Article Text

Abstract
The stomach is traditionally regarded as a hollow muscular sac that initiates the second phase of digestion. Yet this simple view ignores the fact that it is the most sophisticated endocrine organ with unique physiology, biochemistry, immunology and microbiology. All ingested materials, including our nutrition, have to negotiate this organ first, and as such, the stomach is arguably the most important segment within the GI tract. The unique biological function of gastric acid secretion not only initiates the digestive process but also acts as a first line of defence against food-borne microbes. Normal gastric physiology and morphology may be disrupted by Helicobacter pylori infection, the most common chronic bacterial infection in the world and the aetiological agent for most peptic ulcers and gastric cancer. In this state-of-the-art review, the most relevant new aspects of the stomach in health and disease are addressed. Topics include gastric physiology and the role of gastric dysmotility in dyspepsia and gastroparesis; the stomach in appetite control and obesity; there is an update on the immunology of the stomach and the emerging field of the gastric microbiome. H. pylori-induced gastritis and its associated diseases including peptic ulcers and gastric cancer are addressed together with advances in diagnosis. The conclusions provide a future approach to gastric diseases underpinned by the concept that a healthy stomach is the gateway to a healthy and balanced host. This philosophy should reinforce any public health efforts designed to eradicate major gastric diseases, including stomach cancer.
- GASTRIC DISEASES
- GASTRIC MOTILITY
- GASTRIC PHYSIOLOGY
- IMMUNOLOGY
- OBESITY
Statistics from Altmetric.com
Historical introduction
Digestion and the role of the stomach in maintaining health have interested man since early times (reviewed in ref. 1). The ancient Greeks noted the bitter-sour nature of the gastric contents and, in the 16th century, both Paracelsus2 and van Helmont3 believed acid to be present in the stomach and necessary for digestion. Subsequent observations by Reaumur4 and Spallanzani5 suggested the ‘solvent’ effects of gastric juice on animal tissue, but it was not until 1823 that William Prout published his work on the nature of gastric acid secretion.6 The first observations of William Beaumont on his gastric fistula patient, Alexis St Martin, were published only 3 years later in 1826.7 His meticulous observations over almost a decade described gastric digestion in a human during normal life experiences including the effects of stress.
In the early 20th century, control of gastric secretion was explored by ablation of the coeliac axis and vagotomy as therapeutic interventions and this emphasised the complex nature of the control of gastric secretion. This led to a rapid increase in interest and the work of Dale and Laidlaw on histamine,8 which led to the critical discovery by Popielski of histamine's effect on gastric secretion,9 Bayliss and Starling's discovery of secretin10 and Edkins’ treatise on gastrin.11 These discoveries ushered in a new era in our understanding of gastric disease leading to the dramatic advances in the pharmacological management of peptic ulcer disease with the discovery of the H2-receptor antagonists by Sir James Black in 1972.12 The emphasis on acid-related disease preoccupied research in the middle and latter half of the 20th century until the groundbreaking discovery of Helicobacter pylori in 1983 by Marshall and Warren.13 This was counterintuitive to then current thinking, where the stomach was considered microbiologically sterile, despite the many observations of numerous bacterial populations in gastric juice among others by Jaworski14 and the Nobel Prize–winning (1908) contribution of Metchnikoff for his work with Lactobacillus and gut immunity.15 Some clinicians at that time already advocated aspiration of gastric juice in search of flagellated bacilli in patients with suspected gastric ulcer disease, followed by prescription of high doses of bismuth salts.16
The importance of the gastric microbiome and the relationship to H. pylori is revolutionising our understanding of gastric cancer, and especially prevention. Neurohormonal research is leading to a better understanding of the control of appetite, food absorption, metabolism and obesity. Increasing evidence suggests a potential role of the gastric microbiome in several mechanisms of digestion.
This review builds on the inspiring and insightful research directions of our predecessors and brings together the latest clinically relevant information in current areas of gastric research with a particular emphasis on establishing and maintaining a healthy stomach.
Gastric hormones
Hormones regulate several important physiological functions within the stomach, including secretion and motility. The abnormal production of some of these hormones is associated with the development of various gastric diseases. However, several hormones (gastrin, somatostatin and ghrelin) and regulatory peptides are produced by cells within the stomach itself. Several other hormones that are secreted in the more distal portions of the GI tract (eg, cholecystokinin, glucose-dependent insulinotropic peptide) also regulate gastric function but are beyond the scope of this review.
One important hormone secreted by the stomach is ghrelin. Ghrelin is orexigenic (increases appetite) and serum concentrations of this hormone are elevated before a meal and suppressed postprandially. It is produced by endocrine cells in the stomach and regulates appetite by crossing the blood–brain barrier to bind to receptors located on cells in the hypothalamus, as well as by signalling through vagal afferent nerve fibres.17 Ghrelin plays a major role in the short-term regulation of food intake, and exogenous administration stimulates the appetite in humans and rodents. Antagonism of this system has been investigated as a potential treatment for obesity, but results to date have been disappointing.18 The possibility of using ghrelin-related molecules to treat conditions such as cancer cachexia is also under investigation.19 The stomach also produces small amounts of the anorexigenic hormone leptin, although the main source of leptin is adipose tissue. Therapeutic manipulation of the ghrelin/leptin system has not yet found a role in routine clinical practice.
The other main hormone produced by the stomach is gastrin, which continues to be of major current relevance to clinical gastroenterologists in view of its association with various GI diseases. Gastrin is secreted by G cells, which are predominantly located in the antrum of the stomach. It has well-known functions in regulating gastric acid secretion (reviewed in ref. 20). Gastrin is secreted in response to food ingestion and binds to CCK2 receptors on gastric enterochromaffin-like cells, stimulating them to release histamine, which in turn binds to H2-receptors on parietal cells to stimulate them to secrete hydrochloric acid. The secretion of gastrin is inhibited by somatostatin, which is secreted by D cells within the stomach and the intestine. Recent evidence suggests that gastrin also regulates other important cellular pathways in the stomach including cell proliferation, migration, invasion, angiogenesis and apoptosis (recently reviewed in ref. 21) and a recent in vitro study showed that gastrin plays a key role in maintaining gastric stem cells.22 Some of these effects arise as a result of the altered expression of proteins that regulate tissue remodelling including members of the matrix metalloproteinase,23 tissue inhibitor of metalloproteinase24 and urokinase plasminogen activator25 families.
