Article Text

Abstract
Knowledge of the immunological events necessary to control hepatitis B virus (HBV) infection has accelerated in recent years, but their translation towards therapeutic strategies able to achieve a durable HBV suppression has been challenging. The scenario of how HBV deals with the host immunity is presented and used to discuss how the immune response can be harnessed to potentially achieve infection control.
- Immunotherapy
- immunopathology
- hepatitis C
- hepatitis B
- Helicobacter pylori
- acid-related diseases
- non-ulcer dyspepsia
- genetic polymorphisms
- gastric neoplasia
Statistics from Altmetric.com
- Immunotherapy
- immunopathology
- hepatitis C
- hepatitis B
- Helicobacter pylori
- acid-related diseases
- non-ulcer dyspepsia
- genetic polymorphisms
- gastric neoplasia
Introduction
An efficient control of virus infections requires the coordinate action of both innate and adaptive immune responses. Innate immunity has evolved to rapidly recognise viral nucleic acids, viral proteins and tissue damage. It induces an antiviral state on infected cells by producing type I interferons (IFN), decreases the pool of infected cells by directing natural killer (NK) cell-mediated killing of viral infected cells, and supports the efficient maturation and site recruitment of adaptive immunity through production of pro-inflammatory cytokines and chemokines.1 These mechanisms reduce virus spread until the adaptive branch of immunity takes the stage. Adaptive immunity acts through functional maturation and expansion of distinct B and T cell clones able to specifically recognise the infectious agents, a process that necessitates time. This process leads to the control of infection and generates a memory response which increases the host ability to block subsequent infections with the same pathogens.
The different pathogens cause an assortment of clinical conditions, and they have evolved different strategies to escape host immunity; hepatitis B virus (HBV) has undoubtedly developed its own peculiar tactics.2
While most viruses enter a logarithmic phase of propagation immediately after infection (eg, hepatitis C virus (HCV), HIV, human cytomegalovirus (HCMV), influenza virus, dengue virus), HBV infection is characterised by a delayed amplification of HBV replication and spread. Febrile symptoms occur immediately in many acute viral infections, while acute HBV infection is mainly asymptomatic. Finally, while viral persistence is frequently associated with low viral load and protein expression (eg, HCV, HCMV, Epstein–Barr virus), chronic HBV infection is typically characterised by the production of extremely high quantities of viral proteins (HBsAg, HBeAg).3 ,4 We will summarise how HBV deals with host immunity and we will discuss how we can harness the immune response to potentially achieve infection control.
Innate immunity against HBV: lack of recognition or active inhibition?
Our knowledge of anti-HBV innate immune responses is still hampered by technical limitations. Data obtained during natural infection in patients are limited by the difficulty in recruiting patients at the earliest presymptomatic stages of acute HBV infection (reviewed in Bertoletti et al 2). In addition, an in vitro infection system for HBV in non-transformed hepatocytes is not widely available, its infection efficiency is poor and the level of HBV replication is low.5 ,6 Animal models of HBV infection can certainly provide good physiological data, but are hampered by ethical issues and high costs (chimpanzees), by a scarcity of reagents to analyse immunological events (woodchucks),7 ,8 and by technical difficulties to standardise the number of human hepatocytes grafted in chimeric mice.9
Taking into consideration these experimental limitations, an overall scenario of limited or even absent activation of innate immunity seems to be the hallmark of acute HBV infection. Pro-inflammatory cytokines are often undetectable during the early phases of infection (within the first 30 days) in humans infected with HBV and, when present, their production is of lower magnitude and delayed kinetics compared with HCV and HIV infected patients.10 A lack of induction of type I interferons was also seen in a cohort of 21 acute HBV patients, eight of whom were sampled from the inception of the viral expansion phase.11 These observations are in line with the lack of flu-like symptoms experienced by most patients, and support seminal results obtained in HBV infected chimpanzees, showing lack of induction of IFN-related genes during the phase of viral entry and expansion.12 In sharp contrast with these findings, rapid up-regulation of innate response genes was observed in animals infected with HCV,13 clearly indicating how two viruses with identical tropism utilise different strategies in their interaction with the host.
