Article Text

Abstract
Background Escherichia coli strains harbouring the pks island (pks+ E. coli) are often seen in human colorectal tumours and have a carcinogenic effect independent of inflammation in an AOM/IL-10−/− (azoxymethane/interleukin) mouse model.
Objective To investigate the mechanism sustaining pks+ E. coli-induced carcinogenesis.
Method Underlying cell processes were investigated in vitro and in vivo (xenograft model) using intestinal epithelial cells infected by pks+ E. coli or by an isogenic mutant defective for pks (pks− E. coli). The results were supported by data obtained from an AOM/DSS (azoxymethane/dextran sodium sulphate) colon cancer mouse model and from human colon cancer biopsy specimens colonised by pks+ E. coli or pks− E. coli.
Results Colibactin-producing E. coli enhanced tumour growth in both xenograft and AOM/DSS models. Growth was sustained by cellular senescence (a direct consequence of small ubiquitin-like modifier (SUMO)-conjugated p53 accumulation), which was accompanied by the production of hepatocyte growth factor (HGF). The underlying mechanisms involve microRNA-20a-5p, which targets SENP1, a key protein regulating p53 deSUMOylation. These results are consistent with the expression of SENP1, microRNA-20a-5p, HGF and phosphorylation of HGF receptor found in human and mouse colon cancers colonised by pks+ E. coli.
Conclusion These data reveal a new paradigm for carcinogenesis, in which colibactin-induced senescence has an important role.
- E. Coli
- Colon Carcinogenesis
- Gene Expression
- DNA Damage
- Cell Proliferation
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Colibactin-producing E. coli have been found in 50–60% of human colorectal tumours compared with 20% in controls.
-
Colibactin-producing E. coli induce DNA damage, cell cycle arrest, mutations and chromosomal instability.
-
Colibactin-producing E. coli promote colorectal cancer in a murine AOM/IL-10−/− (azoxymethane/interleukin) mouse model.
What are the new findings?
-
Colibactin-producing E. coli indirectly enhance tumour growth by inducing the emergence of senescent cells secreting growth factors.
-
The hepatocyte growth factor (HGF) is the main mechanistic link between pks+ E. coli-induced senescence and tumour growth.
-
The mechanism underlying colibactin-induced senescence involves microRNA-20a-5p, which targets SENP1, a key protein in the regulation of p53 SUMOylation.
-
The expression of microRNA-20a-5p, SENP1, HGF and activated HGF receptor in human colon cancers is influenced by the presence of pks+ E. coli.
How might it impact on clinical practice in the foreseeable future?
-
The data support a new paradigm for colorectal carcinogenesis in which colibactin-producing bacteria favour the emergence of senescent cells secreting growth factors. The colonisation of colon tumours by pks+ bacteria may affect the prognosis of colorectal cancer and therefore the management of curative and/or preventive treatments. Targeting colibactin production may be a strategy to restrain the production of pro-tumourigenic factors.
Introduction
Cancer is defined as uncontrolled cell proliferation caused by accumulated genetic and epigenetic mutations.1–3 The origin of these mutations can be multifactorial and 20% of cancers worldwide are estimated to be attributable to infections.4 ,5 Many cancers occur in tissues with a high exposure to microbiota, especially colorectal cancers (CRCs), the fourth most common cancer with more than one million cases a year.6 The high density of microorganisms seen in the colon (1013 commensal bacteria colonising the colon vs from 102 for the duodenum to 108 for the distal ileum) is associated with a greater risk of cancer than from microorganisms in the small intestine.5 The role of microbiota in colon cancer has been shown by studies in which mice genetically susceptible to CRC, or mice with chemically induced chronic inflammation, developed significantly fewer tumours under germ-free conditions than when harbouring conventional microbiota.7 ,8 The role of microbiota in the development of CRC might be related to the procarcinogenic features of the intestinal bacteria, which may initiate cancer development and/or have a competitive advantage in the tumour microenvironment that allows them to play a role in disease progression.9 ,10 However, few putative procarcinogenic bacteria have been identified. The Enterococcus faecalis species produces superoxide, which can induce DNA damage and chromosomal instability in colonic epithelial cells.11 Enterotoxigenic Bacteroides fragilis produce the fragilysin toxin, which is genotoxic,10 and causes cell proliferation by cleavage of the tumour suppression factor E-cadherin,12 and increases tumourigenesis by triggering production of interleukin-17.13 Finally, colonic malignancies allow Streptococcus gallolyticus subsp gallolyticus to colonise established colorectal tumours, leading to tumour development by induction of the proinflammatory cyclo-oxygenase 2 pathway.14
Escherichia coli are the predominant aero-anaerobic Gram-negative bacteria in the normal intestinal microbiota. As a commensal, E. coli coexist harmoniously with their mammalian host, promote normal intestinal homeostasis and rarely cause disease. However, some virulent E. coli that have acquired pathogenicity islands can colonise the human gastrointestinal tract and induce disease. Mucosa-associated E. coli, which are more frequently identified in colon tissue from patients with adenocarcinomas than in controls, can have procarcinogenic features15 ,16 that promote downregulation of DNA mismatch repair proteins.17 In addition, various toxins produced by pathogenic E. coli modulate the cell cycle of host cells such as cytotoxic necrotising factor, cycle-inhibiting factor, cytolethal distending toxins and colibactin.9 ,18 Colibactin is a genotoxin produced by E. coli harbouring the pks island, which encodes enzymes responsible for its synthesis.19 Colibactin induces double-strand DNA breaks, cell cycle arrest and megalocytosis.19 Colibactin-induced DNA damage is associated with mutations and chromosomal instability,20 lesions often seen in cancers. Interestingly, pks-harbouring E. coli (pks+ E. coli) strains were recently reported in 50–60% of human colorectal tumours versus 20% in controls21 ,22 and, as shown with an AOM/IL-10−/− (azoxymethane/interleukin) mouse model, pks+ E. coli have a carcinogenic effect independent of inflammation.21
In this study, we investigated the mechanisms underlying pks+ E. coli-induced carcinogenesis. We report that pks+ E. coli infection induces cellular senescence, characterised by a high production of growth factors that promote proliferation of uninfected cells and, subsequently, tumour growth.