Elevated fasting serum gastrin concentrations can result from an appropriate physiological response to gastric hypochlorhydria. It may also occur from autoimmune or H. pylori-induced chronic atrophic gastritis, pharmacological inhibition of gastric acid secretion or from an ectopic source, such as a gastrinoma. The latter condition leads to the Zollinger–Ellison syndrome (ZES), which is characterised by florid peptic ulceration and diarrhoea as a result of increased gastric acid secretion. However, the magnitude of the hypergastrinaemia associated with hypochlorhydric states is variable. For example, many patients who take regular proton pump inhibitors (PPIs) have normal fasting serum gastrin concentrations, while others develop hypergastrinaemia. The latter group includes patients with concomitant atrophic gastritis and/or impaired gastric emptying.26 It is possible that components of the gastric microbiome may additionally affect fasting serum gastrin concentrations and that these bacteria may act as co-factors with hypergastrinaemia to influence susceptibility towards tumour development in the stomach.26 Further research on this topic is needed.
Evaluating the cause of hypergastrinaemia can sometimes be difficult, but is essential for guiding management, particularly of patients with ZES. In some cases, a secretin stimulation test may be helpful.27 Although the measurement of serum gastrin concentrations appears to be straightforward, recent data suggest that some of the commercially available ELISA kits used to perform this assay can yield varying results, and therefore, results need to be interpreted with caution in certain clinical situations.28 Interpretation also needs to take into account factors such as PPI use and the presence or absence of H. pylori infection.27
Hypergastrinaemia has been associated with gastric tumour development. Transgenic hypergastrinaemic (INS-GAS) mice spontaneously develop gastric adenocarcinoma with increasing age, and this process is accelerated by Helicobacter felis or H. pylori infection29 ,30 and by other components of the gastric microbiome.31 The precursor hormone progastrin may also influence gastric antral tumourigenesis by expanding CCK2 receptor positive antral stem cells.32 Chronic atrophic gastritis induced by H. pylori infection in humans is a precursor of gastric cancer development and is associated with hypergastrinaemia. However, the evidence that gastrin or its precursor peptides play a major, direct role in the development of human gastric adenocarcinoma remains controversial.33 Measurement of fasting serum gastrin concentration in combination with the pepsinogen I/II ratio has been proposed as a non-invasive screening tool for detecting chronic atrophic gastritis and is used for this purpose in several regions, but a recent report has questioned the accuracy of this approach.34
There is, however, strong evidence that gastrin plays a major stimulatory role in the development of both type I and II gastric neuroendocrine tumours (NETs). Type I gastric NETs arise in the setting of autoimmune atrophic gastritis and pernicious anaemia, whereas type II gastric NETs are associated with a gastrinoma, usually in the setting of multiple endocrine neoplasia-1. The crucial importance of hypergastrinaemia in causing these conditions has major therapeutic implications. Antrectomy or gastrinoma resection to remove the source of hypergastrinaemia,35 as well as treatment with the orally bioavailable CCK2 receptor antagonist drug, netazepide, leads to resolution of some type I gastric NETs in humans.36 ,37 The role of CCK2 receptor antagonist drugs in various other conditions that are associated with hypergastrinaemia warrants further investigation.
Neurogastric physiology
Altered motility and increased sensitivity to mechanical distention have been implicated in the generation of symptoms from the stomach, especially in patients with functional dyspepsia (FD) and gastroparesis.
Gastric sensitivity
The GI tract conveys information to the brain through mechanosensitive and nutrient sensing pathways and both may induce perception. Traditionally, sensing of the presence of nutrients is considered to be volumetric by the stomach and nutritive in the intestine.38–40 This concept implies a dominant role for mechanoreceptors in gastric nutrient sensing and no role for gastric chemosensing in food-related conscious perception.38 ,39
On the other hand, the stomach is not totally devoid of chemosensing capacity: the presence of amino acids and peptones in the gastric lumen stimulates gastric acid secretion through enhanced gastrin release from G-cells, which express the pH-sensitive and peptone-activated calcium sensing receptor.40 To date, such a pathway has not been implicated in perception of gastric filling or the generation of satiation signals in man.
Interdigestive motility
In the interdigestive state, upper GI motility is characterised by the recurrent contractility pattern of the migrating motor complex (MMC). Although well preserved throughout mammalian species, the role of the MMC has remained unclear. Recent observations have identified a role for gastric phase III of the MMC in signalling the return of hunger after meals.41 Surprisingly, in man, motilin rather than ghrelin was identified as the initiator of phase III and the associated increase in hunger.42 The clock or luminal sensing mechanism that controls release of motilin from the duodenum remains to be identified.43
Intragastric pressure as a measure of gastric accommodation
Gastric accommodation is a relaxation of the proximal stomach, triggered by the arrival of nutrients in the oropharynx, stomach or duodenum, which allows storage of the ingested meal (figure 1).44

Schematic outline of neural pathways and receptors involved in the control of the accommodation reflex in man. The identity of the nutrient sensors that trigger activation of the accommodation reflex and of other potentially involved neurotransmitters and receptors requires additional studies. CNS, central nervous system; cGMP, cyclic guanosine monophosphine; NO, nitric oxide; VIP, vasoactive intestinal peptide.
The classical concept that “gastric accommodation serves to prevent a rise in intra-gastric pressure (IGP) during food intake” was recently challenged. In studies in man, nutrient ingestion induces an initial drop in intra-gastric pressure (IGP), followed by gradual pressure recovery until maximal satiation (figure 2A).45 ,46 The drop in IGP is mediated by nitric oxide and contributes to gastric nutrient volume tolerance while the rise in IGP from nadir determines satiation.45 ,46 These changes are sensitive to a number of mechanical and pharmacological interventions, suggesting a potential target for therapeutic interventions aimed at controlling meal volume.45 ,47–50

(A) Example of intragastric pressure response and satiation score evolution during intragastric nutrient infusion. Nutrient infusion induces an initial drop in pressure, followed by gradual recovery associated with rising satiation scores. (B) Schematic conceptual model of gastric mechanoreceptors, relative to the muscular compartment. The four panels represent the modelled differential behaviour of ‘in series’ tension receptors and ‘in parallel’ elongation receptors during distension or contraction.