The apparent lack of induction of IFN-I response was interpreted as an ability of HBV to escape innate recognition.3 This could be the result of the replication strategy of HBV which uses a transcriptional template (cccDNA) that is sequestered within the nucleus of infected cells, where it might not be detected by the innate DNA sensing cellular machinery, and produces polyadenylate viral mRNA that resembles the normal cellular transcripts. Moreover, newly transcribed genomes are protected within viral capsides in the cytoplasm.3
Different lines of evidence have challenged this view and argue that HBV can actually be sensed by the innate immunity.14 For example, HBV replication in HepaRG cell lines (a cell line that is physiologically closer to normal hepatocytes and is permissive of HBV infection) activates IFN-β and other interferon-stimulated genes.15 In addition, acute infection with high doses of woodchuck hepatic virus (WHV) can induce immune genes immediately after infection,8 and Kupffer cells, despite not replicating the virus, seem to be able to sense HBV with up-regulation of interleukin-6 (IL-6) production.16 A note of caution about the physiological relevance of these data is however necessary because of the over-expression of HBV constructs in HepaRG cells to trigger antiviral innate immunity15 and the exceedingly high dose of WHV used to infect the animals.8 These are important parameters to consider since they might influence the results. For example, recent data on the mechanisms of intracellular DNA sensing suggest that the intracellular sensing machinery is triggered by the quantity rather than the ‘quality’ of DNA.17 Furthermore, the quantity of the infecting viral inoculum is known to be an important parameter that influences the outcome of infection in relation to the different triggering of the host immunity induced by different virus doses.18 ,19
Nevertheless, in support of the ability of the innate immunity to sense and react to HBV, a transient though slight activation of IFN-α genes was also detected in human hepatocytes infected by HBV in chimeric mice.20 Unfortunately, the mechanisms responsible for sensing HBV within the infected cells have not been elucidated yet, and which molecular components of the virus—HBV DNA, RNA or viral proteins—are actually recognised by the pattern recognition receptors (PRR) triggering the antiviral response is still undefined. The answer will be most likely found among the growing family of PRR able to discriminate intracellular pathogen DNA.21 In addition to cytosolic DNA receptors, like DAI, RIG-1, DHX9 (helicase) or AIM2 (schematic representation in figure 1), the recent discovery of immune interferon-16 (IFI-16) as an innate immune DNA receptor present in the nucleus is particularly interesting.17 IFI-16 senses the presence of the episomal circular DNA of Kaposi sarcoma-associated herpes virus in the nucleus of infected cells.22 It will be very interesting to test whether such a mechanism can also work in HBV infected cells.

Innate immune sensing of DNA. (A) DNA recognition by cytoplasmic DNA receptors. (i) AIM 2 mediates caspase-1 dependent pro-IL-1b and pro-IL-18 cleavage and secretion of their proactive forms. (ii) Four known cytoplasmic DNA sensors are represented (DAI, IFI-16, Rig-1, helicases DHX9/DH36). They initiate the signalling cascade that leads to the activation of transcription factors, such as IFN regulatory factor 3 (IRF3) and 7 (IRF7) and nuclear factor-kB (NF-k) that translocate in the nucleus to induce expression of interferons and pro-inflammatory cytokines. (B) Recognition of extracellular DNA. The receptor for advanced glycated end products (RAGE) can bind CpG-rich DNA and transport it to a TLR-9 compartment (endosomes). It signals trough Myd88 to induce IFN-α production. (C) Recognition of nuclear DNA. IFI-16 can sense viral DNA and activates transcription factors, such as NF-kB via the adaptor protein STING.17 ,21
Innate immunity against HBV: can HBV suppress the innate immunity?
How can we reconcile the opposite evidence of intracellular sensors of innate immunity capable of detecting HBV infection with the substantial weakness of the classical type I IFN/ pro-inflammatory response in acutely HBV infected patients and animal models? To explain such a conundrum, several studies have investigated the possibility that HBV can actively suppress instead of escaping the innate immunity through the action of different viral proteins. Recent work has, for example, shown the potential ability of HBV polymerase to inhibit IFN-β induction by interfering with interferon regulatory factor (IRF) signalling.23 ,24 Mechanistically, polymerase was shown to interact with DDX3, a transcriptional factor of the IFN-β promoter (figure 2).23–30 Since DDX3 acts downstream of the activation cascade triggered either by Toll-like receptor (TLR)-3 (trough TRIF) or by RIG-I (trough IPS-1), the results of these studies show that polymerase can block the activation mediated by these two receptors recognising dsRNA in the endosomes (TLR-3) or in the cytosol (RIG-I). Since the inhibition of IFN-β production mediated by HBV polymerase was obtained in HepG2 cells where the TLR-3 receptor was over-expressed and the TLR-3 and RIG-I triggering was obtained with heterologous inducers (poly I:C for TLR-3 and Sendai virus for RIG-I), caution is necessary in the evaluation of the physiological role of this mechanism.
