunohistochemistry in cases displaying a positive MLH1 methylation status at this level. (C) MGMT promoter methylation in tumour and adjacent mucosa of patients with MSI CRC determined by pyrosequencing. The fraction of methylated cells before and after microdissection (μ) is indicated as a percentage in each case. For adjacent mucosal samples, the fold increase in the fraction of methylated cells following laser microdissection is indicated. ND, not determined.
Correlation between MLH1 and MGMT methylation status determined by MSP in neoplastic and adjacent normal mucosa of patients with MSI CRCs
We also investigated the MLH1 methylation status in both neoplastic and adjacent mucosa in 14 patients with MSI CRCs (figure 7B). As already published,33 our data confirmed that MLH1 is methylated in the microenvironment of some sporadic MSI CRCs displaying MLH1 methylation in tumour cells, whereas in other tumours a positive MLH1 methylation status is restricted to the neoplastic cells. As expected, all cases displaying an unmethylated MLH1 status in neoplastic cells also displayed an unmethylated MLH1 status in adjacent mucosa. In this small series of samples, a positive MGMT methylation status was observed in CRCs that were or were not methylated for MLH1, suggesting that these two events are not mutually exclusive (figure 7B).
Quantification of MLH1 and MGMT methylation in epithelial neoplastic and adjacent mucosa using pyrosequencing
We further investigated MGMT and MLH1 methylation status using pyrosequencing in the neoplastic and adjacent mucosa of four patients with MSI CRCs who displayed positive profiles with MSP (P111, P113, P114, P90; see figure 7C). This was performed before and after microdissection of the neoplastic and adjacent mucosal samples to remove non-epithelial cells. As expected, the presence of infiltrating lymphocytes and/or other inflammatory cells prevented the preparation of ‘pure’ epithelial cells from these patients (data not shown). Under these conditions, microdissection of the adjacent mucosa led to a 3- to 28-fold increase in the fraction of MGMT and MLH1 methylated cells in these patients (figure 7C). In tumour cells, an enrichment of MGMT and MLH1 methylated cells was also observed using this approach, but not of the same magnitude (figure 7C). These data support the notion of epigenetic similarity between tumour and adjacent epithelial cells in terms of their methylation status.
MGMT expression and MMR defects in MSI CRC
There was no evidence for different patterns of MMR protein expression in MSI CRC that showed loss (n=58) or normal expression of MGMT (n=35) by immunohistochemistry. Indeed, we observed 67.2% (39/58) versus 51.4% (18/35), 24.1% (14/58) versus 45.7% (16/35), 5.2% (3/58) versus 2.9% (1/35) and 3.4% (2/58) versus 0% (0/35) of these tumours with MLH1, MSH2, MSH6 or PMS2 deficiencies, respectively (p=0.13). Similar results were observed when sporadic, inherited or IBD-related MSI CRCs were considered separately (data not shown).
The MMR genes for which loss of protein expression was observed by immunohistochemistry were sequenced in a series of 14 and 12 MSI CRCs showing loss of or conserved MGMT expression, respectively. These included 15 HNPCC-related CRCs, nine sporadic MSI CRCs from patients with MLH1 methylation and two IBD-associated MSI CRCs. As expected, no MMR gene mutation was detected in sporadic MSI CRCs from patients without IBD displaying MLH1 methylation, whereas 14 deleterious MMR mutations were diagnosed in patients with HNPCC and IBD (p<0.001), including five transitions, five transversions and four small insertions/deletions (supplementary table 1 online). Of interest, the five transitional events were all diagnosed in MSI CRCs showing loss of MGMT expression. Three were confirmed to be somatic events in the tumours that were not present in the patients' germline DNA (data not shown).
BRAF and KRAS mutations in MSI CRC according to MGMT status
A total of 50 MSI CRCs were screened for the V600E hotspot transversion in BRAF and for transitions in KRAS codons 12 and 13. As expected, HNPCC-related tumours showed only KRAS mutations (n=6/14; 42.9%), whereas BRAF mutations occurred almost exclusively in sporadic MSI CRCs (n=18/25; 70.8%). BRAF and KRAS mutations were mutually exclusive, except in one case. Reduced or total loss of MGMT protein expression was not associated with an increased frequency of KRAS or BRAF mutations (supplementary table 1 online).
