Article Text

Abstract
Objective In human chronic liver disease, there is association between ductular reaction (DR) and fibrosis; yet, the mechanism triggering its onset and its role in scar formation remains unknown. Since we previously showed that osteopontin (OPN) is highly induced during drug-induced liver fibrosis, we hypothesised that OPN could drive oval cells (OC) expansion and DR and signal to hepatic stellate cells (HSC) to promote scarring.
Results In vivo studies demonstrated increased OPN expression in biliary epithelial cells (BEC) and in OC in thioacetamide (TAA)-treated mice. OPN ablation protected mice from TAA and bile duct ligation-induced liver injury, DR and scarring. This was associated with greater hepatocyte proliferation, lower OC expansion and DR along with less fibrosis, suggesting that OPN could activate the OC compartment to differentiate into BEC, which could then signal to HSC to enhance scarring. Since TAA-treated wild-type mice and cirrhotic patients showed TGF-β+ BEC, which were lacking in TAA-treated Opn−/− mice and in healthy human explants, this suggested that OPN could regulate TGF-β, a profibrogenic factor. In vitro experiments confirmed that recombinant OPN (rOPN) decreases hepatocyte proliferation and increases OC and BEC proliferation. To evaluate how BEC regulate collagen-I production in HSC, co-cultures were established. Co-cultured BEC upregulated OPN and TGF-β expression and enhanced collagen-I synthesis by HSC. Lastly, recombinant TGF-β (rTGFβ) and rOPN promoted BEC proliferation and neutralisation of OPN and TGF-β reduced collagen-I expression in co-cultured HSC.
Conclusions OPN emerges as a key matricellular protein driving DR and contributing to scarring and liver fibrosis via TGF-β.
- Cell Matrix Interaction
- Extracellular Matrix
- Fibrogenesis
- Hepatic Fibrosis
- TGF-Beta
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
In human chronic liver disease, there is association between ductular reaction (DR) and fibrosis; yet, the mechanism triggering its onset and its role in scar formation remains unknown.
-
It still remains enigmatic why fibrillar collagen deposition does not necessarily occur at the immediate sites of lobular injury. It is rather paradoxical that damage to hepatocytes within lobules induces fibrosis predominantly in portal areas.
-
When the liver parenchyma is damaged, oval cells (OC) become a source of regenerating hepatocytes, biliary epithelial cells (BEC) and draining ductules in order to restore the functional liver mass.
-
Proliferating BEC could be a source of molecules that activate extracellular matrix deposition and secrete mediators, which could attract and activate hepatic stellate cells (HSC) and portal fibroblasts; yet, these factors are largely unknown.
What are the new findings?
-
Osteopontin (OPN) expression increases during liver injury and under oxidative stress conditions.
-
Genetic ablation of Opn increases hepatocyte proliferation and decreases OC expansion, DR and fibrogenesis in response to thioacetamide treatment or bile duct ligation.
-
BEC signal to HSC via OPN and TGF-β to induce collagen-I deposition, hence creating a profibrogenic loop.
How might it impact on clinical practice in the foreseeable future?
-
Preventing or limiting DR and the induction of profibrogenic factors that BEC release during the progression of liver disease, such as OPN and TGF-β, represents a novel first line of defence to prevent fibrogenesis in portal areas. In addition, these studies addressing the interaction of BEC with HSC reveal potential novel signalling mechanisms that could be simultaneously targeted for therapeutic benefit.
-
Myofibroblast activation and periportal fibrosis may not simply be a consequence of DR but rather are likely to be important players amplifying the alternative regenerative pathway since HSC and portal fibroblasts also secrete factors that could promote DR.
Introduction
Although considerable progress has been made in our understanding of the pathogenesis of hepatic fibrosis, it remains enigmatic why fibrillar collagen deposition does not necessarily occur at the immediate sites of lobular injury. It is rather paradoxical that damage to hepatocytes within lobules induces fibrosis predominantly in portal areas. Recent studies have suggested that altered hepatic regeneration along with oval cells (OC) reaction could play a major role in periportal fibrosis, a recognised event highly associated with disease progression.1 ,2
While in normal liver, replacement of necrotic and apoptotic hepatocytes occurs by replication of quiescent adjacent hepatocytes within the lobule, this primary pathway is easily altered by a variety of insults such as toxins, viral infection, steatosis, oxidative stress and alcohol consumption.2–,4 When periportal hepatocytes are damaged and their proliferation is impaired, a pool of OC arises acting as a secondary proliferative pathway.5 ,6 OC are bipotential cells residing primarily in the periportal region, and when the liver parenchyma is damaged they become a source of regenerating hepatocytes, biliary epithelial cells (BEC) and draining ductules in order to restore the functional liver mass. 4–,7 A by-product from the activation of this alternative proliferative pathway is ductular reaction (DR)2 ,8; a reactive lesion at the portal tract interface comprising small biliary ductules with the associated stroma, bile plugs and inflammatory cells.9
Several studies have pointed at the possible involvement of the biliary epithelium in fibrosis associated with chronic liver disease.10 ,11 Indeed, DR has been tagged ‘the pace-maker of portal fibrosis’ since proliferating BEC are a source of molecules that activate extracellular matrix (ECM) deposition and secrete pro-inflammatory and chemotactic cytokines, which attract and activate hepatic stellate cells (HSC) and portal fibroblasts.12–,14 These and other, yet to be discovered mediators trigger a fibrogenic response and contribute to the progression of liver fibrosis. Moreover, inflammatory cells, by secreting matrix metalloproteinases, degrade the ECM that physiologically surrounds bile ducts. This pathological alteration of the liver milieu serves as a powerful stimulus to increase myofibroblast proliferation and fibrillar collagen deposition, hence contributing to the fibrogenic response to liver injury.
While some current therapies have proven beneficial, dissecting key profibrogenic events, pathways and/or mediators of disease progression is vital. Several studies, including our own, have indicated that osteopontin (OPN) is significantly upregulated during liver injury, particularly in OC, BEC, hepatocytes and HSC.15–,19 OPN, a matricellular soluble protein and a matrix-bound phosphoglycoprotein, can remain intracellular or can be secreted to the extracellular space allowing autocrine and paracrine signalling within tissues.20 ,21 OPN functions as adaptor and modulator of cell–cell and cell–matrix interactions.21 Moreover, OPN regulates cell migration, adhesion and ECM invasion due to its ability to bind integrins via its RGD motif or to CD44 via a cryptic site (SVVYGLR) exposed after proteolytic cleavage18 ,22; thus, it plays a relevant role in tissue remodelling, cell survival and chemoattracting inflammatory cells.23
Upon the onset of liver injury, OPN is highly induced. Since sustained impaired hepatocyte proliferation leads to OC activation and the resulting DR could promote portal fibrosis, we hypothesised that OPN could play a significant role driving DR, which could contribute to the pathogenesis of liver fibrosis by regulating collagen-I deposition by HSC. To test this hypothesis, we used two in vivo models of liver fibrosis where significant DR occurs, thioacetamide (TAA) administration and bile duct ligation (BDL), and correlated Opn ablation with the extent of hepatocyte proliferation, DR, BEC proliferation and fibrosis. In vitro studies evaluated the role of OPN in regulating BEC proliferation and motility and identified BEC-derived OPN and TGF-β as inducers of HSC collagen-I protein expression.