Role of gastric dysmotility as a cause of symptoms in dyspepsia and gastroparesis
Historically, delayed gastric emptying has been considered the major mechanism underlying symptoms in FD and gastroparesis. Recent studies in diabetic and idiopathic gastroparesis showed a poor correlation of the pattern and severity of symptoms with the severity of delay in gastric emptying.51 ,52 This was confirmed in a recent integrated statistical analysis of prokinetic therapy trials in idiopathic and diabetic gastroparesis.53 Studies in both idiopathic and diabetic gastroparesis have identified other mechanisms, including visceral hypersensitivity and impaired gastric accommodation as stronger determinants of the symptom pattern and severity.54 ,55 However, a recent controlled trial targeting visceral hypersensitivity with nortriptyline in idiopathic gastroparesis also failed to demonstrate meaningful symptomatic benefit.56 Identification of the mechanoreceptors in the stomach whose inappropriate activation generates symptoms in disordered motility may unravel novel therapeutic targets for symptom control.38 ,39 ,57
Identity of gastric mechanoreceptors
Gastric filling and the accompanying pressure changes are likely to play a major role in gastric satiation signalling and sensitivity to distention (figure 1). In humans, low-volume distension of the proximal stomach induces sensations of satiety and higher volume distention leads to discomfort, nausea and pain.58 Animal studies have shown that gastric distension triggers stretch-sensitive (‘in parallel’) as well as tension-sensitive (‘in series’) mechanoreceptors that convey information to the brain via vagal and splanchnic nerves.38–41 ,59 The response of different mechanoreceptors to distention or relaxation varies according to the type (figure 2B). Studies using isobaric and iso-volumetric distensions of the proximal stomach support the hypothesis that gastric mechanosensitivity in man relies mainly on tension-sensitive (‘in series’) mechanoreceptors.38 ,39 ,60 ,61 Hence, gastric smooth muscle relaxation is expected to decrease activation of these mechanoreceptors.
Enhancing accommodation in FD
Based on the putative involvement of tension-sensitive mechanoreceptors, decreasing gastric smooth muscle tone may decrease symptoms induced by gastric filling, for instance, in patients with impaired gastric accommodation. Pharmacological studies in healthy volunteers identified the 5-HT1 receptor as a target for enhancing gastric relaxation62 ,63 (figure 1). This approach was evaluated in FD, showing a beneficial symptomatic effect of enhancing gastric accommodation by the anxiolytic 5-HT1A agonist buspirone in a pilot study64 and by tandospirone in a multicentre trial.65 In both studies, anxiolytic effects did not explain the symptomatic benefit on FD symptoms. Acotiamide is a combined muscarinic autoreceptor antagonist and cholinesterase inhibitor, which also targets impaired accommodation, among other mechanisms.66 In a phase III study in Japan, acotiamide was superior to placebo in improving functional dyspeptic symptoms,67 leading to approval for this indication in Japan. A phase III programme is ongoing in Europe.
Role of impaired accommodation in the overlap between dyspepsia and GORD
In health and disease, transient lower oesophageal sphincter relaxations (TLESRs) are the main mechanism underlying reflux events. TLESRs occur mainly during the postprandial period, triggered by gastric distension, which activates mechanoreceptors in the proximal stomach. Gastric accommodation is the physiological response to meal-induced gastric distention, and so its relation to TLESR and reflux events has been studied in health and in patients with GORD.68 Using IGP as a marker of gastric accommodation, a significant negative correlation was found between meal-induced accommodation and the occurrence of TLESRs and reflux events in the first postprandial hour, both in health and disease.68 This mechanistic observation suggests that accommodation and TLESRs are closely linked, probably through activation of tension-sensitive mechanoreceptors that also trigger TLESRs (figure 3). This may help explain the frequent overlap between dyspepsia and GORD. Moreover, treatment targeting impaired accommodation has the potential to be beneficial not only to FD but also to patients with GORD.

Schematic outline of gastric events and their relationships in response to nutrient ingestion. TLESR, transient lower oesophageal sphincter relaxation.
Emerging role of gastric nutrient sensing in the perception of gastric filling and control of hunger and satiation
The concept of gastric nutrient perception being exclusively mechanosensitive or volumetric is challenged by recent data. First, animal studies have shown the expression of taste receptors on ghrelin cells in the stomach, and bitter taste receptor agonists can alter ghrelin release, gastric motility and food intake in mice.38 ,69 Second, preliminary evidence in man shows that intragastric administration of a bitter taste receptor agonist inhibits the drop in IGP and the amount of food ingested until maximum satiation.70 Transient receptor potential channels are involved in luminal sensing and can influence gastric motility and nutrient tolerance in man.50 ,71 The identity and expression of gastric nutrient sensors in man requires further study, and the effect of activation or inhibition on gastric sensorimotor function may lead to novel therapeutic approaches.
The stomach in appetite control and obesity
Control of appetite
The vagus nerve innervates regions of the GI tract involved in calorie intake, satiation and digestion,72 and it serves as a crucial link between the brain, brainstem and gut. The afferent fibres of the ventral and dorsal vagal trunks in the abdomen are involved in mediating satiation and, as a result, regulating appetite. Vagal afferents are stimulated by change in viscus tension induced by food passing through the GI tract. The vagus nerve is also stimulated by hormonal mediators activated by mechanical and chemical stimuli. In the stomach, ghrelin secretion inhibits afferent vagal fibres to increase appetite (orexigenic), whereas leptin secreted in the stomach stimulates vagal fibres and induces satiety – an anorexigenic effect. Other anorexigenic hormones, such as CCK, GLP-1 or PYY, are released in the small intestine.
Efferent vagal neuronal fibres control much of the GI motor and secretory functions involved in food digestion and absorption. Partial vagotomy, or total sub-diaphragmatic vagotomy, or intermittent vagal nerve electrical stimulation performed to inhibit vagal function in humans73 decreased food intake and induced early satiety and weight loss. The vagus nerve plays a dual role, interacting with anorexigenic and orexigenic pathways that are altered in obesity74 and may contribute to body weight and glycaemic control.