Comparative analysis of clinicopathological features associated with MSI and MSS CRC according to MGMT expression status
For both MSS and MSI tumours, no significant associations were seen between the loss of MGMT expression in tumour cells and clinical variables, regardless of the clinical context (table 3).
Clinicopathological features of patients in relation to tumour mgmt expression
Discussion
Based on recent molecular findings, most field cancerisation has been explained by the presence of cells with genetic alterations, in accordance with a multistep oncogenic process. In the initial phase, a stem cell acquires genetic alterations and forms a patch with genetically altered daughter cells.34 As a result of subsequent genetic alterations, the lesion gradually becomes a field and is thought to be a critical step in epithelial carcinogenesis. Such clones have a growth advantage and proliferate to acquire more genetic alterations, ultimately leading to the development of one or more tumours within a contiguous field of preneoplastic cells. MGMT shows aberrant promoter methylation and/or loss of expression in both tumour cells and normal-appearing mucosal cells of patients with CRC and is therefore thought to be involved in creating a field defect in these tumours.3 The present results confirm this working hypothesis. They also indicate that the MGMT field defect depends on the underlying clinical context in which CRC arises, since it was observed only in patients with sporadic CRC. In other clinical contexts, MGMT inactivation is still often observed in the adjacent tumour microenvironment, even in the absence of a ‘true’ MGMT field defect as defined by loss of MGMT expression in both normal-appearing adjacent mucosa and distant mucosa.
MGMT inactivation has until now been proposed as a plausible predisposing factor for CRC without taking into account the molecular heterogeneity of this disease.3 6 22 Through investigation of a large, retrospective series of CRCs enriched with MSI tumours, the present data clearly indicate an additional and even preferential role for MGMT inactivation in the initiation of MSI colon carcinogenesis. MSI CRC was found to occur more frequently than MSS CRC within patches of adjacent mucosa that lacked MGMT expression. Nevertheless, one limitation of this study is that MGMT expression was investigated by a non-quantitative immunohistochemical method. As described previously, a heterogeneous pattern of MGMT expression was observed within tumours and normal tissues.3 35–38 We therefore decided to set stringent criteria by considering that MGMT expression was lost when at least 50% of nuclei were unstained (0/1 labelling). Using this criterion, the MGMT expression level correlated with MGMT promoter methylation status in the neoplastic and normal adjacent mucosal tissues of patients with MSI CRC. Nonetheless, loss of MGMT expression often occurred in the absence of MGMT promoter methylation, notably in patients with IBD, as reported in previous studies.39–42 This suggests that MGMT inactivation might occur due to inflammation of the colonic mucosa which leads to enhanced production of O6-methylguanine DNA adducts. In this context, it is possible that MGMT deficiency in the mucosa adjacent to a tumour might be the consequence rather than the cause of inflammatory neoplasms and hence be associated with MSI due to the frequent inflammatory features observed with this tumour phenotype. We therefore investigated further the association between inflammation and the existence of an MGMT field defect in CRC (see supplementary table 2 online).43 This allowed us to reject the hypothesis since in both MSI and MSS CRCs, inflammatory features were unlikely to be associated with the MGMT field defect.
Some tumours displaying MGMT promoter methylation were also observed here to express MGMT protein. One explanation for this could be the high sensitivity of PCR-based techniques. Indeed, hypermethylation may be detected even when it occurs in only a small subset of tumour cells, as shown here in some tumour and adjacent ‘normal’ mucosal samples when analysed for MGMT (and MLH1) methylation status using the sensitive and quantitative approach afforded by pyrosequencing. Another explanation is that the MGMT promoter region most strongly linked with gene silencing was not targeted in the methylation analysis by MSP, as described for other genes such as MLH1.44 45 Our pyrosequencing analysis of another part of the MGMT promoter highlights the fact that MGMT methylation status may be different in distinct regions of the promoter, as described in glioblastoma.46 Additional studies are required to identify the promoter region whose methylation correlates with the loss of MGMT protein expression in CRC.