Experimental procedures
Mice
Opn−/− mice (B6.Cg-Spp1tm1Blh/J) and their matching wild-type (WT) littermates (C57BL/6J) were obtained from the Jackson Laboratories (Bar Harbor, Maine, USA). A targeting vector containing the neomycin resistance cassette and the Herpes simplex virus thymidine kinase gene was used to disrupt exons 4 through 7 of the targeted gene. The targeted mutation deleted the coding region of the Opn gene.24 The resulting chimeric animals were backcrossed to C57BL/6J for 10 generations. 129sv Opn−/− mice, generated with a different targeting strategy, along with their matching WT littermates were donated by Dr Denhardt (Rutgers University, New Jersey, USA)25 and were used in validation studies described in online supplementary figure S2. In both cases, mice were generated by intercrossing Opn+/− mice and littermates were used in all experiments.
Induction of liver injury
Ten-week-old male WT mice and their Opn−/− littermates and two in vivo models of liver injury were used in this study. In the first model, drug-induced liver injury was provoked by treatment with TAA (300 mg/L, Sigma, St. Louis, Missouri, USA) in the drinking water for either 2 or 4 months while controls received equal volume of water. Mice were sacrificed 48 h after TAA withdrawal. In the second model, cholestasis was induced by ligating the common bile duct, whereas controls were sham operated. Mice were sacrificed 8 days later. All animals received humane care according to the criteria outlined in the ‘Guide for the Care and Use of Laboratory Animals’ prepared by the National Academy of Sciences and published by the National Institutes of Health. Protocols were approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee.
Please see online supplementary experimental procedures.
Results
OPN expression increases during liver injury and under oxidative stress conditions
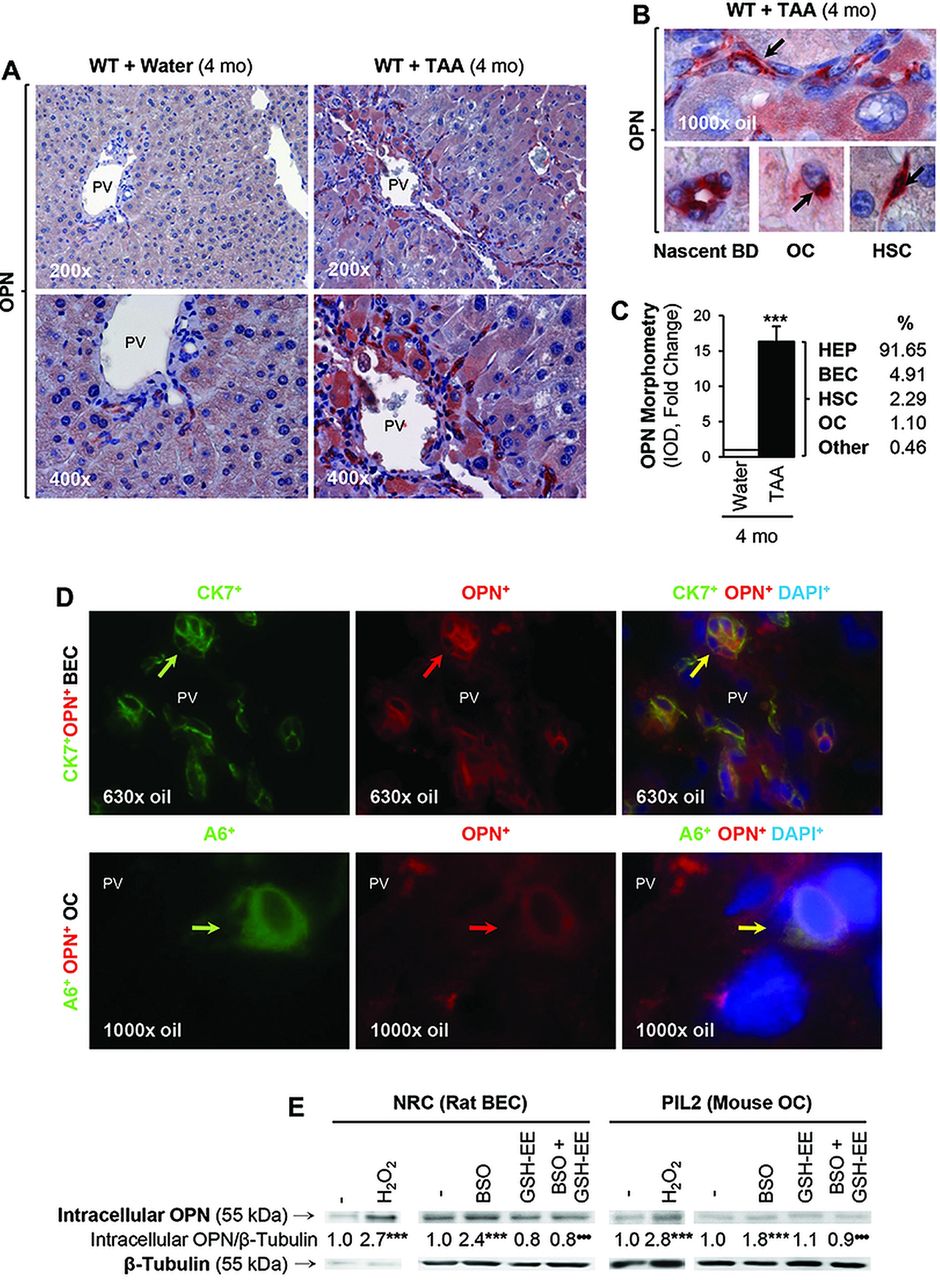
We have previously described correlation between OPN induction and the extent of liver fibrosis in chronic CCl4-treated mice and in Hepatitis C virus patients.15 To further identify the source of OPN during liver injury, we used a well-established in vivo model to induce liver fibrosis in mice based on chronic TAA administration. TAA undergoes cytochrome P450 metabolism leading to significant generation of reactive oxygen species (ROS), hepatocyte necrosis, inflammation and DR. Immunohistochemical (IHC) analysis revealed OPN induction in hepatocytes, BEC, OC and HSC in TAA-treated WT mice (figure 1A,B), and the morphometric analysis showed a 16-fold increase in OPN protein in TAA-treated WT mice compared with control mice (figure 1C). Whereas expression in control WT mice was almost entirely hepatocyte and BEC-derived, a significant increase was detected in OC, BEC and HSC from TAA-treated WT mice (figure 1C). There was co-localisation of OPN+ staining with CK7+ or CK19+ (not shown) (both biliary epithelial cell markers) (figure 1D, top), with A6+ (an OC marker) (figure 1D, bottom) and with α-smooth muscle actin (α-SMA+) (a HSC activation marker, shown in ref. 15) in the TAA model. Furthermore, OPN expression was elevated in NRC (rat BEC) and in PIL2 (mouse OC) by H2O2 treatment, a prooxidant typically generated during TAA metabolism, and by L-buthionine sulfoximine (BSO), which depletes glutathione, whereas co-treatment with glutathione-ethyl ester to replenish glutathione stores blunted the BSO-mediated induction of OPN (figure 1E). These data proved upregulation of OPN in response to TAA treatment and to oxidative stress.