Methods
Bacterial strains
The laboratory E. coli strain DH10B hosting the pBeloBAC 11 bearing the pks island (pks+ E. coli), or hosting an empty pBeloBAC 11 (pks− E. coli) were used.19 The clinical pks+ E. coli strain (CCR20) was isolated from a human colon tumour.22 The isogenic mutant of E. coli CCR20 designated CCR20Δpks was generated as indicated in the online supplementary section. Bacteria were grown at 37°C in Luria-Bertani (LB) medium. Bacterial inoculums were assessed at optical density 620 nm using a UV-1800 spectrophotometer (Shimadzu) and controlled by bacterial spreading on LB agar plates.
Cell culture and infection
Intestinal epithelial cells were grown following ATCC guidelines and infected at a multiplicity of infection (MOI) of 500, as previously described.19 After 3 h of infection, cells were washed three times with phosphate-buffered saline, and culture medium supplemented with 200 µg/mL gentamicin was added to kill remaining bacteria. The culture medium was then changed every 2 days. To prepare the conditioned medium (CM), 1.5×105 cells were infected as described above and at 1, 2 or 4 days after infection, the medium was removed, washed three times with phosphate-buffered saline and replaced by serum-free medium. Twenty-four hours later, the medium was collected and used as CM to culture 5.0×104 uninfected cells. For growth factor inhibition, conditioned media were supplemented with 4 nM hepatocyte growth factor (HGF) inhibitor (JNJ-38877605, Selleckchem), 4 nM fibroblast growth factor (FGF) inhibitor (PD173074, Selleckchem) or 0.1 mg/mL of anti-HGF, anti-granulocyte macrophage colony-stimulating factor (anti-GM-CSF), anti-FGF neutralising antibody (Sigma-Aldrich) or rat IgG isotype control (Biolegend). For all the xenograft experiments, cellular viability was assessed using trypan blue dye.
P53 status
HCT116, Int-407 and IEC-6 harbour a wild-type (WT) p53 gene, whereas HCT8, Caco2 and HT-29 harbour a mutated p53.
Tumourigenesis assay using in vivo infected cells
2×106 HCT116 cells mixed with pks+ E. coli or pks− E. coli (MOI=20) were embedded in growth factor-reduced Matrigel (Becton Dickinson) to obtain a tumour easily measurable with a calliper and to decrease the bias due to growth factors present in Matrigel. The cells were then immediately subcutaneously injected (200 µL) into the dorsal flap of 5-week-old nude mice (Charles River). Each mouse received one xenograft. Three hours after injection, the mice received intraperitoneally 150 mg/kg of broad-spectrum antibiotic imipenem (MSD), and absence of bacteria was confirmed by plating tumour homogenates and blood on LB plates 24 h after treatment.
Tumourigenesis assay using in vitro infected cells
HCT116 cells were infected in vitro as described above with pks+ or pks− E. coli. pks+ E. coli-induced senescence was checked 5 days after infection using a senescence-associated β-galactosidase (SA-β-gal) assay as described below. Cells were trypsinised 5 days after infection, and 0.4×106 previously infected cells were mixed with 1.6×106 uninfected HCT116 cells. The resulting cellular mixes were embedded in growth factor-reduced Matrigel (Becton Dickinson), subcutaneously injected into the dorsal flap of 5-week-old nude mice and the tumour size was measured as previously described.37 Each mouse received one xenograft.
AOM/DSS murine model
Colon cancer was induced in C57/B6 mice (6 weeks old, Charles Rivers), as previously described,23 with some modifications. Mice were intraperitoneally injected with AOM (10 mg/kg body weight) and treated with streptomycin in drinking water (2 mg/mL) for 2 days to facilitate bacterial colonisation.24 One day after the end of the antibiotic treatment, they received by gavage 109 colony forming units of a clinical pks+ E. coli strain (CCR20) or its isogenic mutant (CCR20Δpks), and were maintained on regular diet and water for 5 days. They were subjected to two cycles of 2% dextran sodium sulfate (DSS) treatment23 and then killed. Colonic tumours were counted and measured with a dissecting microscope, then Swiss-rolled and fixed for histology. Two macroscopic tumours from each mouse were collected and pooled for western blot and quantitative real-time PCR. In parallel, bacterial colonisation was assessed by counting CCR20 and CCR20Δpks in faeces, healthy colonic tissues and malignant colon tumours by spreading on selective agar (bioMérieux).
Human biopsy specimens
Biopsy specimens of colon adenocarcinomas were collected in a previously published study,22 from colon resections, which were required for the treatment of patients. Ethical approval for the study was granted by the Clermont-Ferrand research ethics committee. Verbal informed consent to participate in the research was obtained from all patients included in the study in accordance with the French bioethics law (Act No 2004-800 of 6 August 2004). Samples were taken on resected colon at the site of malignant tumours. Pathological analysis confirmed the neoplastic features of the samples. Age and sex of patients, tumour node metastases stage, neoplastic grade and inflammatory score are given in online supplementary table S1. We included randomly selected biopsy specimens colonised by pks+ E. coli (n=8), for which the functionality of pks island was confirmed in vitro.19 The biopsy specimens colonised by pks− E. coli (n=9) were also randomly selected.
Senescence-associated β-galactosidase staining
Senescent cells were visualised using the Senescence Cells Histochemical Staining Kit (Sigma-Aldrich) according to the manufacturer's instructions. In vivo β-galactosidase staining was performed as previously described.25
Senescence-associated β-galactosidase activity
Human biopsy specimens and mouse tissues (∼10 mg) were lysed using an ultra-turrax homogeniser (IKA) in a reaction buffer (200 µL containing 10 mM potassium ferricyanide, 300 mM NaCl, 4 mM MgCl2 and 0.2 M phosphate buffer pH6.0). After centrifugation, 25 µL of supernatants was supplemented with an equal volume of 2 mg/mL 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (Sigma-Aldrich) and hydrolysis was monitored at 420 nm. Enzymatic activity was normalised against protein concentration. SA-β-gal activity was correlated (r2=0.898) with the number of SA-β-gal-positive cells counted with the Senescence Cells Histochemical Staining Kit (Sigma-Aldrich).