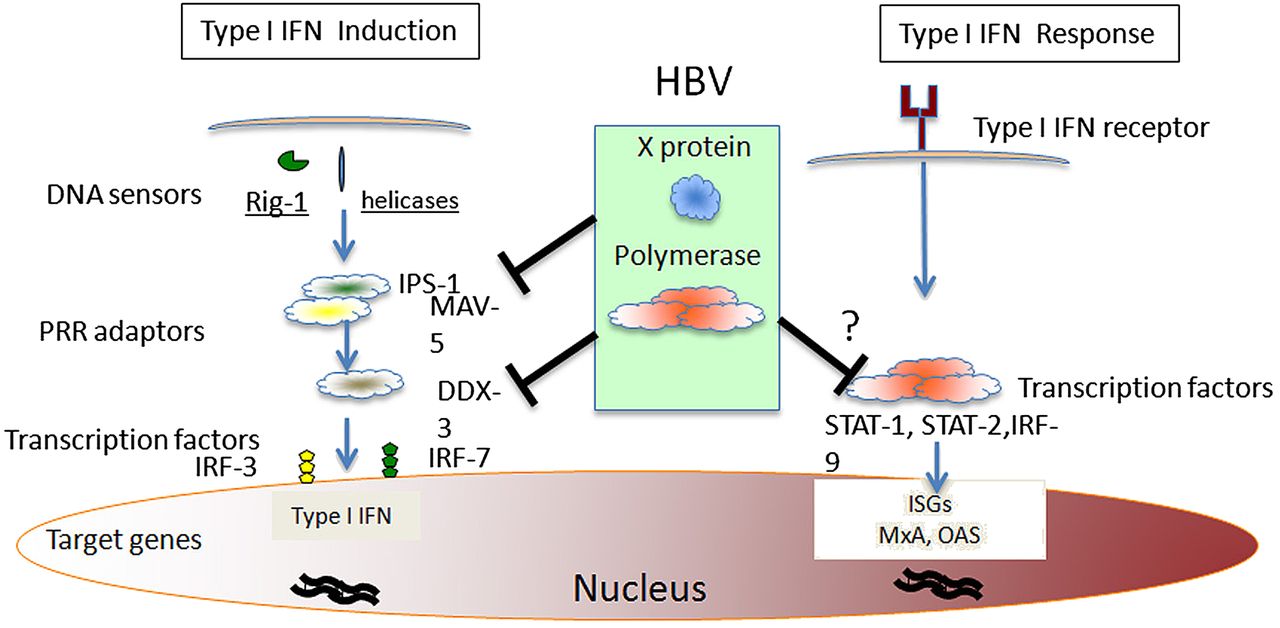
Molecular targets of different hepatitis B virus (HBV) proteins in host innate responses. Induction of intracellular type I IFN-production might be suppressed by HBV polymerase trough DDX-3 interaction23 ,24 or by HBV x protein through down-regulation of the mitochondrial antiviral signalling protein MAV-5.25–27 IFN-α induced response can be blocked by HBV polymerase.28–30 A potential mechanism is the block of STAT-1 nuclear translocation. No data are available on the potential mechanisms responsible for the suggested ability of HBsAg and HBeAg to inhibit innate immune response.
Furthermore, the HBV x protein has been reported to actively inhibit the innate immunity (figure 2) by interfering with signalling mediated by the cytosolic sensory molecules (RIG-I, helicases) through interaction with the β interferon promoter stimulator 1 (IPS-1) protein, also known as mitochondrial antiviral signalling (MAVS) protein or virus-induced signalling adaptor (VISA), which is essential for the induction of type I IFN.25–27 Also in these experimental models, intracellular IFN-β production was activated by heterologous inducers (poly dAT:dAT or poly I:C or vescicular stomatitis viruses), RIG-I and IPS-1 were over-expressed in HepG2 cells, while HBx/IPS-1 interaction was detected by over-expressing HBx. All these studies performed on HBV transfected cells are important to reveal potential clues on the interplay between HBV and innate immunity, and suggest that HBV, through its polymerase and x proteins, may affect IFN-β production by interfering at multiple sites of the intracellular signalling triggered by HBV infection. The real efficiency of these inhibitory mechanisms mediated by HBV proteins is however difficult to quantify and it will necessitate the use of a natural infection system.
HBV capacity to suppress innate immunity could not be only limited to the inhibition of type I IFN induction but could also extend to blocking its antiviral activity, thereby inhibiting the effect of IFN-α or -β produced by other non-infected parenchymal cells, including dendritic cells (DC) and Kupffer cells, or administered exogenously. Recent work in chimeric mice has, for example, shown that HBV infecting human hepatocytes prevents IFN-α mediated signalling by inhibiting nuclear translocation of STAT-1 (figure 2) and thus interfering with transcription of interferon stimulated genes (ISG).20 The cause of this inhibition could be related to the activation of intracellular IFN-type I response triggered directly by HBV. This activation could interfere with the response to exogenous IFN-α, as reported in hepatitis C infected patients.31 Alternatively, HBV interference with IFN-α signalling may be explained by the capacity of HBV proteins to affect STAT-methylation,28 and HBV polymerase has been shown in vitro to inhibit not only IFN-type I induction23 ,24 but also IFN-α and IFN-γ mediated cellular responses.29 ,30 Nevertheless, the relevance of such inhibitory phenomena is challenged by the lack of viral interference in the HBV/HCV co-infection model32 and by the fact that HBV/HCV co-infected patients respond to IFN-α therapy in vivo.33
Innate immunity inhibition in HBV infection can also result from the action of HBV proteins (HBsAg, HBeAg) actively secreted by HBV during its replication cycle. Secretory HBV proteins have been reported to abrogate the TLR-induced innate response34 and to modulate the surface expression of TLR-2.35 This effect should be mediated only by the secretory form of the HBV proteins since intracellular expression of envelope, core and e proteins did not inhibit the TLR-3 mediated triggering of IFN-β activity.23
The possibility that the secretory HBV proteins (HBsAg and HBeAg) can interact and modulate the inflammatory liver environment directly or through the induction of immunosuppressive cytokines, such as IL-10, is appealing since it could explain the evolutionary necessity of the virus to produce such a large quantity of secretory proteins. Anti-HBe+ patients manifest more aggressive liver disease, a clinical observation that could reflect the ability of HBeAg to down-modulate the pro-inflammatory liver environment. The production of HBeAg and HBsAg has been historically associated with an ability of the virus to interfere with adaptive immune mechanisms; in support of this, hepatocytes expressing both HBcAg and HBeAg have been reported to be more susceptible to cytotoxic T lymphocytes (CTL)-mediated lysis than hepatocytes expressing only HBcAg, suggesting that precore variants unable to produce HBeAg should have a selection advantage over wild type viruses able to produce both HBcAg and HBeAg.36 An additional effect of the secretory HBV proteins on innate immunity cannot however be dismissed.