The simple association between loss of MGMT expression in adjacent mucosal and tumour tissues shown here in MSI CRC is not a formal demonstration that the two tissues are clonally related. This observation applies to all detected molecular abnormalities that are not constitutional but characterise acquired field defects associated with tumours. Nevertheless, by using microdissection we clearly demonstrated in some patients that methylated cells were epithelial, thus confirming the epigenetic similarities between tumour and adjacent mucosal tissues in these cases. Functional approaches have also been used to show that MGMT inactivation in the microenvironment of a tumour is causal for an MSI-driven malignant process. First, MGMT deficiency in vitro favours the selection of MMR-deficient cells through methylation tolerance.9 Secondly, genetic inactivation of Mgmt combined with MMR haplo-insufficiency favours the development of MSI neoplasms in Mgmt−/−Mlh1+/− mice following treatment with N-methyl-N-nitrosurea (MNU).9–15 The present results on primary human tissues strengthen these observations at the clinical level.
Overall, our data support the idea that an MGMT field defect is more frequently associated with MSI than MSS CRCs. An MGMT field defect has already been reported to be associated with KRAS transitions in the microenvironment of these tumours.3 6 7 Of note, KRAS mutations and MSI have been shown by several groups to be inversely related in CRCs,32 47 48 thus explaining why we did not observe a positive association between these two molecular events in our study. From our data, we therefore propose a model based on the hypothesis that an MGMT field defect might be a triggering event for cell transformation through two alternative and mutually exclusive mechanisms—that is, KRAS mutations that allow cells displaying an MSS or MSI-low phenotype to become malignant49 or, more frequently, the development of MMR deficiency that leads to MSI (see a presentation of this model in figure 8). Since an MGMT field defect was found to be associated with both sporadic and inherited MSI CRCs, our data indicate that loss of MGMT expression might promote the initiation of MSI tumours by favouring either MLH1 promoter methylation or other defects through different mechanisms including transitional events in the coding sequence of MMR genes, as reported here in some HNPCC- and IBD-related MSI CRCs or as previously described for KRAS mutation in MSS CRCs.3 6 7 The quasi-systematic loss of MGMT expression observed here may explain the occurrence of these tumours at a young age in patients with IBD, in contrast to MSI CRCs that are generally observed in elderly women. Therefore, MGMT inactivation might be considered an early biomarker of MSI tumours prior to MMR inactivation. Whatever the mechanism underlying MGMT silencing in the normal-appearing mucosa of patients with MSI CRC, these observations highlight the importance of early detection and monitoring of the MGMT field defect. This applies particularly to patients who are genetically predisposed to developing MSI CRCs or who are at increased risk for their development.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