Statistical analysis
Statistical analyses were performed with R software (V.2.13.2, http://www.r-project.org/). A p value < 0.05 was considered statistically significant. Redundancy analyses (RDAs) were performed with the Vegan package (V.2.0-2, http://cran.r-project.org/web/packages/vegan/index.html). Significant differences were investigated by 1000 permutation tests.
Results
Colibactin-producing E. coli infection increases tumour growth
Intestinal epithelial cells HCT116 mixed with E. coli DH10B strain producing colibactin (pks+) or not (pks−) were subcutaneously injected into nude mice. Three hours later, a broad-spectrum antibiotic (imipenem) was administered to kill bacteria. This single and transient exposure to pks+ E. coli significantly increased both tumour growth (figure 1A) and the number of Ki67-positive cells (a cellular proliferation marker) (figure 1B). Nude mice were then subcutaneously injected with HCT116 cells infected in vitro in combination with uninfected HCT116 cells (20% and 80%, respectively) and collected 5 days after infection. Xenografts containing cells infected by pks+ E. coli in vitro grew more rapidly and harboured a higher number of Ki67-positive cells than those containing pks− E. coli-infected cells (Figure 1C,D). Tumours composed of pks− E. coli-infected cells exhibited a similar development to that seen with tumours composed of uninfected cells (figure 1A,C), suggesting that pks− E. coli DH10B have no significant effect on tumour growth.
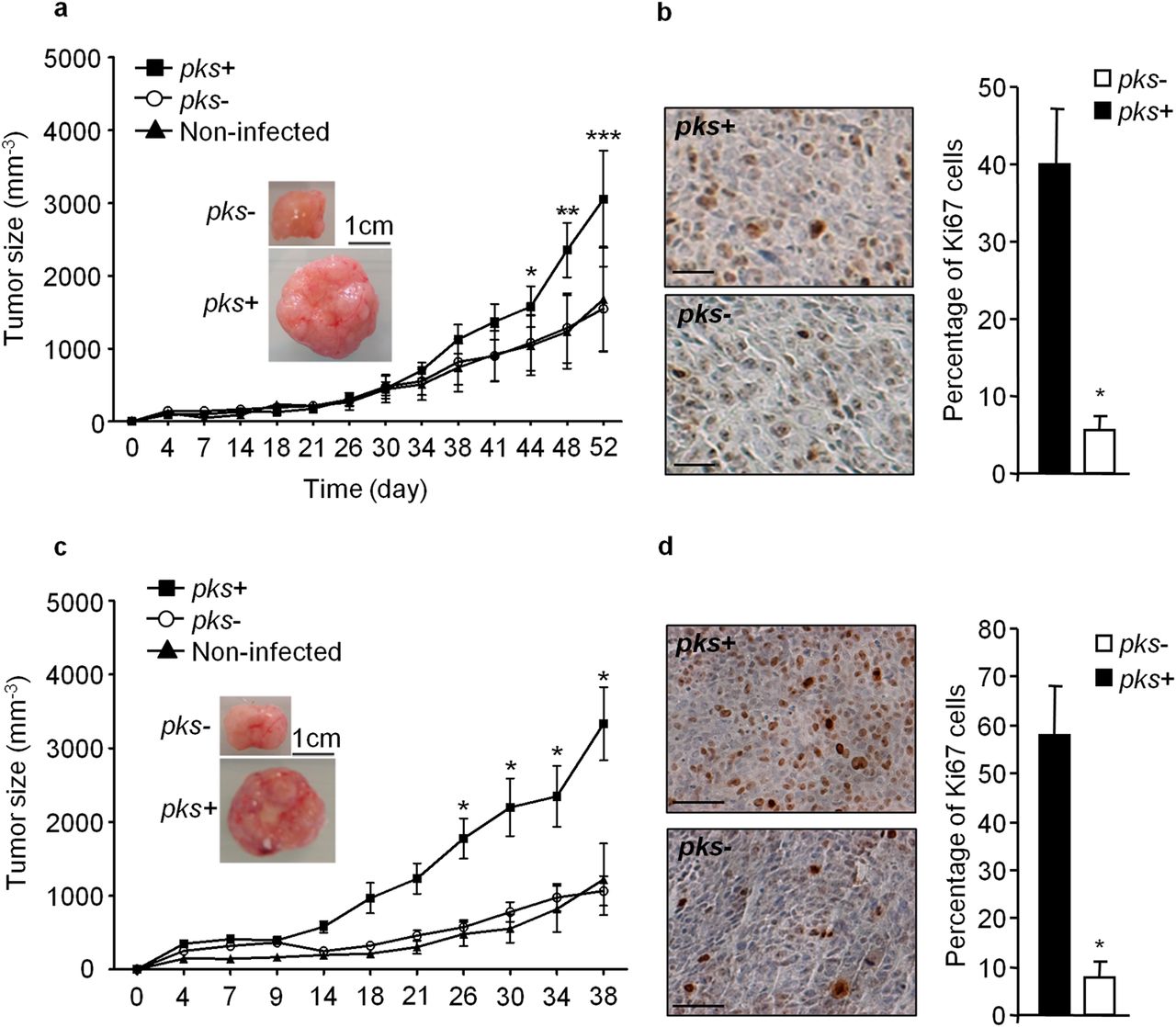
Infection with pks+ E. coli promotes tumour growth and cell proliferation. (A) 2×106 HCT116 cells mixed with pks+ E. coli or pks− E. coli (MOI=20) were embedded in Matrigel, and immediately subcutaneously injected into the dorsal flap of 5-week-old nude mice. Three hours after the injection, the mice received intraperitoneally 150 mg/kg of the broad-spectrum antibiotic imipenem to kill bacteria. Tumour size was monitored during the indicated period (n=5/group; **p<0.01; ***p<0.001, by analysis of variance (ANOVA)). Representative photographs of tumours are presented (scale bar: 1 cm). (B) Representative Ki67 immunohistochemical staining in xenograft 52 days after infection. Scale bars: 100 µm. The bar graph on the right represents measurement of Ki67 positive cells. Data are presented as means±SEM (*p<0.05, by Mann–Whitney test). (C) 0.4×106 HCT116 cells, infected in vitro by pks+ or pks− E. coli and collected 5 days after infection, were mixed with 1.6×106 non-infected HCT116 cells and subcutaneously injected into nude mice. Tumour size was monitored during the indicated period (n=10/group; *p<0.05, by ANOVA). Representative photographs of tumours are presented (scale bar: 1 cm). (D) Representative Ki67 immunohistochemical staining in xenograft 38 days after infection. Scale bars: 100 µm. The bar graph on the right represents measurement of Ki67 positive cells. Data are presented as means±SEM (*p<0.05, by Mann–Whitney test).
