Article Text

Abstract
Objective The mammalian commensal gut microbiota is highly diverse and displays an individual-specific composition determined by host genotype and environmental factors. The temporal development of host–microbial homeostasis in the digestive tract is recognised as a major function of the immune system. However, the underlying cellular and molecular mechanisms are just beginning to come to light. Nucleotide-binding, oligomerisation domain 2 (NOD2) recognises bacterial muramyl dipeptide and is regarded as a pivotal sensor molecule of the intestinal barrier. The aim of this study was to investigate its influence on the development and composition of the intestinal microbiota using a Nod2-deficient mouse model.
Methods The dynamics of faecal and ileal microbial composition were investigated in Nod2+/+and Nod2−/− mice on a C57BL/6J background. We assessed microbial diversity and composition using 16S ribosomal RNA gene-based clone library sequencing and high throughput pyrosequencing and quantified the observed changes by real-time PCR. Changes in the major bacterial phyla were investigated in human samples by quantitative real-time PCR.
Results We found that adult Nod2-deficient mice display a substantially altered microbial community structure and a significantly elevated bacterial load in their faeces and terminal ileum compared to their wild-type counterparts. Interestingly, we demonstrate that these findings are also present in weaning mice, indicating a profound influence of Nod2 on the early development and composition of the intestinal microbiota. We demonstrate that NOD2 genotypes also influence the microbial composition in humans.
Conclusions Our results point to an essential role of Nod2 for the temporal development and composition of the host microbiota, both in mice and in humans, which may contribute to the complex role of NOD2 for the aetiopathogenesis of Crohn's disease.
- Inflammatory bowel disease
- NOD2
- microbiota
- crohn's disease
- IBD basic research
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Nucleotide-binding, oligomerisation domain 2 (NOD2) is an intracellular sensor for the bacterial cell wall component (muramyl dipeptide), and regulator of the commensal gut microbiota in mice.
Nod2-deficient mice display diminished bacterial killing ability.
Loss of function variants in the human NOD2 gene are associated with an increased susceptibility for Crohn's disease.
Altered intestinal microbial community structure is a feature of Crohn's disease in humans.
What are the new findings?
Nod2-deficient mice display an increased load of commensal microbiota, and altered microbial composition.
The microbial composition of Nod2-deficient mice is altered already at an early weaning stage.
Humans carrying NOD2 variants (SNP13) also have significantly increased loads of Bacteroidetes and Firmicutes.
How might it impact on clinical practice in the foreseeable future?
The data form the basis for a clinical understanding that NOD2 variants may affect the microbial community structure development early in life.
The results may govern genotype-guided preventive approaches in babies at risk aiming at the establishment of a regular intestinal flora.
Introduction
The mammalian gastrointestinal tract harbours an extremely complex microbial ecosystem including bacteria, protozoa and fungi. A total of about 500–1000 different bacterial species are estimated to be present in the human gastrointestinal tract, with a density of 1011 bacterial cells per gram wet weight in the distal large bowel contents, outnumbering eukaryotic cells by an order of magnitude. Host genotype and several other factors such as diet, age, stress and diseases can significantly influence the composition of the microbiota.1 Several studies support the importance of host genotype in determining microbial community structure in humans,2 3 as well as in animal models.4 5
It is becoming increasingly clear that the close symbiotic relationship between hosts and a resilient microbiota is a vital part of the homeostasis of intestinal physiology.6 The resident microbiota is crucially involved in diverse processes such as the production of short-chain fatty acids, metabolism of nutrients and organic substrates,7 the development of intestinal epithelium,8 protection against foreign pathogens and, importantly, the maturation and development of the immune system.
Recently, Rakoff-Nahoum et al (2004) demonstrated that tonic stimulation of the Toll-like receptor (TLR) system by the commensal microbiota in the intestinal epithelium is required for normal regeneration of the intestinal lining after mucosal injury.9 Experimental interference with the commensal microbiota (ie, antibiotic treatment) or the host recognition system (ie, genetic ablation of the universal TLR adaptor MyD88) leads to an increased susceptibility of the epithelial layer to inflammatory noxa. These results demonstrated for the first time a prime importance of the intimate cross-talk between the intestinal microbiota and the innate immune signalling of intestinal epithelial cells (IECs) for resistance to intestinal inflammation.9 Similarly, TLR2 is associated with lineage specific modification of the caecal tissue attached microbiota in mice lacking TLR2 receptors and an increased susceptibility to specific opportunistic bacteria.10
Nucleotide-binding oligomerisation domain (NOD)-like receptors (NLRs) represent ancient sentinels of the host innate immune system, and genetic variants in NLR genes are associated with complex chronic inflammatory barrier diseases.11 The Nod2 gene is an intracellular sensor for the bacterial cell wall component muramyl dipeptide, and loss of function variants in the human Nod2 gene have been associated with an increased susceptibility for Crohn's disease, a human chronic inflammatory bowel disease associated with a severely altered intestinal microbial community structure,12–14 Recently, Nod2 was found to be an important regulator of the commensal gut microbiota in mice.15 Nod2-deficient mice display diminished bacterial killing ability, increased loads of commensal bacteria and are less effective in preventing the colonisation of pathogenic bacteria. However, the direct influence of host innate immune factors on the composition and dynamics of the resident intestinal microbiota has yet to be fully described.