A final component of innate immunity, NK cells, needs to be discussed. NK cells can recognise and kill viral infected cells, and their effector function is determined by the balance between positive and negative signals released from activating and inhibitory receptors. Loss of major histocompatibility complex (MHC) class I on the surface of virally infected cells, along with up-regulation of host or pathogen encoded ligands that signal cell stress, optimise NK cell recognition. NK cells can also be directly activated by cytokines induced in viral infections, such as IFN-I, IL-12 and IL-18.37
What role do NK cells play in HBV infection? Even without an appropriate induction of pro-inflammatory cytokines directly triggered by HBV infection, NK cells remain well poised to respond to acute HBV infection in the liver. Hepatocytes express very low levels of MHC class I,38 such that any up-regulation of cellular stress ligands able to engage NK activatory receptors should be able to induce local NK cell effector function. Furthermore, NK cells are extremely abundant in the normal liver, constituting 30–40% of intrahepatic lymphocytes. In chimpanzees, the clearance of HBV infected hepatocytes by adaptive immunity was found to be preceded by an increase in intrahepatic IFN-γ and tumour necrosis factor-α (TNF-α),39 which could be produced by NK cells. However, subsequent experiments showed a critical role for T cells rather than NK cells in HBV control in this model.40 Studies of patients around the time of first detection of surface antigen and HBV-DNA revealed an increase in the number of circulating NK cells,41 ,42 but their activation and effector function was suppressed as viral load increased, and only peaked once viraemia had resolved.11 This inhibition of NK cell activation and effector potential was temporally associated with an induction of IL-10, again raising the possibility that HBV can actively evade immune responses. However, little data are available regarding the involvement of NK cells in the immediate response to infection, and only recent results obtained in woodchucks infected with high WHV dose (1011) show an activation of a gene related to NK cell activation immediately after infection (8–12 h).8 It therefore remains possible that NK cells contribute to HBV control in the earliest lag phase of infection, but such a hypothesis still awaits proper demonstration.
The involvement of NK cells in chronic HBV infection has been investigated in a number of recent studies43–45 suggesting that they could play a role in liver damage during reactivation, even though secretion of immunomodulatory cytokines, like IL-10, can suppress their cytokine secretion.45 In contrast, the NK cytotoxic potential in CH-B patients seems to be intact or even augmented.46 Thus, the NK cell role in HBV persistence and the mechanisms that regulate NK lysis of HBV-infected cells are still controversial.2
Importantly, the recent correlation of NK cell activation with IFN-α treatment success in HCV infected patients47 suggests the need to test whether a similar mechanism can also be operative in IFN-α treated chronic hepatitis B (CHB) patients. Similar studies investigating the early NK cell activation in CHB patients responding to IFN-α therapy could clarify the importance of NK cells in HBV control and support therapeutic strategies aimed at activating intrahepatic NK cells through the use of cytokines (IFN-α, but also IL-12 and IL-18) administered directly or produced by activating Kupffer cells with TLR-agonist.48
Overall, it seems clear that a better understanding of the ability of the innate immune response to control HBV infection during natural infection awaits data generated in more physiological systems. It is, for example, important to remember that analysis of the anti-HBV efficacy of IFN-α in HepG2 or Huh-7 hepatocyte-derived lines may be influenced by the fact that such transformed cells have defects in IFN-signalling pathways; and, as we discussed, over-expression of viral DNA or proteins or of different pathogen recognition receptors on hepatocytes like cells can make the interpretation of the results obtained in such models difficult. At the moment, the majority of data seem to indicate that during natural HBV infection induced by low infectious dose and characterised by the typical kinetics of HBV amplification, innate immune activation of an intracellular antiviral response is absent or weak, possibly due to a combination of inefficient triggering and active HBV-mediated suppression of the innate immunity. Interestingly, the inability to detect innate immune activation is not only restricted to acute infection, but it is observed also during HBV reactivation after therapy withdrawal.49
As a possible consequence of the limited and transient activation of the innate immunity during primary HBV infection, the mechanisms adopted by HBV to suppress the antiviral and pro-inflammatory effect of the innate immunity, even though present, do not seem to be particularly robust since, for example, exogenous TLR-mediated activation can suppress HBV replication in HBV transgenic mice50 and in HBV transfected HepG2 and Huh7 cells.51 It therefore seems logical, as a therapeutic strategy, to consider the possibility to stimulate the weak innate immune response to improve its antiviral effect. In this respect, the therapeutic efficacy of TLR-receptor stimulation with TLR-agonist has been reported in HBV infected woodchucks52 and chimpanzees.53 To further maximise intrahepatic innate immune activation, strategies to specifically deliver antiviral cytokines to the infected liver or to target activation of intrahepatic Kupffer and NK cells should be explored. The discovery of peptides able to specifically bind hepatocytes54 and the production of antibodies with HBV-infected cell specificity55 might provide the tool to target cytokines and TLR-agonist into the HBV infected liver.