HCT116 cells were infected in vitro with pks+ or pks− E. coli for 3 h and cellular proliferation was assessed 24 h after infection. We observed a ∼20% drop in cell proliferation of pks+ E. coli-infected cells compared with pks− E. coli-infected cells (see online supplementary figure S1). This decrease was probably due to colibactin-induced cell cycle arrest.19 The drop in cell proliferation seen in vitro and the pro-proliferative effect seen in the xenograft mouse model in response to pks+ E. coli infection suggested an indirect effect of bacteria on tumour growth in vivo. We therefore hypothesised that cells infected with pks+ E. coli secrete factors that induce proliferation of uninfected cells. We assessed the ability of conditioned medium (CM) derived from HCT116 cells infected with E. coli for 3 h to promote cell proliferation. CMs were obtained by collecting serum-free supernatant of HCT116 cells at days 1, 3 or 5 after infection. Cells incubated with CM derived from pks+ E. coli-infected cells at 5 days after infection proliferated as fast as cells incubated with a medium containing 10% fetal bovine serum, whereas CMs derived from pks− E. coli-infected cells did not affect cell proliferation (see online supplementary figure S2). Similar results were obtained using various human intestinal epithelial cells (Int-407, HCT8, Caco2, HT-29; see online supplementary figure S3) and IEC-6 non-transformed cells (see online supplementary figure S3). These results confirm an indirect effect of colibactin-producing E. coli on tumour growth.
Colibactin-producing E. coli induce senescence of intestinal epithelial cells
Colibactin is known to induce megalocytosis and cell cycle arrest,19 which are features of cellular senescence.26 SA-β-gal activity was therefore assessed in infected cells. pks+ E. coli infection induced an increase in the number of SA-β-bal positive megalocytes 5 days after infection (figure 2A,B and see online supplementary figure S4A). Similar results were obtained with various human intestinal epithelial cells and non-transformed cells (see online supplementary figure S4B). In contrast, the number of SA-β-gal positive cells induced by pks− E. coli was similar to that of non-infected cells (figure 2A,B), indicating that the induction of senescence was due to colibactin synthesis. Of note, we did not find a significant increase in either apoptosis or necrosis of pks+ E. coli-infected cells compared with pks− E. coli-infected cells, as assessed by lactate dehydrogenase release and caspase 3 activation (data not shown).

Colibactin-producing E. coli induce cellular senescence. (A) HCT116 cells were infected with pks+ E. coli or pks− E. coli, and SA-β-gal positive cells were assessed 5 days after infection. The three figures are at the same magnification (scale bars: 100 µm). (B) Percentage of SA-β-gal positive cells determined 5 days after infection. Data are from three experiments performed twice and are presented as means±SEM (n=6/group; ***p<0.001 vs uninfected cells, by Mann–Whitney test). (C) Whole-cell lysates were analysed by immunoblotting experiments 5 days after infection. (D) 2×106 HCT116 cells mixed with pks+ E. coli or pks− E. coli (MOI=20) were embedded in Matrigel, and immediately subcutaneously injected (200 µL) into the dorsal flap of 5-week-old nude mice. Three hours after injection, the mice received intraperitoneally 150 mg/kg of broad-spectrum antibiotic imipenem. SA-β-gal activity was assessed in tumours at 10 days after infection (n=5/group; **p<0.05, by Mann–Whitney test). (E) Expression levels of growth factors mRNA were assessed by qRT-PCR in mice tumours 10 days after infection. Data are presented as means±SEM (n=5/group,*p<0.05; by Mann–Whitney test). (F) HCT116 cells were cultured in presence of conditioned media derived from pks+ E. coli or pks− E. coli-infected HCT116 cells and supplemented with growth factor inhibitors. Cellular growth was assessed by a MTT assay after 24 h incubation (n=8/group; **p<0.01; NS, not significant vs pks−, by Mann–Whitney test). Data are from three experiments performed twice and are presented as means±SEM. (G) 0.4×106 HCT116 cells collected 5 days after pks+ or pks− E. coli infection, were mixed with 1.6×106 non-infected HCT116 cells and subcutaneously injected into nude mice. The mice received 3.4 µg/kg hepatocyte growth factor (HGF) inhibitor or vehicle twice a week. Tumour size was monitored during the indicated period (n=5 mice/group: ***p<0.001 vs other groups, by two-way analysis of variance). Representative photographs of tumours are presented (scale bar: 1 cm).
At day 5 after infection pks+ E. coli-infected cells exhibited an accumulation of phospho-p53, p21Cip and Rb, and a decrease in E2F-1 expression (figure 2C), a protein expression profile characteristic of senescent cells,27 confirming that pks+ E. coli induce senescence of human intestinal epithelial cells. To investigate whether pks+ E. coli promote cellular senescence in vivo at early time points in tumour development, nude mice were subcutaneously injected with HCT116 cells mixed with E. coli as described in figure 1A and tumours were analysed at day 10 after implantation. Tumour sizes were comparable at this early point in animals injected with cells infected by pks+ E. coli and by pks− E. coli (figure 1A). However, tumours derived from pks+ E. coli-infected HCT116 cells showed a significant increase in SA-β-gal activity compared with tumours derived from pks− E. coli-infected cells (figure 2D).