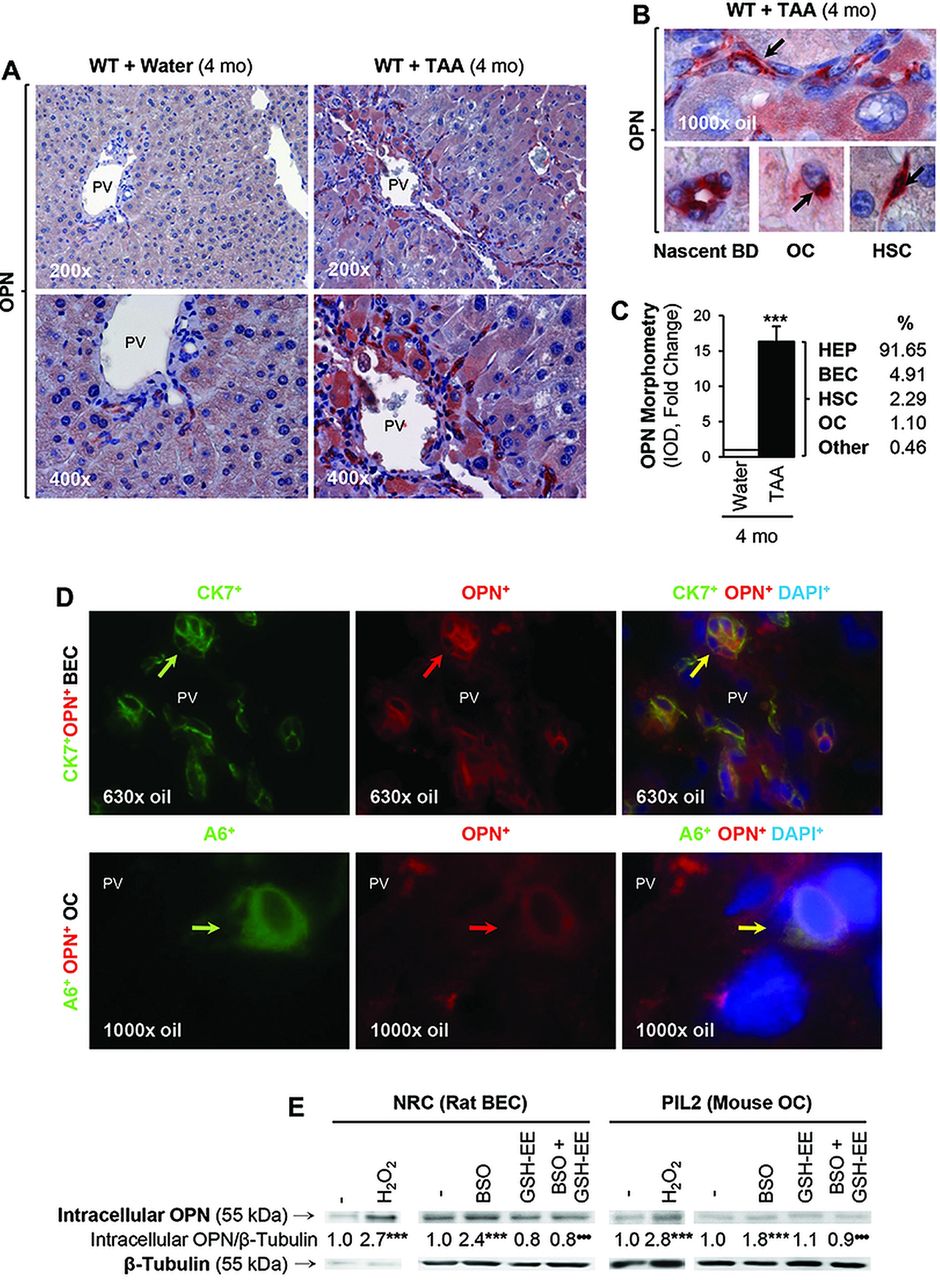
Osteopontin (OPN) expression increases during liver injury and under oxidative stress conditions. C57BL/6J wild-type (WT) were administered thioacetamide (TAA) in the drinking water or isovolumetric amounts of water for 4 months. Immunohistochemistry shows OPN expression in 4 months control and TAA-treated WT mice (A). A closer look at hepatic stellate cells (HSC), biliary epithelial cells (BEC) and oval cells (OC) is shown in (B, black arrows). The OPN morphometric analysis from control and TAA-treated WT along with the percentage of OPN induction in each cell type in the TAA group is shown in (C). Immunofluorescence analysis shows co-localisation of OPN staining with CK7 (a BEC marker) and with A6 (an OC marker) following TAA treatment (D). Western blot analysis for intracellular OPN and β-tubulin in NRC (rat BEC) and PIL2 (mouse OC) cells in the presence of two prooxidants (H2O2 and buthionine sulfoximine (BSO)) and of an antioxidant (GSH-EE, glutathione-ethyl ester) (E). Results are expressed as average values. Experiments were performed in triplicates four times. n=8; ***p<0.001 for TAA versus water or for prooxidant versus control; •••p<0.001 for BSO+GSH-EE versus BSO.
TAA-induced chronic liver injury is greater in WT than in Opn−/− mice
To decipher the role of OPN in the progression of liver disease, we tested whether chronic TAA treatment could lead to differences in the extent of liver injury in WT and in Opn−/− mice. After 2 and 4 months of TAA administration, the haematoxylin and eosin (H&E) staining showed increased necrosis, inflammation and DR at the portal interface in WT compared with Opn−/− mice (figure 2A,B). Serum alanine aminotransferase activity was higher in TAA-treated WT than in Opn−/− mice (figure 2C). The scores for centrilobular and parenchymal necroinflammatory activity were greater in TAA-treated WT than in Opn−/− mice (figure 2D,E). Hence, OPN increases TAA-induced chronic liver injury.