Since the potential rapid release of pro-inflammatory cytokines (such as IL-12, IL-18 and TNF-α) might potentially trigger liver cell injury, the study of the effect of cytokines and TLR-receptor activation on the liver microenvironment should be extended to identify the balance between viral control and liver injury and a careful evaluation of new therapies in animal models will be necessary.
Adaptive immunity against HBV: acute and chronic HBV infections
While the role played by innate responses in initial HBV control is still debated, quality and strength of early adaptive immunity are generally seen as a representative model of the immune response that must be mounted to control a virus infection efficiently.
HBV-specific CD4- and CD8-mediated responses become generally detectable immediately after the start of the exponential increase in HBV replication, which follows an initial phase of negative or weakly positive HBV-DNA lasting for about 4–7 weeks after infection.41 ,42 Therefore, HBV-specific T cell responses are timely activated and only apparently delayed relative to the time of infection because of the initial quiescence of HBV,41 ,56 which does not provide sufficient stimulation to prime and expand HBV-specific T cell responses. These responses are typically multi-specific, Th1 oriented, polyfunctional and much stronger than those detectable in chronic infection.2–4 ,57
In self-limited infections, HBV-DNA falls by more than 90% within 2–3 weeks after the peak of viral replication and before detection of liver damage, indicating that a large quantity of virus is eliminated without the need of liver cell destruction by non-cytopathic mechanisms sustained by cytokines, including IFN-γ and TNF-α, secreted by CD8 T cells.39 ,58 Intrahepatic recruitment of HBV-specific CTL, that is facilitated by the secretion of chemokines (like CXCL-10) and by platelet activation,59–62 also leads to killing of infected hepatocytes with subsequent recruitment of antigen non-specific cells that amplify hepatocellular damage (which is initially triggered by HBV-specific mechanisms).
As predicted by the accepted models of T cell differentiation,63 ,64 when infection is successfully controlled, maturation of T cell memory occurs efficiently, as indicated by the increasing expression of CD127 molecules and the decline of programmed death (PD)-1 on HBV-specific CD8 cells following resolution of infection.65 ,66 This stage is, however, preceded by a functional HBV-specific CD8 T cell impairment which is detectable at the peak of disease when the majority of HBV-specific CD8+ T cells are activated but are poorly able to proliferate and functionally exhausted.11 ,67 This functional decline has been reported to be associated with a peak of IL-10 production,11 but could also be caused by the increased levels of arginase released by dying hepatocytes.68 By depleting the essential amino acid L-arginine, arginase contributes to the down-regulation of the CD3ζ-chain on T cells and thus causes T cell receptor signalling impairment, resulting in the suppression of T cell functions.69 These mechanisms of acute phase suppression can represent a homeostatic process common to many virus infections,70 finalised to avoid excessive immunopathology and to favour T cell contraction.
While infections acquired in the adult life are generally self-limited, most infections acquired at birth or perinatally become chronic. However, irrespective of the time in life when infection occurs, HBV persistence is always associated with defective HBV-specific CD4 and CD8 T cell functions. The intensity of the T cell response appears inversely correlated to viraemia levels,65 ,71 ,72 thus suppression of HBV-specific T cell responses is more profound in highly viraemic patients and T cells are more dysfunctional within the liver than in the periphery.73
The defective T cell function is probably maintained primarily by the effect of the prolonged exposure of T cells to high quantities of viral antigens and by the tolerogenic features of hepatocytes and liver resident cells.74–77
These two combined mechanisms can result in the deletion of HBV-specific T cells or in their functional inactivation (exhaustion), which is characterised by an increased expression of negative co-stimulatory molecules and a dysregulation of co-stimulatory pathways, such as PD-1/PD-L1,65 ,73 ,78 ,79 which affect the quality and intensity of the antiviral T cell response. Lack of T cell help80 can also contribute to the defective functionality of HBV-specific CD8 T cells present in chronic patients.