Senescent cells are metabolically active and usually acquire a senescence-associated secretory phenotype (SASP).26 ,28 SASP was therefore investigated at day 5 after infection. Interestingly, pks+ E. coli-infected cells expressed significantly higher levels of the growth factors HGF, FGF, GM-CSF, bone morphogenetic protein 4 (BMP4) and vascular endothelial growth factor (VEGF) than pks− E. coli-infected cells (see online supplementary figure S5A). Similar results were obtained with five human intestinal epithelial cell lines and non-transformed cells (see online supplementary figure S5B). Of note, expression levels of FGF, GM-CSF and HGF only were also increased in xenografts 10 days after pks+ E. coli infection compared with xenografts infected with pks− E. coli (figure 2E).
To identify the relevant secreted mediators involved in cell proliferation, HCT116 cells were cultured with conditioned media supplemented with growth factor inhibitors. HGF pathway inhibitor, in contrast to vehicle or FGF inhibitor, abrogated in vitro the pro-proliferative activity of conditioned media derived from pks+ E. coli-infected cells (figure 2F). Similar results were obtained using neutralising antibodies; HGF antibodies, in contrast to GM-CSF or FGF antibodies, blocked the pro-proliferative effect (see online supplementary figure S6). As seen in vitro, HGF inhibitor significantly blocked the growth of xenograft obtained from cells infected with pks+ E. coli (figure 2G). Together, these data show that the pro-proliferative effect of pks+ E. coli-induced senescent cells is mainly mediated by HGF.
SENP1 downregulation and p53 SUMOylation are key features of pks+ E. coli-induced senescence
The SUMOylation of p53 has emerged as a key determinant of cellular senescence.29 Using HA-tagged small ubiquitin-like modifier 1 (SUMO1)-expressing cells infected with E. coli, we observed that pks+ E. coli increased the SUMOylation of p53 (figure 3A) and that this modification was associated with a downregulation of SENP1 (figure 3B), a negative regulator of p53 SUMOylation.30 To confirm the role of SENP1 in pks+ E. coli-induced senescence, HCT116 cells were transfected with a vector encoding SENP1.31 Overexpression of SENP1 significantly decreased the number of senescent cells induced by pks+ E. coli infection to a level similar to that found with pks− E. coli-infected cells (figure 3C). This effect was dependent on SENP1 enzymatic activity since the expression of an inactive mutant of SENP1 (C602S) did not protect against pks+ E. coli-induced senescence (figure 3C). In addition, overexpression of wild-type SENP1, unlike overexpression of the inactive SENP1, abolished the pks+ E. coli-induced accumulation in SUMO1-conjugated p53 (figure 3D). During these experiments, the genotoxic effect of pks+ E. coli was not affected (see online supplementary figure S7) and conditioned media derived from pks+ E. coli-infected cells overexpressing functional SENP1 did not promote HCT116 cell proliferation (see online supplementary figure S8). These data show that SENP1 downregulation and subsequent p53 SUMOylation are key features in pks+ E. coli-induced cell senescence.

SUMOylation controls senescence upon infection with pks+ E. coli. (A) Small ubiquitin-like modifier (SUMO)-conjugated p53 was assessed in HCT116 intestinal epithelial cells in response to infection by pks+ E. coli or pks− E. coli. HCT116 cells transiently transfected with a vector encoding SUMO1 HA-tagged protein, were lysed 5 days after infection and then immunoprecipitated using an antibody specific for HA. The amount of co-immunoprecipitated p53 was examined by western blot. (B) Immunoblot analysis of SENP1 expression in HCT116 cells 5 days after infection. (C, D) HCT116 cells transiently transfected with an empty vector or a vector encoding SENP1 (SENP1 WT) or an inactive mutant (SENP1 C602S) were infected with pks+ E. coli or pks− E. coli. (C) The bar graph represents the percentage of SA-β-gal positive cells 5 days after infection. SUMO-conjugated p53 was assessed by co-immunoprecipitation and western blot analysis as indicated in panel A. Data are from three experiments repeated twice and are presented as means±SEM (n=6/group; NS, not significant; ***p<0.001, by Mann–Whitney test). (D) SUMO1-conjugated protein patterns were assessed 5 days after infection.
Colibactin-producing E. coli decrease SENP1 expression via miR-20a-5p and c-Myc
Among the microRNAs previously reported to be deregulated during senescence,32 in silico predictions showed that miR-20a-5p potentially targets SENP1 (see online supplementary figure S9). Interestingly, miR-20a-5p expression was significantly upregulated in pks+ E. coli infected HCT116 cells compared with pks− E. coli infected cells (figure 4A). Furthermore, transfection of cells with mature miR-20a-5p decreased SENP1 expression at both mRNA and protein levels (Figure 4B,C). The SENP1 mRNA 3′-UTR was cloned downstream of the luciferase gene and this construct was transfected into cells in the presence of miR-20a-5p or miR-control. MiR-20a-5p significantly repressed luciferase activity (figure 4D), suggesting the binding of miR-20a-5p to the SENP1 mRNA 3′-UTR. These results indicate that pks+ E. coli infection induces miR-20a-5p expression, which in turn downregulates SENP1 expression by targeting SENP1 mRNA 3′-UTR.