Thioacetamide (TAA)-induced chronic liver injury is greater in wild-type (WT) than in Opn−/− mice. C57BL/6J WT and Opn−/− mice were administered TAA in the drinking water or isovolumetric amounts of water for 2 or 4 months. Hematoxylin and eosin (H&E) staining shows more necrosis (white arrowheads), inflammation (white arrows) and ductular reaction (white open arrows) in WT than in Opn−/− mice both at 2 months (A) and at 4 months (B) of TAA treatment. On the far right in (B), the large image depicts the H&E staining from a TAA-treated WT mouse (necrosis (white arrowheads), inflammation (white arrows), increased biliary epithelial cells (BEC) (white open arrows) and newly formed bile ducts (white circles)). The small insets at the bottom show a magnified picture of a cord of BEC on the left and of a proliferating bile duct (BD) on the right. Plasma alanine aminotransferase activity (C). The centrilobular and parenchymal necrosis (D) and inflammation (E) scores are higher in TAA-treated WT than in Opn−/− mice. Results are expressed as average values±SEM. n=8; *p<0.05, **p<0.01 and ***p<0.001 for TAA versus water; •p<0.05 and •••p<0.001 for Opn−/− versus WT.
OPN decreases hepatocyte proliferation in vivo and in vitro
To analyse whether the different response to TAA-induced chronic liver injury was due to impairment of hepatocyte proliferation, in vivo and in vitro approaches were used. Since the progenitor cell compartment is preferentially activated when hepatocyte proliferation is inhibited, changes in DR may be secondary to impaired hepatocyte proliferation in WT compared with Opn−/− mice. Because proliferating cells are defined as showing Ki67+ nuclear staining, we analysed the expression of Ki67 by IHC (figure 3A). The hepatocyte Ki67 index was ∼3% in TAA-treated WT, whereas it was ∼12% in Opn−/− mice (figure 3B). In addition, p21+ IHC showed increased hepatocyte replicative arrest in TAA-treated WT compared with Opn−/− mice (not shown). In vitro studies demonstrated that recombinant OPN (rOPN) decreased primary hepatocyte proliferation without altering cell viability (figure 3C). Overall, the data suggest that OPN decreases hepatocyte proliferation, which could lead to OC expansion and DR in these models.

Osteopontin (OPN) decreases hepatocyte proliferation in vivo and in vitro. C57BL/6J wild-type (WT) and Opn−/− mice were administered thioacetamide (TAA) in the drinking water or isovolumetric amounts of water for 4 months. Immunohistochemistry shows Ki67+ hepatocyte nuclei (black arrows) (A) and the hepatocyte Ki67 index (B) in control and TAA-treated WT and Opn−/− mice. Results are expressed as average values±SEM n=8; ***p<0.001 for TAA versus water; ••p<0.01 for Opn−/− versus WT. Primary hepatocytes were incubated with 0–100 nM recombinant OPN (rOPN) for 24 h. Methyl[3H]thymidine incorporation into the DNA shows a decrease in the hepatocyte proliferation rate by rOPN-treatment (C, top left). Cell viability and light micrographs are shown (C, top right and bottom). Results are expressed as average values±SEM n=3×4 times; *p<0.05 for rOPN versus control (C).