Other mechanisms, like impairment of T cell receptor signalling by CD3ζ-chain down-regulation69 and enhanced T cell apoptosis caused by Bcl2-interacting mediator (bim) up-regulation80 also contribute to alter the T cell function. Interestingly, addition of L-arginine can restore CD3ζ expression and can improve T cell function.69 Since L-arginine depletion due to increased arginase release is more pronounced during liver inflammation,68 the metabolic conditions of the liver microenvironment can contribute to further modulate the already altered HBV-specific T cell function in chronic HBV patients.
The extended T cell family: Th17 and Th22
Analysis of HBV-specific T cells in acute and CHB has been mainly focused on T cells producing IFN-γ, TNF-α and IL-2 (so called Th1/Th0 cytokine profiles). However, an extended family of distinct T cells is now recognised as an integral part of the immune system.81 Some of these T cells (like Treg and T cells producing IL-10 or TGF-β) have immunoregulatory roles, other T cells are more pro-inflammatory (like T cells producing IL-17 or IL-8), or have pro-fibrotic effects (Th2 and T cells producing IL-13), while cells producing IL-22 have been reported to have a hepatoprotective effect.82 ,83
What is the role of these specific families of T cells during hepatitis B? Many studies have shown that Treg, Th17 and Th22 cell frequencies are higher in patients with chronic hepatitis than in healthy subjects.84–87 T cell populations able to produce IL-17 and IL-22 are often enriched in the intrahepatic environment and can carry CD161 and CXCR-6 receptors, a phenotypic profile that is enriched in tissue-homing T cells.88 These correlations do not however clarify the role of such cells in the pathogenesis of HBV infection. Treg cells can suppress, at least in vitro, HBV-specific T cell functions,86 ,89 but such an effect is also observed in patients that are perfectly able to control the virus.90 Furthermore, Treg frequency is correlated to ALT levels,86 suggesting that Treg might preferentially have an anti-inflammatory effect without playing a role in HBV persistence.
Th17 cells, a cell population which can play an important role in the pathogenesis of autoimmunity and other inflammatory diseases, are detectable at higher frequency in CHB patients with severe liver damage.85 In contrast, in HCV infection the intrahepatic frequency of T cells producing IL-17 has been linked to mild hepatic disease.88 A potential explanation of such controversial data is the inherent plasticity of cytokine production by T cells. Th17 producing cells can co-express IL-22, a cytokine that should have a prominent hepatoprotective role,82 even though a recent work shows that IL-22 has a major pro-inflammatory role in HBV-transgenic mice.91
It is currently unknown what the necessary stimulus is to trigger T cell production of IL-17 and IL-22 during HBV infection. Even though HBV-specific IL-17 producing cells were initially reported,85 more recent data failed to detect IL-17-producing HBV-specific T cells in acute and chronic HBV patients, both in the periphery and in the intrahepatic environment.92 In contrast, HBV-specific T cells maturing in the intrahepatic inflammatory environment can produce CXCL-8 (IL-8),92 a cytokine that has a pro-inflammatory effect93 and that can contribute to the development of liver pathology through the recruitment of granulocytes.94 Overall, with the exception of some interesting correlative analysis, we are far from having a clear understanding of the impact that T cells with regulatory, pro-inflammatory and hepatoprotective effects have on the control of HBV infection and on the pathogenesis of chronic viral hepatitis. Although such cells are not HBV-specific, they should theoretically play an important role in the modulation of liver inflammation during viral hepatitis; thus, they deserve a better characterisation since modulation of their effect may be exploited for future immunotherapies.
Reconstitution of the T cell function
The functional alterations of virus-specific immunity in chronic hepatitis B patients suggest that their functional restoration could result in the resolution of infection. Antigen persistence is a major factor driving the functional alteration of HBV-specific T cells, and thus antigen removal to allow T cell resting can be an essential requirement for the functional reconstitution of antiviral T cell responses.2 ,63 ,95 To what extent is the virus-specific T cell dysfunction reversible in chronic active infections?