The roles of the proximal stomach and ghrelin in appetite control are also illustrated by the effects of bariatric procedures. Thus, isolation of the gastric cardia/fundus and exclusion of the distal stomach from ingested food after Roux-Y gastric bypass may initially limit caloric intake by induction of nausea (and rarely vomiting), thereby discouraging overeating. In addition, stimulation of the gastric mechanical and chemical receptors, rapid emptying of the remaining stomach and release of ghrelin may also contribute to the induction of weight loss.75 ,76
Ghrelin (see section ‘Gastric hormones’) is the most relevant gastric hormone involved in appetite. It is produced from the pre-pro ghrelin gene and undergoes cyclical changes in blood concentrations during fasting and postprandially, reaching highest levels during fasting. Acyl-ghrelin (AG) is metabolised by the ghrelin activating enzyme, ghrelin-O-acyltransferase, to deacyl-ghrelin (DAG). AG and DAG have different physiological effects: AG increases gastric emptying and appetite, whereas DAG decreases gastric emptying, induces postprandial fullness and improves insulin sensitivity.77
Gastric motility, sensation and satiation in obesity
In the vast majority of affected individuals, obesity involves overconsumption of food relative to calorie requirements. The control of appetite is partly determined by hedonic mechanisms, where food consumption affects brain systems associated with pleasure and reward, such as dopaminergic D2 and opioidergic mechanisms in areas such as the ventral tegmental area and the nucleus accumbens. The second homeostatic mechanisms are centred in the arcuate and paraventricular nucleus of the hypothalamus. Until recently, the focus of medical and behavioural therapy was directed to these central mechanisms including the recently launched medications such as bupropion-naltrexone, phentermine-topiramate, lorcaserin or the GLP-1 receptor agonist, liraglutide. However, these treatment approaches result in an average weight loss of ≤5 kg in clinical trials. The greater effectiveness of bariatric surgery, particularly Roux-en-Y gastric bypass, and sleeve gastrectomy clearly suggests that the stomach may play an important role in the control of appetite and food intake.78
The sensory function of the stomach is, in part, determined by its motor functions such as tone and compliance, and by the rate of emptying. However, studies of gastric emptying in normal weight and obese persons have shown inconsistent results (reviewed in ref. 79). Gastric capacity was larger in obese persons when tested with an intragastric latex balloon filled with water. In contrast, other studies using the barostat or imaging (single-photon emission CT) techniques reported no differences in gastric volume or compliance between non-bulimic obese and lean subjects (reviewed in ref. 79). Recent studies from >500 patients ranging from normal body mass index (BMI) to class III obesity showed that gastric emptying of solids is faster and fasting gastric volume larger in obesity,80 confirming results in a prior, smaller study.81 In addition, other alterations in quantitative GI and behavioural traits associated with obesity were reduced satiation and satiety, altered body image, disorders of affect and reduced exercise.80 Principal component analysis80 identified latent dimensions that accounted for approximately 81% of the variation among overweight and obese subjects, including satiety or satiation (21%), gastric motility (14%), psychological factors (13%) and gastric sensorimotor factors (11%).
Increased body mass and fasting gastric volumes are independently associated with delayed satiation under standard laboratory conditions of food ingestion. Thus, Delgado-Aros and colleagues showed that, across a broad spectrum of BMI, there was an association between higher BMI, higher fasting gastric volume and decreased satiation (figure 4), manifested as reduced symptoms of fullness and a higher maximum tolerated volume of a nutrient drink ingested at a constant rate in a laboratory setting.82 ,83

Caloric intake at maximum satiation by gender and body mass index. There was higher caloric intake at maximum satiation in male subjects compared with women (left). Reproduced from ref. 82.
Figure 4 shows the higher maximum tolerated volume in obese compared with normal or underweight participants; an increase of 50 mL in the fasting gastric volume was associated with 114±32 kcal (479±134 kJ) more ingested at maximum satiation.82 ,83 These findings suggest that individuals with a higher BMI require more food to reach satiation (and, by inference, to signal termination of meal ingestion), and, over time, this results in higher caloric intake and weight gain (figure 4).
Other data support the importance of behavioural adaptation; thus, obese individuals have more severe symptoms of fullness, bloating, nausea and pain when reaching maximal satiation than individuals without obesity, and yet they continue to ingest calories,82–87 consistent with a behavioural adaptation to satiation.88 The additional understanding of the role of the stomach in obesity ushers in a new era when new medications, devices (such as balloons and drains or internal liners) and endoscopic interventions may prove more efficacious than the drugs targeting central mechanisms by targeting the GI functions that are critical for appetite and food intake.78
Immunology of the stomach
Until the discovery of H. pylori, the immunological complexity of the stomach was not recognised nor studied in detail. What has emerged is an appreciation of the multiple interactions that regulate the host and the gastric microbiome including genetics, diet and environmental factors. Of particular interest is the intimacy of the immune–epithelial cell interactions that impact most of the major diseases associated with colonisation.89
In view of the mass of microbial and dietary antigens within the intestinal lumen, it is essential that the host selects for a useful and protective response. This is believed to be achieved by the differential production of cytokines. The gastric immune responses entail a broad representation of innate (epithelial, neutrophil, macrophage, dendritic cell) and adaptive (B and T cell) components. In particular, the heterogeneity and functional consequences of the helper T cell (Th) responses have been studied in detail. Th cells are defined by their cytokine production or function. Th1 (interferon-γ producers), Th17 (interleukin (IL)-17 producers) and regulatory Th cells (Treg, mediate tolerance) are all activated in response to infection. These are important as their cytokines regulate a range of processes from acid secretion to epithelial cell turnover and the production of other cytokines that recruit and activate other immune/inflammatory cells. This response cascade is found, in particular, in H. pylori-infected tissue, leading to an immunological paradox in that H. pylori are broadly immunogenic, but despite these host responses, infection generally persists for life (box 1). A further clue for the understanding of mechanisms of H. pylori colonisation came from two independent genome-wide association studies and their meta-analysis correlating a functional TLR-1 gene polymorphism to be associated with H. pylori.90
While epithelial cells in the uninfected stomach die and can be engulfed by antigen-presenting cells (APC) in the lamina propria, these responses are accelerated in response to infection. The engulfment can include microbial cargo that is processed (figure 5) and leads to the activation of Th1 and Th17 cells. Both of these subsets of helper T cells produce cytokines that stimulate the epithelial and other cells to produce chemokines that recruit and activate neutrophils and/or macrophages capable of producing reactive oxygen species (ROS) or reactive nitrogen species (RNS). These Th cell responses also stimulate B cells as robust gastric IgG and IgA responses are associated with infection. To keep these responses in check, regulatory T cells (Treg) produce mediators that can inhibit all aspects of gastritis. One consequence of Treg is they contribute to persistent infection by curtailing potentially protective responses. Further, the dying cells are replaced by stimulated cells. While epithelial stem cells renew, some data suggest that stem cells derived from bone marrow are capable of seeding the epithelial progenitor pool and these cells appear to be more prone to malignant transformation.

Immunological interactions in response to H. pylori infection. APC, antigen-presenting cells; IFN, interferon; IL, interleukin; TNF, tumour necrosis factor.
Major areas of current research include studies of the mechanisms of persistence and how they may be circumvented (eg, through vaccines), the role of the host response in the outcome of long-term colonisation, particularly cancer, and the implications of long-term colonisation for health and disease.
Several compelling studies confirm that persistent infection accounts for the diseases caused by H. pylori. First, infection in childhood is associated with gastroduodenal ulceration decades later. More recently, persistence has been associated with an increase in Treg.91 ,92 Furthermore, infection of neonatal mice with H. pylori induces Treg that not only decrease gastritis but also confer anti-inflammatory responses in other sites.93 Consistent with this model, the induction of Treg and attenuated gastritis has been observed in paediatric subjects. Depleting Treg using antibodies to CD25 increases gastric inflammation and decreases bacterial burden.91 ,94 Another mechanism implicated in the control of gastritis is induced through the engulfment of apoptotic epithelial cells. This process renders local APC less responsive.95 Given the degree of cell death induced during infection, this process is likely to contribute to novel mechanisms of antigen sampling, as well as impacting inflammation.