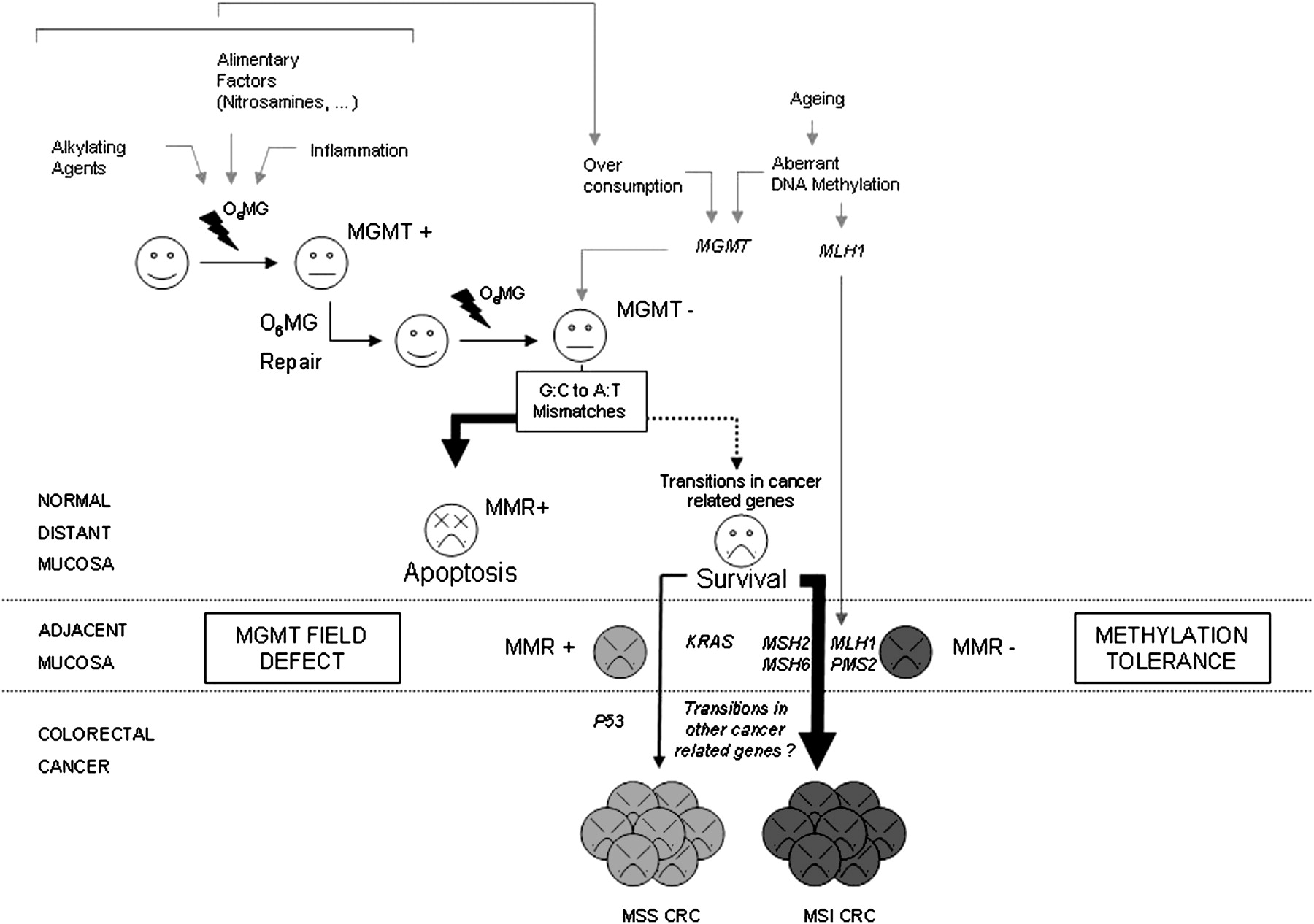
Mutagenic methyl adducts from O6-guanine (O6MG) are formed in the colonic mucosa. While such lesions are usually processed by O6-methylguanine-DNA methyltransferase (MGMT), they accumulate and result in increased G:C to A:T transition mutations in cells that have lost MGMT expression due to different mechanisms. Such mismatches are usually recognised by the mismatch repair (MMR) system that, however, is unable to process such lesions correctly, giving rise to double-strand breaks during replication and apoptosis. Nevertheless, a rescue of cells displaying loss of MGMT expression and their proliferation with daughter cells to form an MGMT field defect associated with colorectal cancers (CRCs) is sometimes observed in that context. It may take place through the occurrence of coding transitions in cancer-related genes such as KRAS or the inactivation of MMR genes allowing tolerance to O6MG (methylation tolerance). According to our data, this last mechanism is likely to be more frequent than and mainly mutually exclusive of the former, and may be responsible for the emergence of a large amount of microsatellite-unstable (MSI) CRCs in different clinical contexts, notably in the general population.
MGMT removes mutagenic and toxic adducts from O6-guanine in DNA, the major site of attack of many carcinogens and alkylating chemotherapeutic agents. Methylating compounds such as nitrosamines are taken up from tobacco50 and food, with volatile nitrosamines being present in foodstuffs and drinks.51 Alkylating agents may also form endogenously within the body, either through normal cellular processes or as a result of transformation mediated via the intestinal flora. The environment created by these methylating compounds may contribute to the development of MSI cancer. Methylating compounds from food or drugs should be avoided in patients who show an increased risk for the development of MSI tumours. Field defects other than those created by the loss of MGMT expression might confer similar or even higher risks for the development of MSI or MSS CRC, and additional research is needed to identify these. CRC is a heterogeneous disease consisting of multiple tumour subtypes characterised by different molecular and morphological features and with varying responses to chemotherapy. The detection and monitoring of field defects may have profound implications for cancer prevention and treatment.
Acknowledgments
We are grateful to Barry Iacopetta, Thécla Lesuffleur, Martine Muleris and Françoise Praz for critical reading of the manuscript. This work was partly supported by grants from the Association de Recherche contre le Cancer (credit number 1041) and from GEFLUC Ile de France.
References
Supplementary materials
Web Only Data gut.2009.194787
Files in this Data Supplement:
Footnotes
Competing interests None.
Ethics approval This study was conducted with the approval of the Human Research Ethics Committee of Saint-Antoine hospital.
Provenance and peer review Not commissioned; externally peer reviewed.