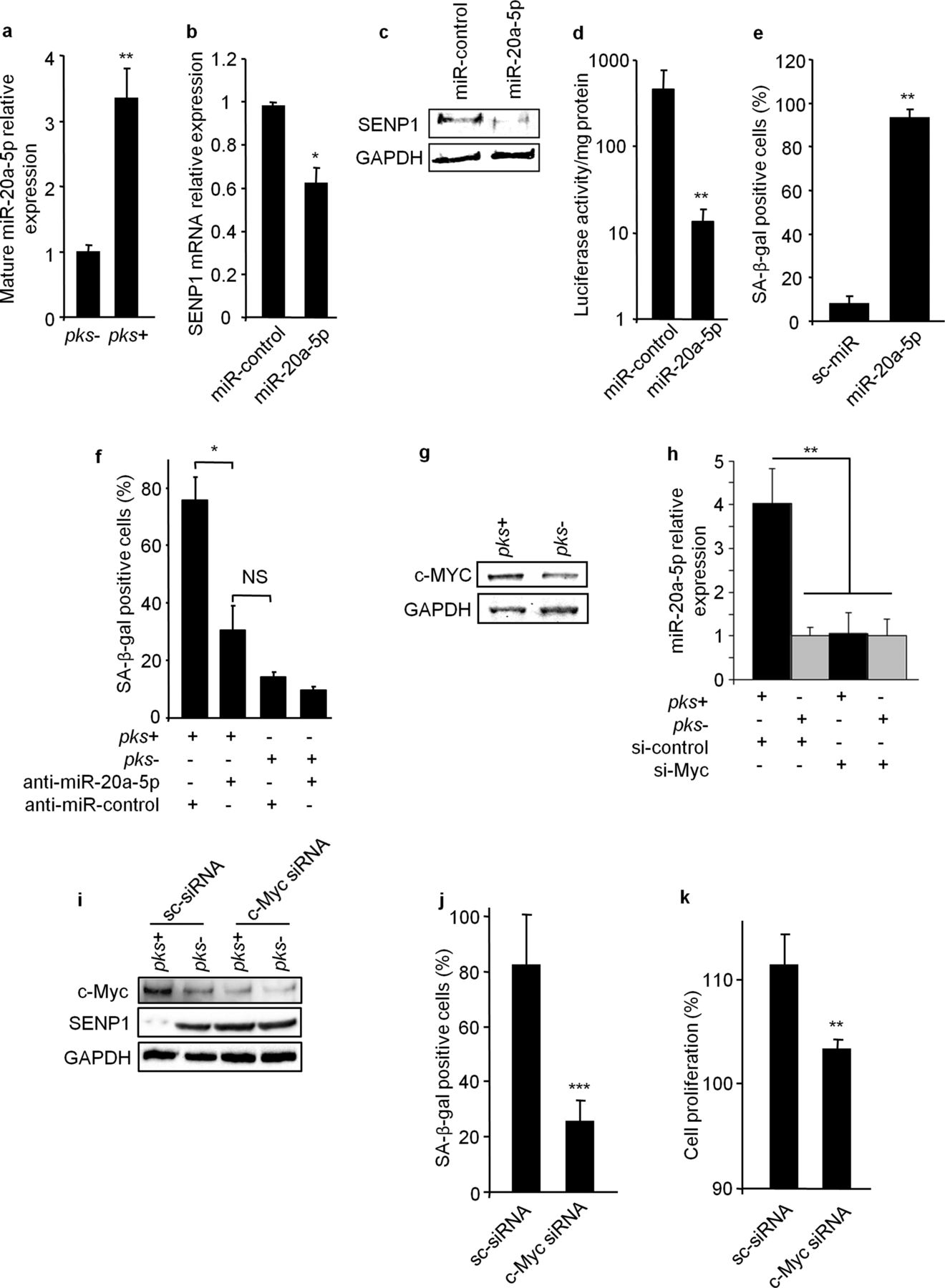
MiR-20a-5p represses SENP1 expression and controls the senescence induced by pks+ E. coli. (A) Mature forms of miR-20a-5p were quantified by qRT-PCR in HCT116 cells 1, 3 and 5 days after infection. (B, C) HCT116 cells were transfected with precursors of miR-20a-5p or miR-control, and SENP1 expression was assessed by (B) qRT-PCR and (C) western blot. (D) HCT116 cells were transfected with a luciferase vector encoding the SENP1 mRNA 3′-UTR in the presence of miR-20a-5p or miR-control. Luciferase activity was determined. (E) HCT116 cells were transfected with the precursor of miR-20a-5p or miR-control and senescence was assessed at day 5 by detecting SA-β-gal positive cells. (F) HCT116 cells were transfected with anti-miR-20a-5p or anti-miR-control and infected with pks+ E. coli or pks− E. coli. Five days after infection senescent cells were assessed by detecting SA-β-gal positive cells. (G) HCT116 cells were infected with pks+ or pks− E. coli and c-Myc expression was analysed by western blot at day 5 after infection. (H) HCT116 cells were transfected with c-Myc siRNA or scramble (sc) siRNA and miR-20a-5p expression level was assessed by qRT-PCR. (I) HCT116 cells transfected with c-Myc siRNA or sc-siRNA were infected with pks+ or pks− E. coli and c-Myc and SENP1 expression levels were assessed by western blot. (J) HCT116 cells transfected with c-Myc siRNA or sc-siRNA were infected with pks+ E. coli, and SA-β-gal positive cells were determined. (K) Conditioned media were derived from HCT116 cells transfected with c-Myc siRNA or sc-siRNA and infected with pks+ E. coli. Conditioned media were used to culture HCT116 cells, and cellular growth was assessed by a MTT assay. All data are presented as means±SEM (n=6/group; NS, not significant; *p<0.05; **p<0.01; ***p<0.001, by Mann–Whitney test (A, B, D, E, J and K) or by analysis of variance (F)).
We next investigated the role of miR-20a-5p in senescence and cell proliferation. HCT116 cells were transfected with mature miR-20a-5p or a miR-control and SA-β-gal positive cells were counted 5 days after transfection. In cells transfected with miR-20a-5p, a significant increase in the number of senescent cells was seen compared with cells transfected with miR-control (figure 4E). In addition, the transfection of an anti-miR-20a-5p in HCT116 cells significantly decreased the percentage of senescent cells and the level of SUMO-conjugated p53 upon pks+ E. coli infection, in contrast to the transfection of an anti-miR control (figure 4F and online supplementary figure S10A, respectively). As observed after pks+ E. coli infection, cells transfected with miR-20a-5p produced a significant level of HGF and their conditioned media induced cell proliferation (see online supplementary figures S11A,B). In contrast, anti-miR-20a-5p abolished pks+ E. coli-induced HGF expression and cell proliferation induced by conditioned media obtained from pks+ E. coli-infected cells (see online supplementary figures S11C,D). These results show that pks+ E. coli induce senescence via miR-20a-5p and, subsequently, can stimulate cell proliferation.