Key immunological concepts during infection with H. pylori
H. pylori infects the host persistently, despite robust immune responses.
Most aspects of innate and adaptive immunity are activated by H. pylori.
Tissue and DNA damage is believed to be largely immune-mediated.
Immunity may require even more exuberant host responses.
To date, two mediators produced by Treg have been implicated in the persistence of H. pylori: IL-10 and adenosine. Deficiency of IL-10 contributes to gastritis and the gastritis decreases H. pylori colonisation, largely through the function of neutrophils. However, when trying to enhance immunity, one must be cautious in attempting to impair Treg or key cytokines such as IL-10 since this could lead to autoimmune disease.
Another interesting discussion that has emerged is the role of the host response in regulating mutations in genes within the Cag pathogenicity island. Solnick et al demonstrated that during infection the CagY gene undergoes in-frame mutations that are driven by the host response.96 These mutations can either lead to a gain or loss of function that impact the ability of the bacteria to induce an IL-8 response. This process could contribute to persistence by attenuating host responses to the infection.
H. pylori represent one species that is widespread in countries cited as having lower rates of asthma or IBD. Epidemiological studies suggest H. pylori confer protection against oesophageal adenocarcinoma or other infections including tuberculosis. Mueller et al have published a series of studies showing that neonatal infection with H. pylori induces Treg that inhibit gastritis, favour persistence but also attenuate airway disease in a murine model of asthma.93 However, these epidemiological and mouse model observations do not provide the proof that the benefits of carrying H. pylori outweigh the risk of gastric cancer, and it is entirely possible that sufficiently attenuated strains can be generated that may be proven beneficial when included in a probiotic mix.
Finally, significant research is being done on understanding the role of host responses induced by H. pylori in the pathogenesis of disease. This direction has been strongly supported by the body of literature associating diseases such as gastric cancer, with polymorphisms in genes that encode proteins that regulate the intensity of the host response.97 Research on the role of inflammation in gastroduodenal disease has largely focused on the control of ROS and RNS and their role in tissue or DNA damage.89 It is also apparent that other factors, including obesity and the associated effects on microbial communities and local metabolism, will impact the onset of gastroduodenal diseases.98 Further, interactions between H. pylori and other microbial triggers that may impact autoimmune gastritis are also areas that are relevant to understanding the impact of the broad GI microbial community on gastric health.
The immune and inflammatory responses in the GI tract are normally well suited to protect the tissue without stimulating excessive inflammation. However, in the stomach, the persistent inflammation induced by H. pylori contributes to disease, particularly in genetically susceptible hosts (box 2). Ongoing research will advance our understanding of the gastric immune system, its interaction with diet and the microbiome will provide greater insights into the maintenance of health and prevention or management of disease.
More recent advances and areas of focus in gastric immunology
H. pylori persistence relies on its niche, antigenic variation and ability to induce host responses that favour tolerance.
Host TLR-1 polymorphisms influence the likelihood of persistent H. pylori colonisation.
Host responses regulate mutations in H. pylori that affect its virulence.
Gastric microbiome
Historically, the prevailing view was that the stomach was essentially sterile because of its acidic milieu. However, with the discovery of H. pylori,13 it is now known that the stomach can support a bacterial community with hundreds of phylotypes,99–101 and while pH values <4 prevent bacterial overgrowth, the acidic milieu is not capable of sterilising the stomach.102 The microbial density of the stomach is 101–103 CFU/g.103–105 The stomach, together with the oesophagus and duodenum, is the least colonised region of the GI tract, thus increasing interest in the role of bacteria in gastric health and disease.
The stomach secretes gastric juice, composed mainly of proteolytic enzymes and hydrochloric acid, providing an environment necessary for denaturing of proteins and facilitating the absorption of nutrients. Gastric acid also restricts the quantity of microorganisms entering the small intestine and reduces the risk of infection by pathogens.106 While the human gastric lumen has a pH of 1–2, the mucus layer establishes a pH gradient that increases the pH to 6–7 at the surface of the mucosa.107 ,108 The mucus layer, which consists of several mucins, forms two sublayers, an inner mucus layer that is firmly attached to the epithelium and a variable mucus layer interfacing with the lumen.109 ,110
To understand the dynamics of the gastric microbiota, it is necessary to consider the site of their isolation. Bacteria, and bacterial DNA when isolated from gastric juice, which forms a formidable barrier for most bacterial colonisation, differ from bacterial isolates adhering to the mucosa. The latter presents a more hospitable environment for colonisation. During abnormal or disease states, this balance may be perturbed. Reduction of gastric acid secretion increases the risk of bacterial overgrowth and also influences the composition of intestinal or oral microorganisms, including those organisms causing disease106 and those with nitrosating ability that are not regularly cultured from a normal, healthy stomach.111
Culture versus culture-independent analyses
Using culture methods, the most common phylum, regardless of H. pylori status, is Firmicutes, followed by Proteobacteria and Bacteroides with Actinobacteria the next most prevalent. The most commonly found genera were Streptococcus, Lactobacillus and Bacteroides spp., Veillonella, Corynebacterium and Neisseria spp., which may reflect bacteria that are more easily cultured. Also, several of these organisms are present in the oral and nasal cavities, raising the possibility that their isolation from the stomach may reflect transient passage rather than colonisation. H. pylori status did not influence the ranking of prevalence when the density of H. pylori was not included. These findings and in vivo rodent studies suggest than non-H. pylori species can promote chronic gastritis and cancer.112 ,31 Culture studies demonstrated the fastidiousness of H. pylori and the limitations of culture, and more recent studies have found Proteobacteria the dominant phylum in subjects infected with H. pylori.100 ,101