TAA-induced DR is increased in WT compared with Opn−/− mice
To determine whether OC expansion indeed occurred, A6+ IHC was performed. Morphometric assessment revealed increased A6+ staining in TAA-treated WT compared with Opn−/− mice (figure 4A). The extent of DR was assessed using the same liver lobe and similar region within the lobe and looking at portal fields of analogous size. For this purpose, we used CK19+ IHC, typically expressed in the ductal epithelium and in newly formed bile ducts.26 Biliary hyperplasia along with a 50% increase in the DR score and in CK19 morphometry was found in TAA-treated WT compared with Opn−/− mice (figure 4B,D). Moreover, proliferation of BEC and bile ducts was widespread and not strictly enclosed within the border of the portal mesenchyme. Overall, the DR was located as a wreath at the periphery of the portal areas and septa, quite distinct from the anatomical bile ducts. In the TAA-treated WT mice, small branches extended into the nearby periportal area, and in the Opn−/− mice, less DR occurred, possibly suggesting that the reaction can precede fibrosis and is not simply a secondary effect. 11 Proliferating BEC were identified as CK7+Ki67+ cells on immunofluorescence (figure 4E). Thus, OPN induces DR and BEC proliferation.

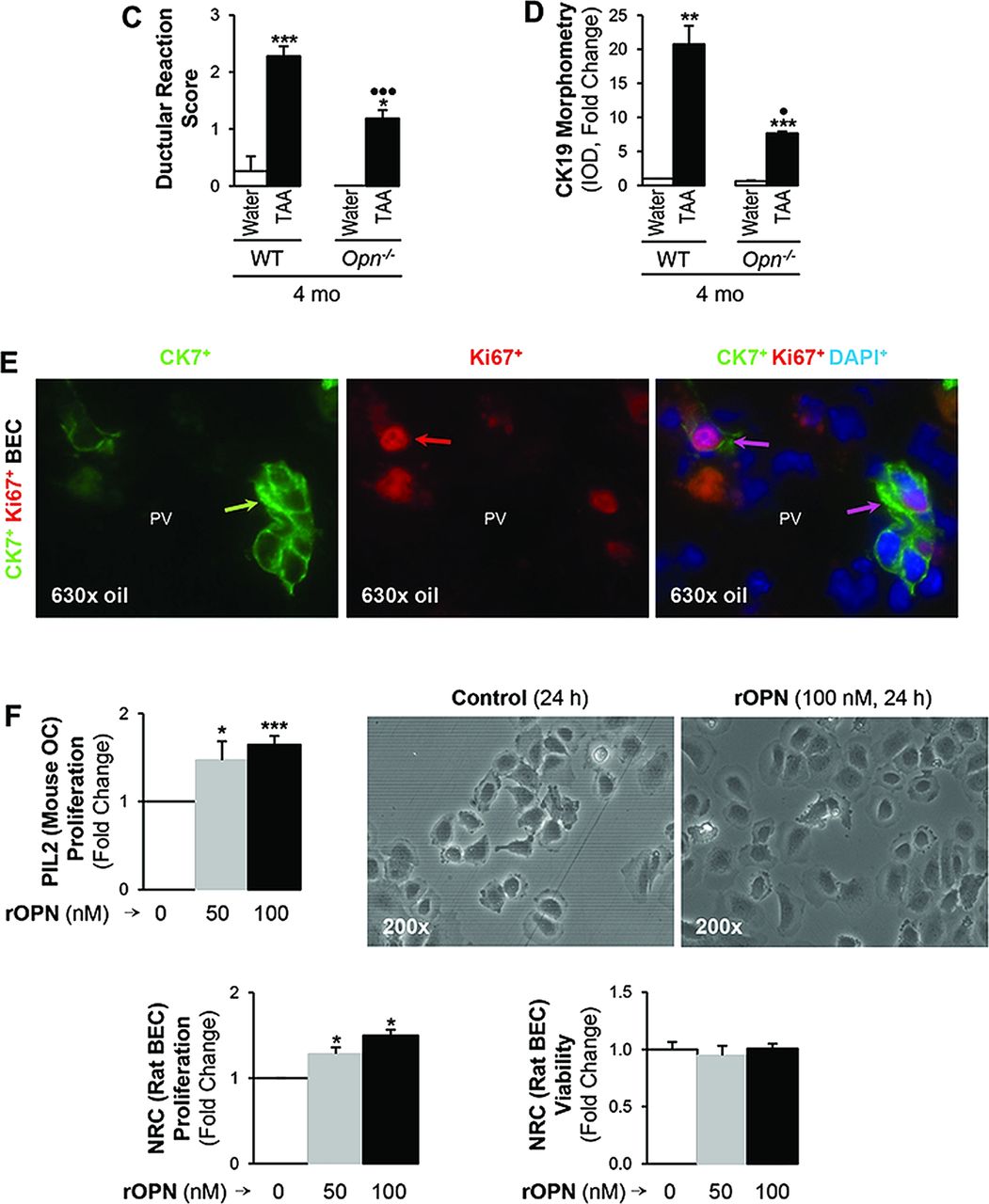
Thioacetamide (TAA)-induced ductular reaction (DR) is increased in wild-type (WT) compared with Opn−/− mice. C57BL/6J WT and Opn−/− mice were administered TAA in the drinking water or isovolumetric amounts of water for 4 months. A6 immunohistochemistry (IHC) (black arrows) and morphometric analysis (A) demonstrate greater activation of the oval cells (OC) compartment in TAA-treated WT than in Opn−/− mice. CK19 IHC (black arrows) shows more DR in TAA-treated WT than in Opn−/− mice. The large image on the right depicts CK19+ staining in biliary epithelial cells (BEC) (black arrows) and in bile ducts (black circles) from a TAA-treated WT mouse. The inset shows a newly formed bile duct (BD). The DR score and the CK19 morphometric analysis for control and TAA-treated WT and Opn−/− mice are shown in (C,D). Immunofluorescence shows co-localisation of CK7 and Ki67-positive staining validating the presence of proliferating BEC (E). Results are expressed as average values±SEM n=8; *p<0.05, **p<0.01 and ***p<0.001 for TAA versus water; •p<0.05 and •••p<0.001 for Opn−/− versus WT. PIL2 (mouse OC) and NRC (rat BEC) were incubated with 0–100 nM recombinant osteopontin (rOPN) for 24 h. Methyl[3H]thymidine incorporation shows that rOPN increases PIL2 (mouse OC) and NRC (rat BEC) proliferation while cell viability remains similar (F). Results are expressed as average values±SEM n=3×4 times. *p<0.05 and ***p<0.001 for rOPN versus control.