In this study, we present the results of over 2700 16S rRNA gene sequences from 34 individual clone libraries analysed by Sanger sequencing and over 57 000 sequences from 14 ileum samples derived by 454 sequencing in an effort to describe the effect of the Nod2 genotype on the intestinal microbial community composition and structure in mice. Compared to wild type, Nod2-deficient mice display a substantially altered microbial community. We demonstrate that a distinct succession of bacterial species composition is present through the course of mouse development, which is disturbed in Nod2-deficient mice. Finally, we show that humans carrying mutations in Nod2 display a similar phenotype, characterised by a significant increase in two major phyla, Bacteroidetes and Firmicutes.
Materials and methods
Animals, housing conditions and sampling of faeces and tissues
C57BL/6J and B6.129S1-Nod2tm1Flv/J (Nod2−/−)16 mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). The Nod2−/− line was back-crossed to C57BL/6J for at least seven generations prior to purchasing mutant mice. Mice were maintained under specific-pathogen free conditions (SPF) upon arrival at the central animal facility of the University Hospital of Schleswig-Holstein, Germany, in individually ventilated cage (IVC) systems from the company Ehret, Emmendingen, Germany. Mice were housed with autoclaved food, bedding, and water. All mice were re-located to autoclaved cages (cage type 2 long, polycarbonate Makrolon) on a weekly basis. This procedure excludes any possibility of exchange of faeces between cages. Animals were tested for possible pathogens according to the established protocol of the Federation of European Laboratory Animal Science Association (FELASA).
To obtain Nod2−/− and Nod2+/+ animals for comparison in our facility, the initial Nod2−/− breeding pairs were back-crossed to C57BL/6J. Nod2−/− and Nod2+/+ littermates from a single heterozygous intercross were then selected to start homozygous intercross lines that were housed separately from this point on. To account for cage (litter) effects, all mice compared in this study were from independent families derived from expansion after the initial intercross. This design ensures that the individual replicate mice of each genotype experience the same amount of potential drift in the microbiota within genotype as between genotype. Only male mice were used in this study. Faeces samples were collected immediately after defaecation and stored at −80°C.
For the analysis of NOD2 in human samples, the biopsy bank of the outpatient clinic of the Department of General Internal Medicine, University Hospital Schleswig-Holstein (Kiel, Germany) was screened for patients with Crohn's disease who were homozygous for the NOD2 SNP13 mutation. Due to the extremely rare occurrence of SNP13 homozygosity in European populations in normal control individuals,17 18 only patients with Crohn's disease were studied. All patient samples (pairs of matching DNA/biopsy) and phenotype information were given pseudonyms prior to the procedure. After determining NOD2 status, 11 homozygous wild-type and three homozygous SNP13 individuals were selected for further study. All biopsies were sampled from macro- and microscopically non-inflamed mucosal area of the terminal ileum (according to the pathological report) and stored in liquid nitrogen until further processing. All patients were in clinical remission as indicated by a Crohn`s disease activity index (CDAI) of <150 and were pretreated with 5-aminosalicylates or azathioprine according to their clinical requirements. No systematic differences in age, disease characteristics and medication were present between patients carrying the NOD2 SNP13 variant or the reference allele. Medication was stable for 4 weeks prior to sampling. The inflammatory activity was independently scored by two investigators. The diagnosis was based on standard criteria using radiological and endoscopic findings in every case.
All patients agreed to participation by giving informed consent at least 24 h prior to the study. Indications for colonoscopy were monitoring of therapy response and cancer surveillance. All biopsies tested negative in specific PCR tests for common intestinal pathogens, including Staphylococcus sp., Salmonella sp., Helicobacter sp., Mycobacterium sp., Tropheryma whippelli, Shigella sp. and Campylobacter jejuni.