Unfortunately, the possibility that T cells chronically exposed to antigen carry permanent changes in their differentiation programme as a permanent ‘signature’ of this long-term contact with antigen cannot be ruled out at present. For example, data in the lymphocytic choriomeningitis virus model of infection indicate that adoptive transfer of dysfunctional virus-specific CD8 cells from a chronically infected to a naïve uninfected MHC compatible animal is not sufficient to restore the T cell memory maturation and differentiation process, even if antigen is totally absent.64 Moreover, these experimental data are confirmed by recent studies in patients with chronic hepatitis C treated with peginterferon-α/ribavirin therapy, indicating that a complete functional T cell restoration cannot be achieved after sustained virologic response, even after long time of complete control of virus replication.96
Control of virus replication in chronic hepatitis B by antiviral therapy can induce only a partial improvement of the T cell function. In most cases such limited recovery can be explained because high concentrations of HBV envelope antigens remain detectable for long time after HBV-DNA suppression. Therefore, it is not surprising that IFN-α therapy cannot restore an efficient antiviral T cell response in the peripheral blood of naïve anti-HBe+ patients with chronic hepatitis B during the first 6 months of therapy (personal communication), despite its known modulatory function on the immune system. In line with this interpretation, a late effect of IFN therapy on proliferative HBV-specific T cell responses has been reported by Sprengers et al 97 and Carotenuto et al,98 who observed significant differences between responder and non-responder patients only during the final weeks of therapy (between weeks 44 and 56). Similar results were reported recently in a cohort HBeAg positive tolerant children with infancy-acquired HBV infection.99 Restoration of HBV-specific T cell responses (both CD4 and CD8) in lamivudine treated HBeAg+ highly viraemic patients is also transient,100–102 and recent studies in nucleoside/nucleotide analogue (NUC) treated anti-HBe+ chronic active hepatitis patients with persistent suppression of HBV replication indicate that despite a gradual and persistent recovery of HBV-specific T cell functions proportional to the HBsAg decline, such recovery remains frequently incomplete even when HBsAg clearance and anti-HBs antibody production are achieved (personal communication).
If antigen reduction is not sufficient to obtain a robust functional recovery, it is likely that additional strategies that directly target the activation defects of HBV-specific T cells should be implemented to achieve a full HBV-specific T cell recovery (summarised in table 1).50–53 ,65 ,73 ,78 ,79 ,100–109 One strategy is to directly target the negative co-stimulatory pathways involved in the pathogenesis of T cell exhaustion. Among the different co-stimulatory molecules, PD-1 is highly expressed on HBV-specific T cells,65 ,73 ,78 ,79 especially within the liver,73 and the effect of PD-1/PD-L1 blocking by anti-PD-L1 antibodies has been reported to be greater on intrahepatic than on peripheral T cells in chronic HBV infection.73 Importantly, different co-stimulatory pathways that have been analysed more thoroughly in human HIV and HCV infections as well as in different animal models of chronic viral infection, including CD137/CD137L, CTLA4/CD86-80, CD27/CD70, CD40/CD40L, CD134/CD134L, CD160/HVEM complex, 2B4/CD48, CD223-LAG3/class II MHC and TIM-3/GALECTIN 9 phosphatidyl serine, seem to be non-redundant in the pathogenesis of T cell exhaustion.110 Based on this, recent studies have been devoted to assess whether combined blockade or stimulation of different receptor/ligand interactions can be synergistic in restoring the HBV-specific T cell function. Thus, the co-inhibitory 2B479 and cytotoxic T lymphocyte antigen-4 (CTLA-4)78 and the co-stimulatory CD137 (personal communication) pathways were tested alone or in combination with PD-1/PD-L1 blockade to optimise functional T cell restoration. Results indicate that the effect of these pathways on HBV-specific T cell exhaustion is non-redundant, with patients responding better to manipulation of one or another pathway and some responding better to combined manipulation. This is promising in the perspective of novel strategies of immunotherapy, but understanding better how different regulatory pathways can act and synergise is essential for the design of immunotherapies able to act selectively on the inhibitory mechanisms directed against HBV-specific responses, avoiding the risks related to generalised reconstitutions of T cells of different specificities.110 In this respect, stimulation of molecules which are selectively up-regulated on activated T cells, such as CD137,111 may be of particular interest as an adjuvant strategy to apply in combination with HBV-specific T cell stimulation, for example with anti-HBV vaccines.