To dissect the effects of OPN on BEC, in vitro experiments were designed. Treatment with rOPN increased PIL2 (mouse OC) and NRC (rat BEC) proliferation without altering cell viability (figure 4F). Challenge with rOPN did not increase the invasive potential of NRC (rat BEC) (see online supplementary figure S1A) but enhanced NRC (rat BEC) motility by 40% (see online supplementary figure S1B). To dissect whether this effect was due to enhanced cell survival, cells were stimulated with rOPN in a time-course experiment and the PI3K-pAkt and pERK1/2 signalling pathways participating in cell survival were analysed27 ,28; yet, the expression of these proteins remained unchanged (see online supplementary figure S1C, and not shown here). These results suggest that rOPN acts mainly by increasing BEC motility.
TAA-induced fibrosis is greater in WT than in Opn−/− mice
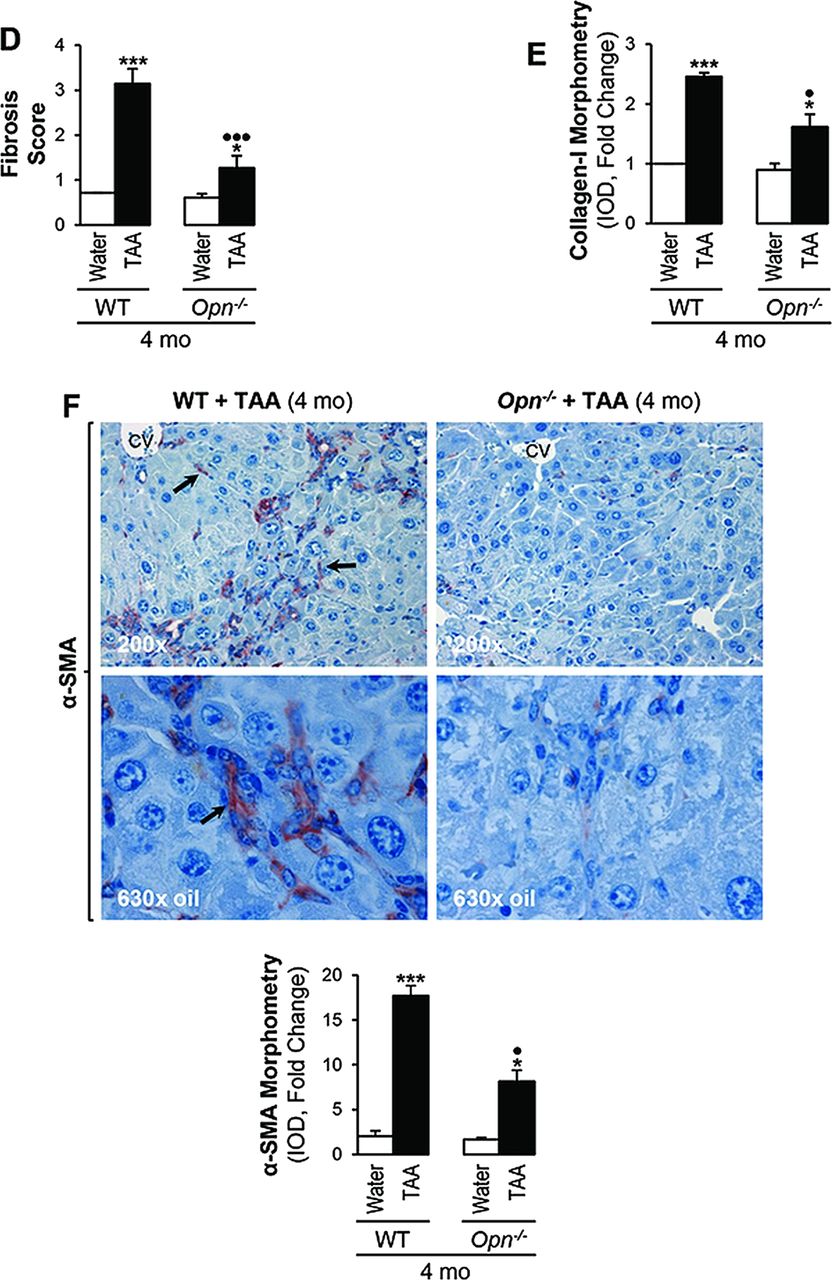
Because the combination of parenchymal cell loss and the activation of the OC compartment could lead to periportal fibrosis, next we analysed the extent of fibrosis in our model taking into account that a protein from the DR, such as OPN, could play a role driving this event. Sirius red/fast green staining and collagen-I IHC showed greater fibrosis in TAA-treated WT than in Opn−/− mice with clear induction of collagen-I deposition, extensive portal fibrosis, bridging fibrosis and an approximately threefold increase in scar thickness (figure 5A,B). Quantitative analysis demonstrated upregulation of collagen-I protein, the Brunt fibrosis score (stage >3 versus 1.2) and collagen-I morphometry (2.5 versus 1.6) in TAA-treated WT compared with Opn−/− mice (figure 5C,E). Likewise, α-SMA+ staining was higher in TAA-treated WT than in Opn−/− mice (figure 5F), suggesting more HSC activation in the presence of OPN.15
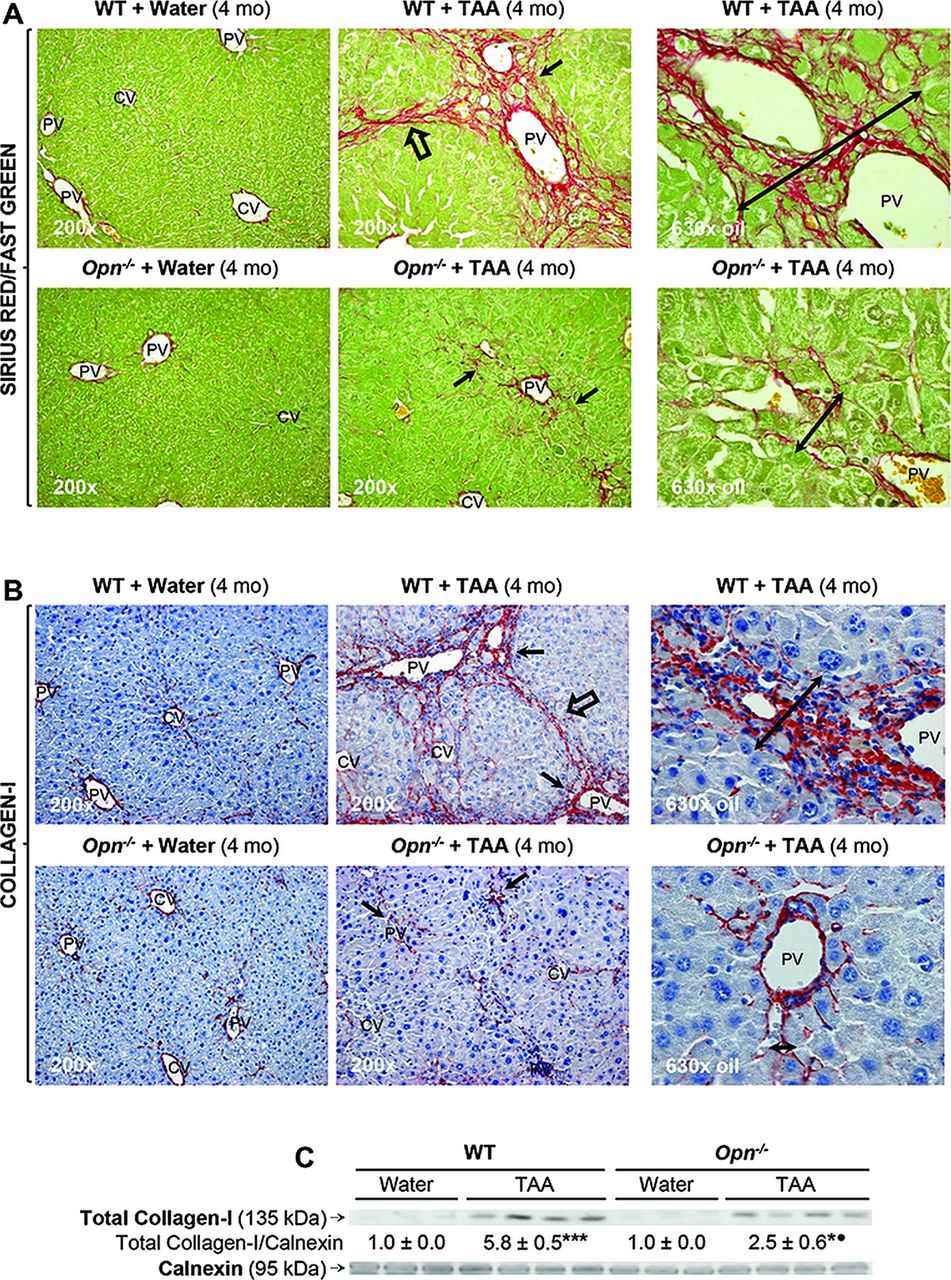

Thioacetamide (TAA)-induced fibrosis is greater in wild-type (WT) than in Opn−/− mice. C57BL/6J WT and Opn−/− mice were administered TAA in the drinking water or isovolumetric amounts of water for 4 months. Sirius red/fast green staining shows more collagenous proteins in TAA-treated WT than in Opn−/− mice (A). Immunohistochemistry depicts more collagen-I in TAA-treated WT than in Opn−/− mice (black arrows) (B). In (A) and (B), scar thickness (double black arrows), portal fibrosis (black arrows) and bridging fibrosis (black open arrows) are higher in TAA-treated WT than in Opn−/− mice. Collagen western blot analysis (C). The Brunt fibrosis score shows stage >3 in TAA-treated WT and ∼1–2 in Opn−/− mice (D). Collagen-I morphometric analysis (E). α-smooth muscle actin staining (black arrows) is greater in TAA-treated WT than in Opn−/− mice (F). Results are expressed as average values±SEM n=8; *p<0.05 and ***p<0.001 for TAA versus water; •p<0.01 and •••p<0.001 for Opn−/− versus WT.
Thus, fibrosis was more distinct in TAA-treated WT mice, where portal-septal thickness positively correlated with DR (figures 5A,E and 4C,E), highly associated with the pathogenesis of liver fibrosis. Collectively, TAA caused more inflammation, necrosis, DR and fibrosis in WT than in Opn−/− mice. These results were also validated in TAA-treated mice on the 129sv genetic background (see online supplementary figure S2).