DNA extraction and amplification of the 16S rRNA gene
DNA extraction was performed with the Fast DNA spin kit as previously described.12 In brief, faecal or biopsy tissue samples were homogenised in 200 μl TL buffer (tissue lysis buffer (200 mM HEPES, pH 7.5, 1 M KCl, 100 mM MgCl2, 1 mM EDTA, 0.2% NaN3)) and 25 μl proteinase K was added before incubating at 55°C for 2 h (PeqLab, Erlangen, Germany). Mechanical force was applied to maximise bacterial cell wall lysis. The 16S rRNA gene coding region spanning variable region V1–V4 was amplified using the broad-range forward primer 8F 5′ AGA GTT TGA TCC TGG CTC AG 3′ and 805R 5′ GGA CTA CCA GGG TAT CTA AT 3′.13 Amplification reactions were performed in a total volume of 50 μl containing 1× PCR buffer, 15 mM MgCl2 (both Qiagen, Hilden, Germany), 0.2 μM of each primer (Carl Roth, Karlsruhe, Germany), 0.2 μM of each dNTPs (Qiagen), 1 U DNA Taq polymerase (Qiagen) and 100 ng of sample DNA. PCR amplification was performed in a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, California, USA) using the following cycling conditions: 95°C for 5 min, 30 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 1 min and a final extension of 10 min at 72°C for PCR product adenylation. Products were checked for the correct size (about 800 bp) by running on 1.5% agarose gels.
Cloning and sequencing
Specific amplicons were purified with the MinElute PCR purification kit (Qiagen) and cloned using the PCR 2.1 TOPO TA cloning kit (Invitrogen, Karlsruhe, Germany). The inserts were amplified using the vector specific primers M13F and M24R as described.12 Sequencing of the inserts was performed on an ABI PRISM 3700 DNA analyser in a final volume of 10 μl using 1 μl Big Dye version 1.1 (Applied Biosystems) at a concentration of 3.2 μM of primer 8F according to the following protocol: 96°C for 1 min, 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min.
Sequence analysis and comparison of 16S rRNA gene clone libraries
The sequences were checked for possible chimera using the program Bellerophon19 and were eliminated from further analysis. Clone sequences of about 800 bases were subjected to the Ribosomal Data Base Project RDP (release 10) classifier for phylogenetic classification. Sequences were aligned with clustal X V 2.0.20 Alignments were checked and corrected manually. Weakly aligned and divergent regions were removed by Gblock v0.91b21 using the recommended parameters for 16S rRNA gene alignments.22
Sequences were grouped according to a 97% similarity threshold into species-level phylotypes23 using the software DOTUR (Distance-based Operational Taxonomical Units and Richness).24 Shannon-Weaver and Simpson diversity indices were also calculated by DOTUR. Clone library coverage (x=1−n/N×100) was calculated by the equation of Good.25 Representative sequences from each phylotype were subjected to the sequence match at RDP II to link our retrieved phylotypes to the nearest clone sequence in the RDP database.
Phylogenetic trees were generated by the PHYLIP software package (version 3.69, distributed by J Felsentein. Department of Genome Sciences, University of Washington, Seattle, USA). Distance matrix files were generated using DNAdist incorporating a Jukes–Cantor model. Phylogenetic trees were generated by the neighbour-joining method and topologies were analysed by 1000 bootstrap replicates.
Differences among the 16S rRNA gene libraries were analysed using ∫-Libshuff,26 SONS (Shared OTUs and Similarity)27 and UniFrac.28 The distance matrix file generated by DNAdist was used as input for ∫-LIBSHUFF. SONS was applied to estimate the similarity between two communities (membership and structure). As recommended, DOTUR-generated list files and manually generated names files were used as input files for SONS.
UniFrac was employed to run Jack-knife Environment Clusters and principal coordinate analysis. A rooted neighbour-joining tree and a manually generated environment label file were imported into UniFrac. Principal component analysis documents the distances between phylotype compositions in 16S clone libraries while cluster analysis groups the clone libraries based on distances.
The software PAST (http://folk.uio.no/ohammer/past) was used for cluster analysis as well as to identify differentially represented OTUs in one or the other genotype/age groups. After ranking and scaling, abundance was converted to the presence or absence of OTUs (‘1’ indicates the presence of an OTU, ‘0’ represents its absence). Euclidean similarity matrices were used to build a dendrogram with the minimal variance algorithm (Ward's method).