MiR-20a-5p is known to be regulated by the transcription factor c-Myc,33 which is involved in DNA damage response.34 We observed that pks+ E. coli infection induced c-Myc expression (figure 4G) and c-Myc binding to the miR-20a-5p promoter (see online supplementary figure S12). Transfection of pks+ E. coli-infected cells with c-Myc siRNA reduced the level of SUMO-conjugated p53 (see online supplementary figure S10B), strongly reduced the expression of miR20a-5p, increased the expression of SENP1, decreased the number of pks+ E. coli-induced senescent cells and consequently, abolished the pro-proliferative effect of conditioned media derived from pks+ E. coli-infected cells (figure 4H–K). These findings suggest that c-Myc plays an upstream role in the cellular signalling cascade induced by pks+ E. coli. Finally, we observed that pks+ E. coli induced c-Myc expression 3 days after infection (see online supplementary figure S13A), followed 2 days later by modifications of miR-20a-5p (see online supplementary figure S13B) and SENP1 expression levels (see online supplementary figure S13A). Altogether, our data demonstrate that pks+ bacteria regulate miR-20a-5p expression and cellular senescence via c-Myc.
Modified expression of miR-20a-5p, SENP1 and growth factors in mouse and human colon tumours colonised by pks+ E. coli
To confirm the reliability of our findings, we explored the impact of a clinical E. coli strain isolated from a human CRC biopsy specimen and harbouring the pks island (CCR20) or its isogenic mutant (CCR20Δpks) in an AOM/DSS colon tumour mouse model (see online supplementary figure S14A). Bacterial colonisation of the intestinal tract (faeces and intestine tissues) was persistent and similar in CCR20- and CCR20Δpks-infected mice (see online supplementary figures S14B,C). CCR20 and CCR20Δpks were predominantly found in colon tumours (see online supplementary figure S14C), as previously reported in human CRC biopsy specimens.15 ,16 CCR20 colonisation significantly increased the number of tumours in comparison with CCR20Δpks colonisation (figure 5A), without affecting the inflammatory score, neoplastic grade or the size of the tumours (see online supplementary figures S14D–F). Furthermore, no significant difference in tumour numbers was seen between CCR20Δpks-infected mice and uninfected mice (figure 5A). In addition, DNA damage (γH2A.x), miR-20a-5p expression level and senescence markers (SA-β-gal activity and p21Cip expression) were significantly higher in tumours isolated from mice colonised by CCR20 than in those isolated from mice colonised by CCR20Δpks (figure 5B–D). Finally, CCR20 induced high levels in tumours of both HGF mRNA and activated HGF receptors (phosphorylated c-Met) (figures 5B,E, respectively). Interestingly, redundancy analysis (RDA) showed a correlation between these factors and discriminated between tumours according to their pks E. coli status (figure 5F).

Clinical pks+ E. coli (CCR20) induced the miR-20a-5p/HGF pathway and consequently promoted tumourigenesis in an AOM/DSS mouse model. (A) Number of tumours per mouse at the end of the AOM/DSS protocol (n=10/group; *p<0.05; **p<0.01; NS, not significant by analysis of variance). (B) γH2A.x, p21cip, and phospho-c-Met (p-c-Met) protein levels in colon tumours were assessed by western blot. The picture presents representative results obtained from four samples in each group (colonised with pks+ E. coli or pks− E. coli). Bar graphs on the right represent the densitometric quantification in the entire tissue collection (n=10/group; *p<0.05; ***p<0.001, by Mann–Whitney test). (C) Mature miR-20a-5p levels in colon tumours from AOM/DSS-treated mice were assessed by qRT-PCR (n=10/group; *p<0.05, by Mann–Whitney test). (D) SA-β-gal activity was assessed in colon tumours from AOM/DSS-treated mice (n=10/group; ***p<0.001, by Mann–Whitney test). (E) HGF mRNA levels in colon tumours from AOM/DSS-treated mice were assessed by qRT-PCR (n=10/group; **p<0.01, by Mann–Whitney test). (F) Redundancy analysis of SA-β-gal activity and expression levels of miR-20a-5p, HGF mRNA, γH2A.x, p21cip and p-c-MET in colon tumours from pks+ E. coli or pks− E. coli colonised mice after AOM/DSS treatment (n=10/group).
To substantiate these results, we investigated human colon adenocarcinomas colonised by pks+ E. coli or pks− E. coli (eight and nine patients/group, respectively). There was no significant difference in tumour node metastases stage, neoplastic grade, inflammatory score, or bacterial colonisation between tumours from the two groups (see online supplementary table S1). However, human biopsy specimens colonised by pks+ E. coli harboured high expression levels of γH2A.x (DNA damage marker), miR-20a-5p, senescence markers (SA-β-gal, p21Cip), HGF mRNA and activated HGF receptors, as found in the AOM/DSS mouse model, and a decrease in SENP1-expressing cells (figure 6A–E). These biological stigmas significantly differentiated human colon adenocarcinomas colonised by pks+ E. coli from those colonised by pks− E. coli using RDA (figure 6F). Overall, these data indicate that the miR-20a-5p/SENP1/senescence/HGF pathway is activated by pks+ E. coli in human and murine colon adenocarcinomas, as seen in vitro, and therefore might contribute to the development of CRC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Modified expression pattern of miR-20a-5p, SENP1 and HGF in human pks+ E. coli-colonised colon tumours. (A) γH2A.x, p21cip, and phospho-c-Met (p-c-Met) protein were assessed by western blot in biopsy samples of human colon cancers. Representative results obtained from four samples in each group (colonised with pks+ E. coli or pks− E. coli) are shown. Bar graphs on the right represent the densitometric quantification in the entire tissue collection (**p<0.01; ***p<0.001 by Mann–Whitney test). (B) Mature miR-20a-5p levels were assessed by qRT-PCR (**p<0.01, by Mann–Whitney test). (C) SA-β-gal activity was assessed in human biopsy samples (n=10/group; *p<0.05, by Mann–Whitney test). (D) HGF mRNA expression levels were assessed by qRT-PCR (*p<0.05, by Mann–Whitney test). (E) SENP1 immunohistochemical staining in a human biopsy samples. Scale bars: 50 µm. (F) Redundancy analysis of SA-β-gal activity, number of SENP1 positive epithelial cells, and expression levels of miR-20a-5p, HGF mRNA, γH2A.x, p21cip and p-c-Met in human biopsy samples colonised with pks+ E. coli or pks− E. coli.