BEC signal to HSC via OPN and TGF-β to induce collagen-I
To determine how DR increases the profibrogenic potential of HSC, co-cultures of NRC (rat BEC) and primary rat HSC were established. HSC co-cultured with NRC (rat BEC) showed co-induction of OPN and collagen-I proteins compared with HSC in mono-culture (figure 6A, left). The increase in OPN in the culture medium was mostly of NRC (rat BEC) origin as shown by the increase in intracellular OPN in NRC (rat BEC) co-cultured with HSC (figure 6A, right). This suggests that HSC may stimulate NRC (rat BEC) to express more OPN and therefore likely amplify the profibrogenic loop. Furthermore, co-cultured NRC (rat BEC) also showed elevated Tgf-β mRNA, whereas HSC did not (figure 6B). To establish whether the increase in NRC (rat BEC) OPN conditioned the upregulation of TGF-β, NRC (rat BEC) were infected with an adenovirus to overexpress OPN followed by co-incubation with non-immune IgG or with an OPN neutralising Ab for 24 h. Overexpression of OPN increased TGF-β protein, and neutralisation of OPN prevented this effect (figure 6C). This was validated in vivo since BEC were TGF-β+ in TAA-treated WT but not in Opn−/− mice (figure 6D), which was confirmed by co-localisation of CK7+ and TGF-β+-positive staining (figure 6E). We validated these findings in human samples since patients with alcohol-induced cirrhosis showed co-induction of OPN and TGF-β in BEC, whereas healthy explants did not (figure 6F, top); hence, it is likely that OPN and, as a consequence, TGF-β upregulate the fibrogenic response.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Biliary epithelial cells (BEC) signal to hepatic stellate cells (HSC) via osteopontin (OPN) and TGF-β to induce collagen-I. OPN and collagen-I protein increase in primary HSC co-cultured with NRC (rat BEC) compared with HSC in mono-culture (A, left). Intracellular OPN increases in NRC (rat BEC) co-cultured with HSC (A, right). Tgf-β mRNA levels in NRC (rat BEC) and HSC in co-culture and in mono-culture (B). NRC (rat BEC) were infected with and adenovirus to overexpress LacZ (Ad-LacZ) or OPN (Ad-OPN) and were co-incubated with non-immune IgG or with an OPN neutralising Ab for 24 h. Overexpression of OPN increases TGF-β protein and neutralisation of OPN blunts this effect (C). BEC are TGF-β+ (black arrows) in thioacetamide (TAA)-treated wild-type but not in Opn−/− mice (D). Co-colocalisation of CK7 and TGF-β positive staining (E). Patients with alcohol-induced cirrhosis show co-induction of OPN and TGF-β in BEC, whereas healthy explants do not (F, top). Treatment with rTGF-β enhances NRC (rat BEC) motility (F, lower left). Blockade of OPN and TGF-β decreases collagen-I expression in HSC in co-culture (F, lower right).
We have previously described the profibrogenic potential of OPN in driving HSC collagen-I upregulation via αvβ3 integrin engagement and activation of the PI3K-pAkt-NFκB signalling pathway.15 Since TGF-β is a well-known profibrogenic factor,29 ,30 the co-culture with HSC increased NRC (rat BEC) Tgf-β mRNA (figure 6B) and treatment with TGF-β increased NRC (rat BEC) motility (figure 6F, lower left); we postulated that a second mechanism for regulating collagen-I expression in HSC could coexist. Hence, we evaluated the effects of neutralising OPN or TGF-β on collagen-I expression in the co-culture system. We demonstrated that blocking OPN and TGF-β in the co-cultures lowered collagen-I expression (figure 6F, lower right). Collectively, these results suggest that increased OPN expression following liver injury could decrease hepatocyte proliferation, induce OC expansion and DR, increase BEC proliferation along with TGF-β expression and stimulate collagen-I deposition in HSC, leading to an enhanced fibrogenic response (see online supplementary figure S3).
BDL induces more liver injury, hepatocyte arrest, DR and portal fibrosis in WT compared with Opn−/− mice
Lastly, since the TAA model showed greater DR in WT compared with Opn−/− mice, next we asked whether OPN ablation could confer protection in a second model of significant portal fibrosis where biliary hyperplasia is a critical trigger due to cholestasis.31 Coagulative necrosis was present in BDL WT, but it was less apparent in Opn−/− mice (see online supplementary figure S4A,B). The DR score and the CK19 IHC and morphometry analysis confirmed the increased DR in BDL WT compared with Opn−/− mice. The latter showed less and smaller bile infarcts (see online supplementary figure S4A,D). Portal fibrosis, the Brunt fibrosis score and collagen-I IHC and morphometry analysis were significantly increased in BDL WT compared with Opn−/− mice (see online supplementary figure 5A,B). The proliferating cell nuclear antigen (PCNA) staining and index revealed decreased hepatocyte proliferation in BDL WT mice compared with Opn−/− mice (see online supplementary figure S5C). Lastly, OPN+ staining, also quantified by morphometry, was found in bile ducts, in myofibroblasts, in OC and in hepatocytes around portal tracts (see online supplementary figure S5D). Similar findings were obtained in BDL mice in 129sv genetic background (not shown). Thus, in a model where significant DR occurs, Opn ablation partially prevents DR and the fibrogenic response.
Discussion
A number of studies have suggested that DR plays a major role in the induction of liver fibrosis, either directly via epithelial-to-mesenchymal transition—although still rather controversial32–,35—or indirectly via activation of neighbouring cells.10 The DR occurring in submassive hepatic necrosis correlates with increased expression of profibrogenic factors and confines close to activated HSC and/or portal fibroblasts.10 ,36–,39 The presence of a fine DR in some patients without portal fibrosis could indicate that it likely precedes the fibrogenic response in many instances. Moreover, the close association between bile ductules and the periportal and septal interface supports a role for DR in the concentric deposition of collagen fibres, which has been shown to develop as a web-like expansion between portal tract branch points.40 The close relationship and the localisation of DR along with activated HSC and portal fibroblasts are quite compelling to suggest that DR could drive scarring.41
It is becoming clearer that OPN is significantly induced during liver injury in humans and in rodents.15–,19 ,42 In the past few years, several groups have studied the potential role of OPN induction in liver fibrosis albeit with conflicting results.17–,19 ,42 ,43 Work by Fickert et al43 suggested that genetic loss of Opn in mice on C57BL/6J background only did not affect the pathogenesis of the 3,5-diethoxycarbonyl-1,4-dihydrocollidine model of sclerosing cholangitis and biliary-type liver fibrosis. Their conclusions were based solely on the presence of CK19+ cells as a readout for DR since no studies were performed to dissect how DR could regulate the profibrogenic potential of HSC and/or portal fibroblasts.43 Yet, the authors indicated that their experimental findings did not allow the direct conclusion that OPN has no role in biliary fibrosis induced under different experimental conditions. Thus, there is a timely need to clarify the role of OPN in driving DR and how this event could enhance the fibrogenic response to liver injury, and this constituted the main goal of our study.