16S rRNA gene pyrosequencing
To analyse the influence of Nod2 genotype on microbial composition and structure in the ileum tissue, the 16S rRNA gene variable region V1–V2 was amplified using the forward and reverse primers 5′-CTATGCGCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG-3′ 5′-CGTATCGCCTCCCTCGCGCCATCAGXXXXXXXXXXCATGCTGCCTCCCGTAGGAGT-3′, respectively.29 The sequence in italics is 454 Life Sciences primer B and A, respectively and the underlined sequence is the broadly conserved bacterial primers. A two-base linker sequence (TC/CA) and a four-base key (TCAG) was added as recommended by Roche. A unique 10-base multiplex identifier (MIDs designated as XXXXXXXXXX) was added to the reverse primer to tag each PCR product. Replicate PCRs were performed for each sample in addition to negative controls. Amplified products were run on an agarose gel and specific bands were excised and purified using the QIAquick gel extraction Kit (Qiagen). The concentration of the purified amplicons was measured using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Equal amounts of PCR products were mixed in a single tube and subsequently sequenced on a 454 GS-FLX using the Titanium Sequencing Kit (Roche, Penzberg, Germany).
Analysis of pyrosequencing reads
Sequence reads were screened and filtered for quality and length with the following criterion, length >200 bp, average quality score >20, no ambiguous bases, a perfect match to the primer/barcode sequence, and no homopolymer stretches >6nt long. Chimeric sequences were detected and removed by Black Box Chimera Checker (described and freely available at http://www.researchandtesting.com/B2C2.html). Samples were binned according to their specific MIDs (barcode).30 High quality sequences were classified by RDP classifier (release 10) up to genera level. For alpha and beta diversity analysis, equal numbers of sequences were randomly selected using a perl script30 and were grouped into operational taxonomic units (OTUs) at the species level threshold of ≥97% sequence identity using Mothur.31 Diversity and shared phylotypes were also calculated using Mothur.31
Phylogenetic trees were built using the FastTree-2 algorithm.32 FASTUnifrac33 was applied to test the dissimilarity of microbial composition between any pair of adult and young Nod2+/+ and Nod2−/− mice, unweighted principal coordinate analysis (PCoA) and jack knife environment cluster analysis was performed on UniFrac matrices to visualise the differences based on phylogenetic structure.
Real-time quantification of the microbiota
β-Actin34 and 16S rRNA gene primers specific to all bacteria, Bacteroidetes, Firmicutes,35 Bacteroides–Prevotella36 and Faecalibacterium prausnitzii37 were used to quantify commensal microbiota at the phylum or genus level (table 1). Real-time quantitative PCR was carried out in a volume of 10 μl on an Applied Biosystems 7900HT real-time PCR system using 384-well plates. Each PCR mixture consisted of 5 μl of Power SYBR PCR Master Mix (Applied Biosystems), 100 nM of each primer, and 5 ng of DNA. The amplification program consisted of (i) pre-incubation at 50°C for 2 min, and 95°C for 10 min; (ii) 45 cycles of denaturation at 95°C for 15 s and annealing at the appropriate temperature (table 1) for 1 min; and (iii) one cycle at 95°C for 15 s, 53°C for 15 s, and 95°C for 15 s.
Primers used for real-time quantification
Results
Gut microbiota composition depends on age
In order to interpret the effects of Nod2 genotype in the context of the temporal development of the intestinal microbiota, we analysed clone libraries generated from faeces at eight stages of development ranging from 1 day to 32 weeks of age. Cluster analysis performed using UniFrac28 clearly reveals distinct groups distinguished by age (figure 1A). The clone sequences were further classified using RDP classifier,38 revealing that the microbial colonisation pattern from the first day to 32-week-old mice is clear at the level of the major phyla: the Proteobacteria are predominant in 1-day-old mice, followed by the Firmicutes in the first 2 weeks and the Bacteroidetes in 4-week-old mice. Mice older than 10 weeks displayed stable microbiota dominated by the Firmicutes and Bacteroidetes (figure 1B), an observation consistent with other metagenomic studies of the intestinal microbiota of adult mice.39

UniFrac jack-knife environmental cluster (A) and distribution of sequences at the phylum level (B) of 16S rRNA gene clone libraries generated from C57BL/6J mice at different ages. Sequences were assigned taxonomically using RDP classifier.