Strategies designed to increase the efficacy of immune-based therapy
A different strategy to restore the HBV-specific T cell function is to manipulate the liver microenvironment that can inhibit the intrahepatic T cell function through the effect of suppressive cytokines, such as IL-10103 ,104 or TGF-β,105or inhibitory Treg cells.106 In this respect, another interesting cytokine that is preferentially produced by follicular helper T cells is IL-21, which has been shown, at least in transgenic mice, to allow control of HBV replication, by likely contributing to reverse the defective HBV-specific T helper cell response.112
All these strategies would essentially attempt to reverse a defective immune response and thus re-programme HBV-specific T cells. However, breaking immunological tolerance has been shown to be difficult in different clinical settings, and a global restoration of efficient HBV-specific T cell responses able to target liver infected cells may be dangerous, and need to be tightly regulated.113 Instead of relying on the functional restoration of HBV-specific T cells, new approaches have been proposed to target HBV-infected hepatocytes by fully functional memory T cells specific for HBV-unrelated antigens, thereby circumventing HBV-specific T cell hyporesponsiveness. To do this, an HBV pseudovirus carrying influenza antigens was generated; since it is able to express influenza antigenic epitopes only in HBV infected cells, it can make HBV infected hepatocytes visible to influenza-specific memory T cells.109
As an alternative, different groups have successfully engineered fully functional HBV-specific T cells, by transfection of normal memory T cells with HBV-specific T cell receptors derived from functionally efficient T cells108 or with chimeric antigen receptors (T-bodies) that fuse the antigen specific determinants of an anti-HBs antibody to the signalling domain of the T cell receptor.114 These newly generated T cell responses might supplement the deleted or functionally deficient HBV-specific T cell responses of chronic patients and the quantity of adoptively transferred T cells could be tightly regulated in vivo. Many obstacles will have to be overcome before technologies such as adoptive transfer of HBV-specific T cells will reach the clinic, but their potential is obvious. Adoptive transfer of Epstein–Barr virus or HCMV-specific T cells is used during viral reactivation of patients under immunosuppressive therapy, and the clinical efficacy of infusing T cell receptor-redirected T cells in patients with human tumours has been demonstrated.115
In conclusion, the increased knowledge of the innate and adaptive immunity during viral hepatitis B has led to the development of new approaches designed to trigger the defective innate immunity or to restore the exhausted adaptive immunity that characterise this infection (figure 3). Some of the sophisticated methods based on reconstitution of HBV-specific T cell immunity through T cell receptor transfer,108 ,114 the use of human monoclonal antibodies targeting HBV infected cells55 or blocking pro-apoptotic pathways to bypass the HBV-specific T cell defects,107 are technically too complex to be implemented, but others, like the use of TLR agonists,50–53 are likely to be tested soon in clinical trials. Current antivirals will continue to be the standard of care for the foreseeable future due to their efficacy and affordability. However, their substantial inability to achieve HBV clearance or sustained HBV control after therapy interruption, and the potential threat of selecting HBV variants,116 advocate their association with new therapeutic strategies aimed at boosting immune-mediated viral control.

{kind=link}
{kind=link}
{kind=link}
How to apply immunomodulation of T cell responses to hepatitis B virus (HBV) therapy in practice? Since T cells in chronic active hepatitis B are exhausted and a primary cause of this condition is their persistent exposure to high antigen doses, the first goal of therapy should be to restore the antiviral T cell function by inducing a decline of antigenaemia with nucleoside analogues. This may be not sufficient and it may need to be implemented with additional strategies to improve protective immune responses, by manipulating negative co-stimulatory pathways involved in the pathogenesis of T cell exhaustion, such as PD1, or suppressive cytokines, such as IL-10 or TGF-β, or inhibitory Treg cells. Once restoration of T cell responsiveness has been at least partially achieved, other immunostimulatory compounds, such as IFN-α or anti-HBV vaccines, which would be most likely ineffective in naïve patients because of functional T cell paralysis, should acquire optimal stimulatory capacity and should thus be considered in order to achieve complete virus control. As an alternative, the overall antiviral T cell function could be improved by engineering fully functional autologous T cells to render them capable of recognising virus infected cells without suffering from the inhibitory effect of the host environment. Both approaches can run the risk of inducing severe liver pathology if the quantity of antigen and the number of infected liver cells is high; for this reason, all these strategies should be applied when HBV replication is persistently suppressed and when HBsAg levels have already consistently declined under nucleoside analogue therapy.
As discussed, inhibition of viral replication with antiviral therapies has been shown to induce a partial restoration of antiviral immunity (figure 3). Nevertheless, nucleoside analogues should be included in clinical trials aimed at testing new immune based therapies. Reduction of HBV antigen expression in hepatocytes and amelioration of liver inflammation is important to create the liver micro-environment where therapeutic strategies designed to stimulate intrahepatic innate immunity or to restore HBV-specific immune responses can be more safely tested. Fatal outcomes are observed in patients after adoptive transfer of immunity or after withdrawal of cytotoxic chemotherapy,117 ,118 but the prophylactic suppression of viral replication with nucleoside analogues should prevent the exacerbation of hepatitis B and liver-related morbidity and mortality.119 ,120
Better understanding of the different aspects of HBV pathogenesis has provided the rationale to modulate immunity with the goal of achieving long-lasting control of HBV. This is therefore a stimulating time for scientists and clinicians who will embrace the challenge to design and test new therapies based on the rational combinations of antiviral and immunomodulatory compounds.
Acknowledgments
We wish to thank the staff of the Viral Hepatitis Laboratory, Singapore Institute for Clinical Sciences, and Unit of Infectious Diseases and Hepatology, Azienda Ospedaliero-Universitaria di Parma for their help and comments. We regret that many important papers could not be cited due to space constraints.
References
Footnotes
-
Funding Fondazione Cassa di Risparmio di Parma, Italy, by an FIRB grant from the Italian Ministry of the University and Research, Protocol RBAP10TPXK and by a programme grant of the Agency of Science Technology and Research (A*STAR), Singapore.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.