In eight studies, using four molecular methods the human gastric microbiota was surveyed: next-generation sequencing technologies,101 ,113 ,114 Sanger sequencing of 16S rDNA,97 ,98 a community fingerprinting method to define a library for Sanger sequencing115 ,116 and the PhyloChip.117 Although there is considerable variation in the gastric microbiome between individuals at the genus level, the most prominent phyla detected in the stomach are Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria and Fusobacteria. The most abundant phyla in H. pylori-infected stomachs are Proteobacteria, Firmicutes and Actinobacteria. In the absence of H. pylori, the most abundant phyla are Firmicutes, Bacteroidetes and Actinobacteria. In humans, H. pylori is the most dominant species in the stomach, comprising 72–99% of sequencing readouts.100 ,101 In the absence of H. pylori, analysis consistently notes the presence of Streptococcus spp., possibly originating from the oral or nasal cavities and which appear to be the most abundant genus.99 ,113 ,114 ,116
Acid suppression therapy affects colonisation dynamics
Hypochlorhydria induced by acid suppression is associated with higher levels of gastric nitrites and an increased risk of gastric cancer.118–120 Chronic H2RA therapy or atrophic gastritis increases the growth of nitrosating bacteria that can convert, depending on the pH, nitrite and other nitrogen compounds in gastric juice to produce carcinogenic N-nitroso compounds.111 These reactions are favoured where pH >4 allows the persistence of nitrites by reducing the antioxidant activity of vitamin C, a powerful inhibitor of nitrosation.121 ,122 PPIs also elevate intragastric pH levels to a greater degree than H2RA.123 Studies have reported a logarithmic relationship between intragastric pH and median bacterial counts in gastric juice and increased risks for enteric infections and bacterial-induced diarrhoea.124 ,125 The microbiota in non-H. pylori-infected achlorhydric stomachs can be sufficiently populated with urease-producing bacteria to result in a false positive urea breath test.126
H. pylori influences gastric colonisation dynamics
The gastric microbiota might be a reflection of transient bacteria from the mouth and oesophagus; however, three separate studies demonstrated that in spite of high intersubject variability the gastric microbiota was distinguishable from microbiota found in the mouth, nose and distal GI tract.101 ,113 ,114 In the absence of H. pylori, the structure and composition most resemble the microbiota reported for the distal oesophagus with unique differences due to the make-up of Proteobacteria.101 ,127
The effects of H. pylori on the gastric microbiota are not fully understood. H. pylori density increases with the onset of gastritis,115 which may reflect changes in the gastric niche that allow H. pylori to outcompete other bacteria.100 Some studies note a strong effect of H. pylori on the composition of the gastric microbiota.101 ,117 In one study, H. pylori accounted for 93–97% of all reads in the infected stomach and substantially decreased the diversity as only 33 phylotypes were observed in H. pylori-positive individuals while 262 phylotypes were observed in H. pylori-negative subjects.101 Importantly, multiple studies report the ability to detect H. pylori sequences at extremely low levels in subjects who were H. pylori negative by other diagnostic means.100 ,113 ,115 ,117 This may reflect a host response, leading to reduction of H. pylori or the presence of non-H. pylori Helicobacters.128
Regarding the uniformity of the microbiota within the stomach, selected studies found no differences in the microbiota of the antrum and corpus in their populations, with the exception of decreased Prevotella in the antrum of patients with gastritis.99 ,100 In contrast, others have noted bacterial differences between subjects and between the antrum and corpus.114
Thus, H. pylori and the associated changes in the stomach alter the ecological niche inhabited by the gastric microbiota. However, the gastric microbiota also compete with H. pylori for a gastric niche and may play an important role in the progression of disease. More studies involving the microbiota–host–environment interactions, including the effect of diet and gender, are needed to fully understand the role of gastric bacteria in human health and disease.
Gastritis and gastritis-associated carcinogenesis
The term ‘gastritis’ defines any histologically confirmed inflammation of the gastric mucosa. It is usually classified as acute or chronic, a clinical distinction that does not imply a different profile of the inflammatory cell population.129
Most gastric mucosal inflammation is self-limiting (clinically acute) and causes no permanent anatomical changes. Chronic gastritis, which is not self-limiting gastritis, has different aetiologies,130 but its worldwide epidemiology overlaps that of H. pylori infection.131 ,132
Histology distinguishes two main phenotypes of gastritis: non-atrophic and atrophic. Gastric mucosal atrophy is defined as the loss of ‘appropriate’ glands, and it is consistently recognised as the ‘cancerisation field’ for non-hereditary (so-called ‘epidemic’) intestinal-type gastric adenocarcinoma.129 ,132 ,133
Longstanding (H. pylori-associated) mucosal inflammation results in various structural changes, including ‘absolute’ loss of the resident glands (‘desertification’ of the gastric mucosa) and/or their replacement by inappropriate (metaplastic) glandular units. Both these situations are consistent with the current definition of gastric mucosal atrophy (figure 6). The metaplastic variant of atrophy has two main phenotypes: pseudo-pyloric metaplasia and intestinal metaplasia (IM). By definition, pseudo-pyloric metaplasia (also called spasmolytic polypeptide-expressing metaplasia (SPEM)) only affects the corpus/fundus. Here, native oxyntic glands are replaced by antral-like mucus-secreting glandular units. In contrast, IM can affect both natively antral (mucus-secreting) and SPEM-metaplastic (pseudo-antralised) oxyntic epithelia. The histological diagnosis of IM relies on the detection of mucin-producing goblet cells and/or Paneth cells. Sophisticated histochemical reactions and/or immune-histochemical stains can further classify all these metaplastic variants, but such methods are time-consuming, expensive and, most importantly, have a negligible impact in routine diagnostic practice.