Nod2 genotype influences bacterial composition in young and adult mice
Having demonstrated that microbial composition substantially changes during development under common environmental conditions, we next investigated the influence of the Nod2 gene on faecal communities using a constitutive murine Nod2 knockout model. Species level (97% similarity) OTUs and diversity indices calculated by DOTUR show a trend of reduced bacterial diversity in Nod2−/− versus Nod2+/+ clone libraries in adults, but the difference was not significant. However, Nod2−/− mice harbour roughly twice the number of OTUs compared to Nod2+/+ mice at 2 weeks of age (Supplementary table S1). This indicates that mice lacking Nod2 expression display a profound difference in bacterial colonisation, perhaps due to a lack of bacterial killing activity,15 40–42 Library coverage (X) of bacterial clones was not significantly different in any of the genotype or age groups, providing a valid reason for inter-group comparison. ∫-LIBSHUFF analysis revealed significant differences between Nod2+/+ and Nod2−/− mice in pair wise comparisons (p value <0.002). In contrast, comparisons between clone libraries within a group (Nod2+/+ and Nod2−/−) displayed no significant differences (Supplementary table S2). As ∫-LIBSHUFF analysis revealed no significant difference between mice within a genotype, we further investigated the differences between Nod2+/+ and Nod2−/− mice by pooling the clone libraries of replicate mice of the same genotype. SONS (shared OTUs and similarity) incorporates OTU-based nonparametric estimators of similarity between communities, including the abundance-based Jaccard similarity index (Jabund), which measures community overlap and may be defined as the probability that an OTU selected at random is found in both libraries in question. Values range from zero (completely different communities) to 1 (identical communities). Values were low and significantly differed from 1 in comparisons between Nod2+/+ and Nod2−/− mice at both 2 weeks of age (0.40±0.20 (SE)) and among adults (0.21±0.09 (SE)), indicating significant differences in community composition.
To evaluate the variability and distances among clone libraries between Nod2+/+ and Nod2−/− mice, environmental distance matrices were calculated by UniFrac. Principal coordinate analysis (PCoA) performed on these distance matrices revealed that a considerable proportion of the variation in microbial community composition can be explained by age and genotype (figure 2B). Nod2−/− mice display a distinct cluster from their Nod2+/+ counterparts. Furthermore, while Nod2+/+ mice were further subdivided according to their age, this effect was completely lost in Nod2−/− mice. The microbiota of 2-week-old mice was more similar to adult mice of the same genotype than it was to mice of the same age, indicating a profound influence on the microbial community. These findings were further supported by cluster analysis based on the presence/absence of OTUs in clone libraries. OTUs influencing this clustering are shown in Supplementary figure S1. OTUs belonging to the phylum Bacteroidetes were present already in2-week-old old Nod2−/− mice, but were absent from the Nod2+/+ mice of the same age.

UniFrac jack-knife (A) and principal coordinate analysis (B) of 16S rRNA gene clone libraries in 2 week and adult faecal samples. Circles represent clusters according to age and genotype.
Because Nod2 is highly expressed in the ileum, which is also a common site of inflammation in Crohn's disease, we further analysed microbial composition and structure in the ileum of Nod2+/+ and Nod2−/− mice. After the application of quality filters (see Materials and Methods), a total of 57 562 16S rRNA gene sequence reads from 2-week-old (n=3) and adult (n=4) Nod2+/+ and Nod2−/− were classified from the phylum to genera level using the RDP classifier tool. A detailed list of classifications and abundances is provided in Supplementary table S3.
As observed in faeces, a trend of reduced diversity was also observed in the ileum of Nod2−/− compared to Nod2+/+ adult mice, but the differences were not significant (data not shown). However, the number of observed phylotypes (species level) was significantly different (p<0.05) in Nod2−/−compared to Nod2+/+ (figure 3A) in both age groups. We further calculated overlapping or shared phylotypes in order to see the phylogenetic relatedness of phylotypes in Nod2+/+ and Nod2−/− groups. A significantly higher number of shared species level phylotypes were observed in libraries from the same genotype (Nod2+/+ or Nod2−/−) than in libraries from different genotypes (Nod2++ vs Nod2−/−) in both age groups, indicating dissimilarity in microbial composition influenced by Nod2 status (figure 3B).

Number of species level phylotypes (97% similarity) in Nod2+/+ and Nod2−/− mice (A). Number of shared phylotypes in Nod2+/+ and Nod2−/− libraries in 2 week and adult ileal samples (B). Intra- and intergroup comparisions depict shared phylotypes in libraries from the same and different genotypes, respectively. Data represent mean of (n=3 from 2 week old, n=4 from adult mice) with SE. Significance was determined by the Student t test.
To determine the overall similarity of ileal microbial composition with respect to age and genotype, we performed principal coordinate (PcoA) and jack knife environment cluster analyses using the UniFrac metric. As observed in faecal communities, a considerable proportion of the variation in composition could be explained by age and genotype, with all four groups displaying distinct clusters (figure 4A,B).

UniFrac jack-knife (A) and principal coordinate analysis (B) of 16S rRNA gene tag sequences (454) in 2 week and adult ileal samples. Circles represent clusters according to age and genotype.