Discussion
Pks+ E. coli were recently shown to be over-represented in human colorectal tumours21 ,22 and to have a carcinogenic effect.21 In addition, it has been reported that genes present in the pks island are significantly induced during development of inflammation and CRC.35 In this study, using mouse models and human CRC biopsy specimens, we show that colibactin-producing E. coli contribute to the emergence of senescent cells, which enhance tumour promotion via growth factor secretion (see online supplementary figure S15). A short time of contact between tumour cells and colibactin-producing E. coli is sufficient to stimulate tumour growth in the xenograft model, suggesting that even a transient colonisation could affect tumour fate.
The toxic effect of pks requires direct bacteria–host cell contact19 and thus an environment in which bacteria can more readily access the epithelium. Consequently, pks+ E. coli might not induce adenocarcinomas in healthy colon epithelium,21 which is protected by a mucus layer and the production of antibacterial peptides. Arthur et al21 developed a model in which inflammation mediated by IL-10 gene knockout alters the natural barrier function, thereby facilitating adhesion of pks+ bacteria to colonic epithelial cells. In the murine AOM/DSS model, the non-genotoxic compound DSS is known to promote inflammation and may therefore favour contact between bacteria and intestinal epithelial cells. In addition, tumoural tissues are known to have altered mucosa barrier function,36–38 suggesting that colon cancer also creates an environment suitable for the adhesion of bacteria to the colonic epithelial cells.
Initiation of cancer requires genomic changes within cells such as point mutations or chromosomal rearrangements, which activate oncogenes or inactivate tumour suppressors.3 While genetic changes are responsible for many aspects of cancer development, they cannot alone, promote tumour growth and progression. Inflammation is known to play a major role in tumour promotion especially in CRC.39 However, like Arthur et al21 we did not find any significant increase in inflammatory score in the colon of pks+ E. coli-infected mice, which suggests that these bacteria may enhance tumour growth by mechanisms other than inflammation.
We observed that pks+ E. coli could sustain tumour development by inducing cellular senescence. Colibactin-producing E. coli-induced senescent cells acquire a SASP characterised by the production of growth factors, as seen for tumour-associated fibroblast.40 We found that HGF produced by senescent intestinal epithelial cells induced proliferation of uninfected cells both in vitro and in vivo. Interestingly, the production of HGF, which is a key determinant of colon cancer progression, a marker of poor prognosis and a target for treatment,41 ,42 was significantly increased in CRC biopsy specimens colonised by pks+ E. coli compared with those harbouring pks− E. coli. This was correlated with an increase in SA-β-gal activity in mouse and human tumours colonised by pks+ E. coli compared with that seen in tumours colonised by pks− E. coli. Most senescent cells undergo cell cycle arrest; however, experiments using intestinal epithelial cells treated with doxorubicin showed that a small fraction of senescent cells escaped from senescence and underwent improper divisions.43 We cannot rule out the possibility that these cells participated in the tumour growth seen in our study.
The mechanism underlying pks+ E. coli-induced senescence involves c-Myc, miR-20a-5p and SENP1 (see online supplementary figure S15). Colibactin-producing E. coli increased miR-20a-5p expression in infected cells (via c-Myc transcription factor), which bound to SENP1 mRNA 3'UTR, resulting in its translational silencing. Interestingly, we observed a similar deregulation of SENP1 and miR-20a-5p expression in human CRC biopsy specimens colonised by pks+ E. coli. SENP1 is a negative regulator of p53 SUMOylation.30 ,44 Accordingly, SENP1 downregulation induced by colibactin-producing E. coli or by transfection of miR-20a-5p triggered an accumulation of SUMO-conjugated p53, which is consistent with the predominant enzymatic activity of SENP144 and known to be a key step in the regulating framework that triggers cellular senescence.29 ,30 However, pks+ E. coli-induced senescence was also found in p53−/− cells suggesting that other pathways, in addition to p53, may be involved in the senescent process.
In conclusion, our data support a new paradigm for colorectal carcinogenesis, in which colibactin-induced senescence has a critical role. Colibactin-producing bacteria induce the emergence of senescent cells, which promote tumour growth by the secretion of growth factors, notably, HGF. Targeting colibactin production may therefore be an efficient strategy to restrain the production of pro-tumourigenic factors.
Acknowledgments
We thank Professor Andrew D Sharrocks (Faculty of Life Sciences, University of Manchester, UK) for kindly providing us with vector-encoding wild-type or mutated SENP1. We are grateful to Marlène Jan for her excellent technical assistance and the ICCF platform of the Université d'Auvergne for technical assistance with histology.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
AC and GD contributed equally.
-
Contributors Study design: AC, GD, RB; acquisition of data and interpretation: AC, GD, RM, EB, LG, MB, CD, PS, PD, JD, DP, HW and RB; drafting of the manuscript: RB, GD and AC; critical revision of the manuscript: RB and AD-M.
-
Funding This work was supported by the Ministère de la Recherche et de la Technologie, the Institut National de la Santé et de la Recherche Médicale (UMR Inserm U1071), the Institut National de la Recherche Agronomique (USC-2018), the Ligue Contre le Cancer and the Centre Hospitalier Régional Universitaire de Clermont-Ferrand, France.
-
Competing interests None.
-
Ethics approval Research ethics committee of University of Clermont-Ferrand, France.
-
Provenance and peer review Not commissioned; externally peer reviewed.