The results from the present work employing two in vivo models of liver injury and using null mice generated with two different targeting strategies demonstrated that genetic ablation of Opn increased hepatocyte proliferation and decreased OC expansion, DR and fibrogenesis. Studies by Petersen et al44 suggested that inhibition of hepatocyte proliferation must take place in order to observe a true OC response. In addition, matrix composition is likely essential for OC activation to occur; hence, it may be possible that a soluble matricellular protein such as OPN bound to the matrix and expressed and secreted by hepatocytes, OC, BEC and HSC upon liver injury, could be a key player mediating these events. Because of their close association with differentiating ductal plate cells, ECM components of the portal mesenchyma are likely controlling DR via cell–cell and cell–matrix interactions. Hence, since mobilisation and homing of OC and DR are modulated by ECM, adhesion molecules and cytokines,45 OPN could act as a key cytokine regulating and enhancing this process. Thus, OPN may be involved in the initial response to the hepatic insult and further downstream driving hepatocyte replication, OC expansion, DR and subsequently fibrosis, and it is also possible that activated HSC may positively feed back to enhance DR. OPN may play a role in injury per se in addition to in DR, but it can create a vicious circle to enhance DR contributing to injury.
Because activation of the OC compartment is histologically detectable when hepatocyte proliferation is suppressed or to compensate for the increased turnover of damaged hepatocytes, it can function as one of the liver's adaptive responses to ROS as it may occur in the TAA model via direct ROS generation by cytochrome P450 metabolism or in the BDL model via glutathione depletion.11 ,46 Oxidative stress significantly impairs the hepatocyte replicative ability due to induction of cell-cycle inhibitors such as the cyclin-dependent kinase inhibitor p21, which blocks G1/S progression,3 hence promoting compensatory mechanisms such as an increase in OC and the DR.5 ,6 ROS such as O2.− and H2O2 and low glutathione levels are likely mediators of this effect.3 Moreover, these species were shown to increase OPN expression in OC, BEC and HSC,15 therefore working in tandem to further enhance DR. Whereas OC lack cytochrome P45047 and have a survival advantage, OC-derived hepatocytes express cytochrome P450s48; hence, they are highly sensitive to drug-induced liver injury as it occurs in the TAA model. Oxidative liver damage, which prematurely precipitates senescence and parenchymal cell loss, likely causes activation of the OC compartment as a compensatory response. It is reasonable to believe that secretion of a soluble protein from the DR, such as OPN, may be a candidate signal for these and other downstream effects (ie, collagen-I upregulation).15 ,49 Indeed, the Ki67 and PCNA indexes confirmed decreased hepatocyte proliferation in treated WT compared with Opn−/− mice. These indexes inversely correlated with the activity scores, the fibrosis stage, collagen-I morphometry4 and TGF-β expression.
In our in vivo models, upon hepatocyte injury, OPN likely plays a key role in activating the replicative pathway of bipotential periportal OC, inducing DR, which serves as a profibrogenic stimulus in WT mice, whereas these events are reduced in Opn−/− mice. The lower OC reaction in treated Opn−/− mice could suggest that disruption of the parenchyma needs to occur before OC activation can take place and thus less scarring also occurs.
Lastly, in line with the above, fibrillar collagen-I was significantly lowered by OPN ablation in vivo. OPN expression in OC and BEC allows paracrine signalling to HSC, while its expression in HSC signals in an autocrine fashion amplifying the fibrogenic response to increase collagen-I15 and perhaps further increase BEC proliferation and TGF-β production, creating an autoregulatory loop. The cell and matrix-binding ability of OPN may also facilitate a proper stromal and fibrillar collagen network organisation. Interdependence between deposited ECM and DR is feasible as OC may require matrix support for migration and anchor in order to differentiate and restore the damaged liver.50 Overall, it is reasonable to propose that OPN may not only drive the fibrogenic response inducing OC activation and DR as a response for the loss of liver mass, but also by subsequently regulating TGF-β production and collagen-I deposition, as previously demonstrated by us,15 and perhaps the increase in ECM and in TGF-β further stimulates DR. Thus, OPN may multitask and create a positive feedback loop in promoting liver fibrosis.
The present study serves to highlight several important aspects. Myofibroblast activation and periportal fibrosis may not simply be a consequence of DR but rather are likely to be important players amplifying the alternative regenerative pathway since HSC and portal fibroblasts also secrete factors that may promote DR. The fate of expanded OC is not constant, and since these cells are bipotential it is expected that the composition of the ECM and the inflammatory milieu may modify the percentage of cells differentiating along hepatocytic or biliary lineage over time. Finally, although the source of collagen-I surrounding OC is probably HSC and/or portal fibroblasts, there is unexpected plasticity in many of the parenchymal and stromal cells of the liver, adding further complexity to the scenario.
Preventing or limiting DR and the induction of profibrogenic factors that BEC release during the progression of liver disease, such as OPN and TGF-β, represents a novel first line of defence to prevent fibrogenesis. In addition, these studies addressing the interaction of BEC with HSC reveal potential novel signalling mechanisms that could be simultaneously targeted for therapeutic benefit.
Acknowledgments
The authors are very grateful to Drs David T. Denhardt (Rutgers University, NJ) for providing the 2A1 Ab and the Opn−/− mice in 129sv background and to Valentina Factor (National Cancer Institute, Bethesda, MD) for her generous gift of the A6 Ab used for OC immunostaining. We are also very thankful to all former and current members from the Nieto Laboratory for their helpful comments and suggestions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
-
XW, AL and XG are contributed equally.
-
Contributors XW, AL and XG significantly contributed to the experimental design and performed most of the experiments. YL, NK, RU and T-ML performed some experiments. MIF evaluated and scored the H&E and immunochemistry. NN directed the project, obtained funding, analysed data and wrote the manuscript. All authors contributed to editing the manuscript.
-
Funding National Institutes of Health. Postdoctoral fellowships from the Basque Government (Spain) (AL), Keio University School of Medicine (Japan) (NK) and the Government of Navarre (Spain) (RU). US Public Health Service Grants 5R01 DK069286, 2R56 DK069286 and 3 R56 DK069286-06S1 from the National Institute of Diabetes and Digestive and Kidney Diseases (NN). US Public Health Service Grant U01 AA021887-01 from the National Institute on Alcohol Abuse and Alcoholism (NN). US Public Health Service Grants 5 P20 AA017067, 5 P20 AA017067-01S1 and 5 P20 AA017067-03S1 from the National Institute on Alcohol Abuse and Alcoholism (NN and MIF).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Any additional data from the study are available to all members from the Nieto Laboratory and to the reviewers from our NIH grant applications.