Relative abundance of Bacteroidetes and Firmicutes in Nod2+/+ and Nod2−/− mice
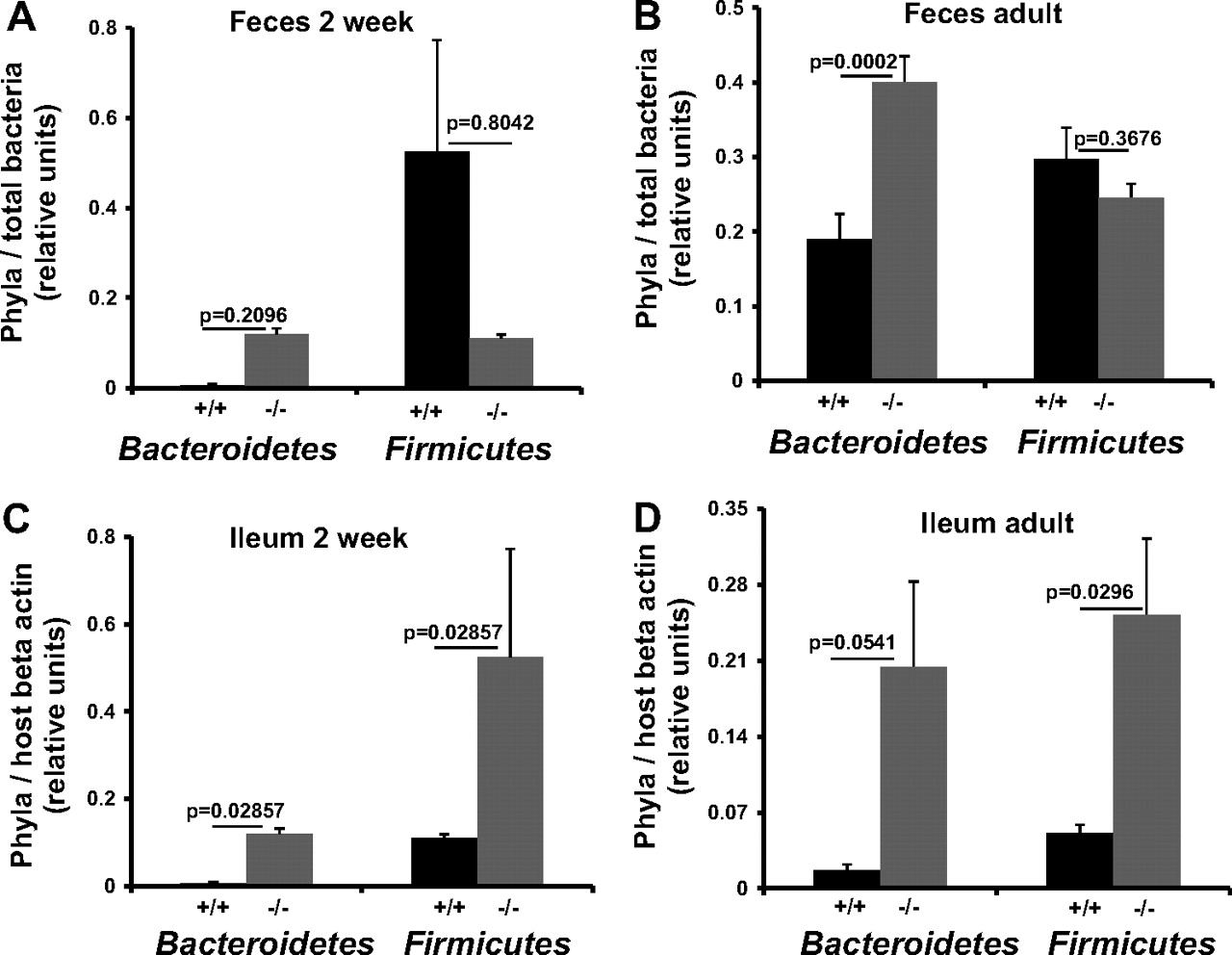
Bacteroidetes and Firmicutes are the two most abundant phyla of the mammalian intestinal microbiota, and the relative abundance of these two phyla is important for normal gut physiology.43 The ratio between these phyla changes at different stages of life, with adults having more Firmicutes than infants and elder individuals,44 and is also influenced by genotype.39 To test if Nod2 status influences this ratio, the relative abundances of these two phyla were determined using real time PCR quantification. No significant difference in either the Bacteroidetes or Firmicutes was observed in the faeces of 2-week-old mice (figure 5A), although both were significantly higher in the ileum (figure 5C). The Bacteroidetes were significantly higher in the faeces of adult Nod2−/− mice (figure 5B) and marginally significant in the adult ileum (figure 5D), whereas the Firmicutes were significantly higher only in the ileum of Nod2−/− mice (figure 5D).