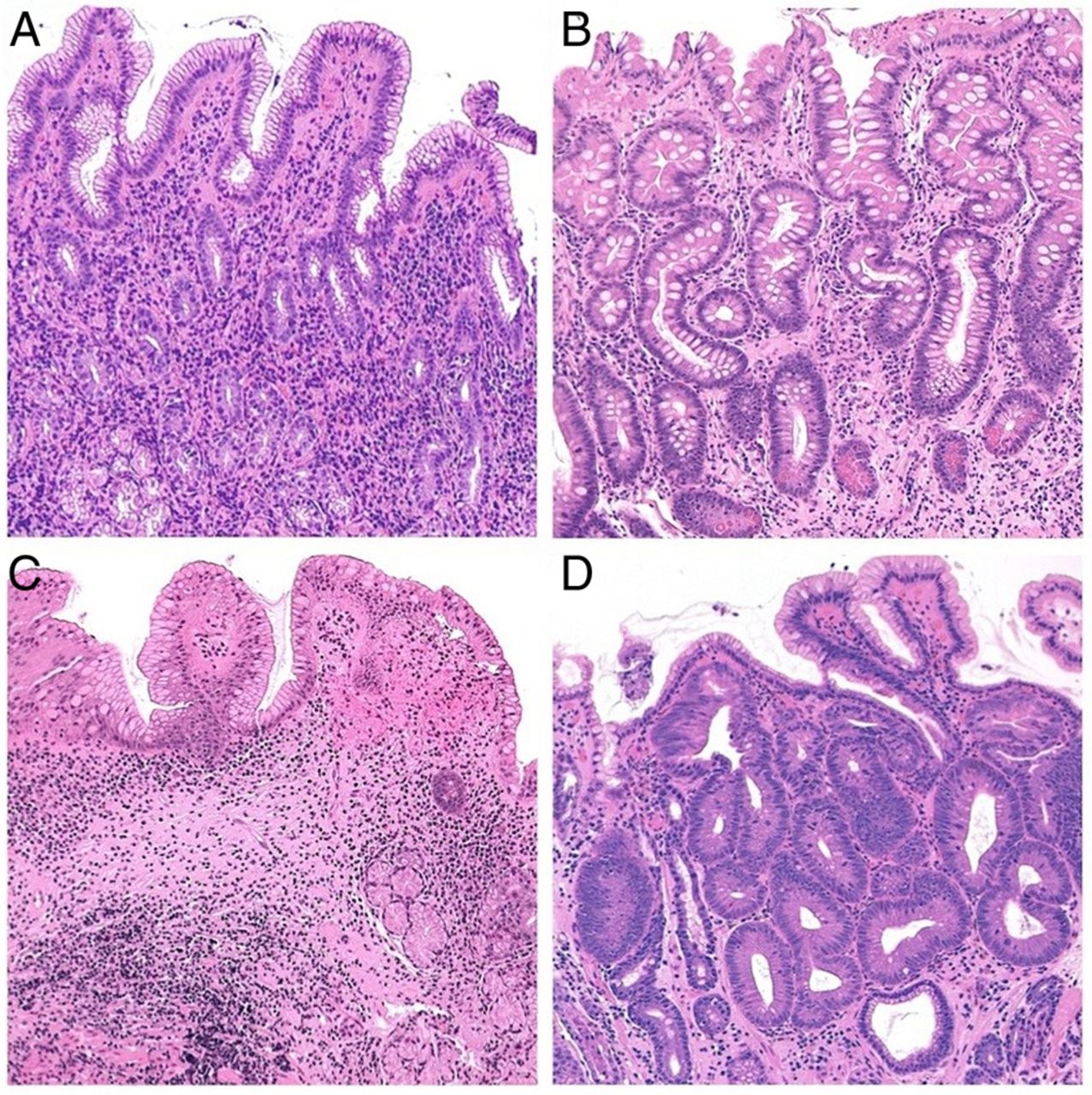
(A) Non-atrophic H. pylori-associated gastritis: The number of the native antral glands is within the normal limits; inflammatory cells are detectable in the lamina propria (H&E; original magnification ×25). (B) Atrophic gastritis with extensive gastric glands intestinalisation (intestinal metaplasia of the native mucus secreting antral glands) (H&E; original magnification ×25). (C) Atrophic gastritis. The population of the resident glands is decreased (‘mucosa desertification’); the glandular units are replaced by extensive fibrosis of the lamina propria (H&E; original magnification ×25). (D) Intraepithelial neoplasia ((IEN), ie, dysplasia) in intestinalised glands. The glandular structures are crowded (back to back appearance), and the dysplastic glandular units show columnar atypical epithelia (H&E; original magnification ×40).
In H. pylori-associated gastritis, atrophic changes occur earlier at the transitional mucosa of the angulus, later involving the distal stomach (antrum-restricted atrophic gastritis), before spreading to the proximal oxyntic mucosa (multifocal atrophic gastritis) or atrophic pan-gastritis.134 It takes years for non-atrophic inflammatory disease to progress to its atrophic counterpart, with a consequent rising prevalence of atrophic gastritis with ageing. The distal-to-proximal spreading of atrophic changes can also be confidently assumed as an indicator of the stepwise progression of the atrophic disease. Consistent with this natural history, Japanese researchers identify oxyntic atrophy as the most advanced stage of H. pylori-associated gastritis.
Metaplastic/atrophic glands are biologically ‘unstable’ and prone to further de-differentiation. This results in ‘neo-epithelia’, which harbours most of the biological traits of neoplastic cells, but these already neoplastic epithelia lack the capacity for invasion, being topographically confined within the basal membrane of the glandular structure (intraglandular neoplasia, or intraepithelial neoplasia, or non-invasive neoplasia, formerly termed dysplasia) (figure 6).135 Further progression of the molecular derangements is coupled with a proliferative advantage, loss of cell-to-cell adhesion and invasiveness, ultimately resulting in (early) invasive adenocarcinoma.136 This oncogenic process, known as the Correa cascade, provides the biological rationale for effective secondary prevention strategies for gastric cancer.133 ,137 Several studies associate gastric atrophy severity/topography with gastric cancer risk.
Built on the seminal experience of the Sydney System's – Histological Division,138 ,139 microscopic gastritis phenotyping demands a topography-based assessment of inflammatory/atrophic changes. Biopsy specimens should therefore be obtained from each mucosal compartment, three from the antrum (including the incisura angularis) and two from the gastric body.130
An alternative for the ‘descriptive’ approach behind the Sydney System has recently been proposed,130 aiming for a more clinically relevant stratification of the gastritis-associated gastric cancer risk (ie, gastritis staging). The recent guidelines on the management of gastric precancerous conditions/lesions recognise the prognostic reliability of the staging approach, but base their recommendations on the topographical ‘spread’ of atrophy/metaplasia.140
Two staging systems are currently in clinical use – operative link on gastritis assessment (OLGA) (table 1) and operative link for gastric intestinal metaplasia assessment (OLGIM)141–143 and both distinguish four stages of gastritis (stages 0–IV) associated with a progressively increasing gastric cancer risk. In the OLGA system, published in 2005,143 gastritis is staged by combining the atrophy scores obtained for the distal stomach and the proximal gastric mucosa.141 The stage indicates the individual likelihood of malignant neoplasia, and most cancer cases develop in patients with stages III and IV.144 The stage of the organic lesions correlates with ‘functional’ gastric mucosa parameters and serum pepsinogens in particular:145 this link between ‘organic’ and ‘functional’ gastric disease may become fundamental to serologically identifying patients with atrophy in whom a second-level endoscopy/biopsy (invasive and expensive) can be performed with the aim of secondary prevention of gastric cancer.
The operative link on gastritis assessment staging system (OLGA)
The simplified OLGIM system focuses only on the IM score/topography in the antral and corpus mucosa141 (table 1). There is still debate on which staging approach is more efficient, but both serve the clinical priority of stratifying gastritis patients by cancer risk.142 ,144–148 Both systems identify stage III/IV patients as being at higher risk and recommend specific endoscopic/biopsy surveillance only for this restricted population. The prognostic value of gastritis staging, already recognised by the Maastricht IV Consensus Conference,149 was recently confirmed at the Kyoto Global Consensus Meeting on H. pylori gastritis.150
Changing epidemiology of gastric disease
Environmental risk factors
The occurrence of a disease is influenced by environmental risk factors that may vary over time, by geography or by demography. H. pylori infection is the major determinant for gastric cancer, gastric ulcer and duodenal ulcer.151 Smoking is also a risk factor for all three diseases152 and increased consumption of dietary salt is a risk factor for gastric cancer and gastric ulcer.153 Aspirin and other non-steroidal anti-inflammatory drugs are risk factors for gastric and duodenal ulcer. On a population level, however, all these influences pale in comparison with H. pylori, and all major epidemiological trends of gastric cancer and peptic ulcer reflect primarily those of H. pylori infection.