Relative abundance of Firmicutes and Bacteroidetes in Nod2+/+ and Nod2−/− mice. Values were normalised to total bacteria and host β-actin. Data represent mean with SE. Significance was determined by the Mann–Whitney U test. (A) Faeces from 2 week Nod2+/+ (n=3) and Nod2−/− (n=2), (B) Faeces from adult Nod2+/+ (n=11) and Nod2−/− (n=16), (C) Ileum from 2 week Nod2+/+ (n=4) Nod2−/− (n=4), (D) Ileum from adult Nod2+/+ (n=4) and Nod2 −/− (n=4).
Nod2 mutations influence the mucosa-attached microbiota in humans
To determine whether the effects of Nod2 on the intestinal microbiota observed in mice are also present in humans, we analysed the tissue-attached microbiota from non-inflamed ileal biopsies and faeces obtained from patients with Crohn's disease who were homozygous for either SNP13 or wild-type NOD2 alleles. DNA extracted from biopsies and faeces was analysed by real-time PCR to quantify the bacterial load using 16S rRNA gene specific primers for the two most dominant phyla, the Bacteroidetes and Firmicutes. The genus Bacteroides and species Faecalibacterium prausnitzii were also analysed due to their importance in gut physiology, either as a suspected disease-associated candidate or of therapeutic importance. Primers detecting the human β-actin (ACTB) gene were used to normalise the quantification of bacterial load relative to host tissue. The load of Firmicutes and Bacteroidetes was significantly higher in biopsies taken from patients homozygous for SNP13, and a similar pattern was observed for Bacteroides. The anti-inflammatory commensal bacterium Faecalibacterium prausnitzii was reduced in SNP13 homozygotes, but the difference is only marginally significant (figure 6A). A similar trend was observed in faecal samples for all groups except for the genus Bacteroides, although no differences were significant (figure 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relative abundance of mucosa-attached (A) and faecal (B) microbiota with respect to SNP13 in humans. Ileal biopsies and faecal samples from patients with Crohn's disease who were homozygous for wild-type or SNP13. Nod2 alleles were quantified by real-time PCR using 16S rRNA gene primers specific to Firmicutes, Bacteroidetes, Bacteroides, and Faecalibacterium prausnitzii. Data were normalised to total bacteria and host DNA (β-actin). Ileal data represent the mean of three SNP13 homozygotes and 11 normal patients with Crohn's disease. Faecal data represent the mean of three SNP13 homozygotes and 10 wild-type patients with Crohn's disease. p-Values were calculated by the Mann–Whitney U test.
Discussion
In this study, we have revealed novel and important aspects of the role of Nod2 for the development and composition of the mammalian intestinal microbiota. A detailed view into the colonisation process during mouse development highlights that microbial composition is not only influenced by genotype, but also by stage of development. Previous studies based on fingerprinting approaches found that the caecal microbiota in SPF mice changes drastically with age, but stabilises after 4 weeks.45 In our study based on 16S rRNA gene clone libraries, the microbial community continued to change after week 4b, fluctuating in the proportions of the three most abundant phyla (Firmicutes, Bacteroidetes and Proteobacteria; figure 1B) until 10 weeks of age, at which point stable Firmicutes-dominated communities became apparent. Earlier culture-based studies found that the caecal microbiota matures as early as 4–5 weeks of age. These differences likely highlight the limitations of culture-dependent methods.46 Our results are also consistent with another recent culture-independent method, denaturing gradient gel electrophoresis (DGGE), which found a predominance of Proteobacteria and facultative anaerobic members of the phylum Firmicutes, the Lactobacillales, in 3- and 7-day-old mice.47 Given the striking differences in microbial composition during development, we analysed the intestinal microbiota between wild-type and Nod2-deficient mice at both the adult stage and at the dynamic time point of 2 weeks of age. It is important to note that studies interrogating microbiota communities in genetically altered mice are prone to systematic errors (eg, by cage effects, drift in independent lines or genetic background). The study design chosen here employed samples of individual male mice from independent families after an initial intercross (see Materials and methods) trying to circumvent cage and female hormonal cycle effects as strictly as possible. Indeed, we found that microbial communities in Nod2-deficient mice are altered in terms of composition and quantity not only in adult mice, but are also drastically altered in weaning mice. This appears to be largely due to members of the phylum Bacteroidetes being present at a much earlier stage of development, possibly in relation to the lack of Nod2-dependent bacterial killing found in wild-type mice.15 40–42
Using real-time PCR quantification, we demonstrate a significantly greater load of Firmicutes and Bacteroidetes in the terminal ileum of Nod2-deficient mice of both ages, in addition to a greater load of Bacteroidetes in adult faeces. Petnicki-Ocwieja et al (2009) found a significantly increased load of the phylum Firmicutes and the genus Bacteroides in terminal ilea, but not in the faeces of adult Nod2-deficient mice.15 The authors reason that this is likely due to Nod2 expression being largely limited to the crypts of the terminal ileum. One main reason our study was sensitive enough to observe an influence of Nod2 expression on the faecal composition is likely a difference in sample size: we analysed 11 Nod2+/+ and 16 Nod2−/− mice compared to one Nod2+/+, two Nod2+/− and four Nod2−/− mice in the aforementioned study. We did, however, observe that the terminal ilea of patients with Crohn's disease in remission homozygous for SNP13 display a significantly increased load of both the phyla Bacteroidetes and Firmicutes and the genus Bacteroides.
While several studies in human patients with inflammatory bowel disease (IBD) have detected a lower number of Firmicutes and and increased abundance of Bacteroidetes being associated with the disease state regardless of the genotype13 48 our data indicate that the influence of a defect Nod2 genotype on the abundance of Bacteroidetes and Firmicutes in the terminal ileum is conserved between mouse and humans.
Bacteroidetes are the second largest group of the gut microbiota; however, they may also be considered as opportunistic pathogens. Bacteroides fragilis is very common in patients with IBD and can make up to 60% of the biofilm mass in these patients.49 A proinflammatory toxin-producing strain of this species, enterotoxigenic B fragilis (ETBF) has also been associated to IBD50 and induces colitis in conventional and germ-free mice.51 Another species from the same group, B vulgates, was found to be the most resistant among all tested bacteria to tissue extracts of from patients with Crohn's disease.52 Our finding of increased levels of Bacteroidetes in Nod2-deficient mice and humans further confirms their importance in the pathogenesis of IBD.
Nod2 represents the first and most well-replicated of the disease genes for Crohn's disease.53 A frameshift mutation leading to a partial truncation of the bacterial sensing leucine-rich region and other single-nucleotide polymorphisms (SNPs) within the leucine-rich region (R702W and G908R) have been associated with increased susceptibility17 18 and result in a diminished nuclear factor kappa B (NF-κB) activation upon stimulation of muramyl dipeptide and decreased release of the chemotactic cytokine interleukin-8, defensins and the immune exclusion molecule DMBT1 (deleted in malignant brain tumour 1),54 55 In conjunction with other proteins such as DUOX2 and GRIM19, Nod2 may itself function as an intracellular anti-bacterial factor which could shape microbial community profiles via local production of reactive oxygen species,42 56 Nod2-deficient mice have an increased susceptibility to oral infection with cytoinvasive Listeria monocytogenes, which correlates with a decreased expression of the murine α-defensin orthologues. In light of these earlier observations, an increased load of the major bacterial phyla of the intestinal microbiota attached to the mucosa in adult patients with Crohn's disease who are homozygous for SNP13 may be of high significance. Our study in mice indicates that a dysbiosis is present in Nod2-deficient individuals already at a very early age and affects the temporal development of a physiological gut flora. The findings suggest that Nod2 is an integral part of a co-evolved interaction between host and intestinal microbiota and highlight the importance of murine models in the annotation of a growing list of genes to be included in future molecular risk maps of disease.57 Deficient Nod2 function may result in a complex defect including defective regulation of defensin expression, impaired bactericidal capacity and inappropriate bacterial colonisation starting early in infancy. Given the early onset of Crohn's disease in Nod2-mutated individuals,58 future studies in human infants may help shed light on the development of this disease.
Acknowledgments
The authors would like to thank Manuela Kramp and Tanja Kaacksteen for excellent technical support.
References
Footnotes
AR, CS, SS and PR contributed equally to this study.
Funding This study was supported by an Infection Network grant of the BMBF (German Ministry for Education and Research) to PR, SO and SS, the DFG grant RO2994/5-1 and the Excellence Clusters Inflammation at Interfaces IRN G and Junior group Evolutionary Genomics and Future Ocean JRG B2.
Competing interests None.
Ethics approval This study was conducted with the approval of the Medizinische Fakultaet der Christian-Albrechts-Universitaet zu Kiel. All patient-related procedures were approved by the University Hospital ethics committee (B231/98).
Provenance and peer review Not commissioned; externally peer reviewed.