Birth cohort phenomena of gastroduodenal disease
Gastric cancer, gastric ulcer and duodenal ulcer are similarly influenced by H. pylori infection and also show similar epidemiological patterns. All three diseases affect mostly adults, although infection with H. pylori is generally acquired during childhood. Therefore, the epidemiological patterns of gastric cancer and peptic ulcer reflect the environmental conditions at the time of acquisition during childhood rather than those at the time of disease. For reasons not yet fully understood, infection with H. pylori increased in all industrialised countries during the 19th century and then declined during the 20th century.151 The rise may have been precipitated by crowded living conditions and poor sanitation during early urbanisation, whereas the decline may have occurred secondary to increasing affluence and standards of hygiene during the 20th century. Consistent with the changing time trends of H. pylori acquisition during childhood, the occurrence of gastric cancer, gastric ulcer and duodenal ulcer rose among successive birth cohorts, that is, generations, born throughout the 19th century, peaked among those born around the turn of the century and then declined in all subsequent generations born during the 20th century (figure 7, left panel).154 This birth cohort pattern is discernible in all health statistics pertaining to incidence, prevalence, physician visits, hospitalisation, surgery, pathology and mortality. The birth cohort patterns of gastric cancer and peptic ulcer similarly affect both genders, different ethnic groups and populations from various regions and countries, such as Japan, Australia, Europe and North America.

Birth cohort patterns of common digestive diseases in mortality data from England and Wales. GCA, gastric cancer; GU, gastric ulcer; DU, duodenal ulcer; Rectum, rectum cancer; Colon, colon cancer
Role of gastric acid secretion
Acid secretion exerts the largest influence of all gastric functions on the occurrence of other diseases outside the stomach. Erosive oesophagitis and oesophageal adenocarcinoma result from gastro-oesophageal reflux. Reduced acid output, associated with chronic gastritis or gastric atrophy following long-term infection with H. pylori, partly protects against reflux disease. The falling infection rates of H. pylori during the 20th century have resulted in a marked concomitant rise of all forms of reflux disease.155 A diminished acid output compromises the acid barrier function against intestinal invasion by infectious organisms. For instance, pharmacological inhibition of gastric acid secretion renders patients more susceptible to the development of C. difficile-induced colitis.156
Long-term time trends of colorectal cancer and UC reveal birth cohort patterns that are strikingly similar to those of gastric cancer and both types of peptic ulcer (figure 7, right panel).157–159 A large body of literature shows that H. pylori infection constitutes a risk for colorectal neoplasm.159 It has been suggested that the reduced acid barrier associated with pangastritis from H. pylori infection could facilitate intestinal invasion by infectious organisms and/or result in dysbiosis of the GI microbiome that might promote the development of colorectal cancer or even UC.160
Role of NSAIDs and antiplatelet agents
Although the long-term time trends of peptic ulcer have not been influenced by the rising usage of non-steroidal anti-inflammatory drugs (NSAIDs), there is no doubt that NSAIDs result in substantial morbidity from ulcer bleeding, perforation or mortality. The occurrence of such adverse events depends foremost on the type of drug and its dose.161 For instance, fenoprofen, diclofenac and sulindac are associated with a relatively low risk; diflunisal, naproxen and indomethacin are associated with a moderate risk; piroxicam, ketoprofen and azaproprazone are associated with a relatively high risk. Multiple other clinical parameters influence the risk.162 NSAID-induced adverse events are 2.5-fold more common in patients with a previous ulcer history and fivefold more common in patients with previous ulcer complications. Old age, smoking and alcohol consumption increase the risk twofold to fourfold. Concomitant use of aspirin or antiplatelet agents with NSAIDs increases the risk fourfold to sevenfold, while concomitant use of selective serotonin reuptake inhibitors, aldosterone antagonists or oral corticosteroids even increase this risk to 7-fold to 13-fold.163 H. pylori and NSAIDs act synergistically on the gastric mucosa.164 Eradication of H. pylori infection prior to initiation of NSAID therapy diminishes the risk for the subsequent occurrence of gastric and duodenal ulcers.165 In addition to the adverse effect on the gastric mucosa, long-term inhibition of prostaglandin synthesis by NSAIDs can also positively affect the development of adenocarcinoma of the upper GI tract. Aspirin reduces the risk for oesophageal and gastric cancer by 30–40%.166–168 To a lesser extent, other types of NSAIDs exert a similarly protective influence.166–168
Gastric and duodenal ulceration
Ulceration is defined as a breach in the mucosa of at least 5 mm in diameter and occurs when there is imbalance between the luminal challenge exerted by the highly acidic and proteolytic properties of gastric juice and the ability of the mucosa to resist it. Ulceration of the duodenum and distal pre-pyloric region of the stomach is associated with high levels of gastric acid secretion, whereas in patients with gastric ulcers, acid secretion is normal or low and these ulcers are thought to be a consequence of impaired mucosal defence.
Causes and treatment of peptic ulcers
H. pylori infection
H. pylori infection is the most important cause of duodenal ulceration. The great majority of patients with duodenal ulcers have the infection and eradicating it usually produces a long-term cure of the ulcer disease.169 However, only about 10–20% of H. pylori-infected subjects develop duodenal ulcers. In these subjects with a duodenal ulcer, gastritis caused by the infection largely spares the acid-producing mucosa of the body of the stomach, which consequently retains normal sensitivity to stimulation by gastrin released by the antral mucosa.170 In addition, the high local concentration of ammonia produced by the organisms’ urease activity impairs acid inhibition of gastrin release causing excessive release of the hormone.171 This combination of increased gastrin release and unimpaired response of the body mucosa to gastrin stimulation results in increased acid secretion, duodenal acid load and, consequently, ulceration. Furthermore, duodenal ulcer patients have an increased parietal cell mass, which further contributes to their high acid output. The high parietal cell mass may also be an important premorbid factor protecting the body mucosa from H. pylori-induced gastritis. H. pylori infection is also a cause of gastric ulcer, and eradicating the infection prevents the recurrence in patients not taking NSAIDs.172 In patients with H. pylori-related gastric ulcers, the gastritis involves the body as well as antral mucosa and induces atrophy and IM. The inflamed and atrophic mucosa secretes subnormal amounts of acid and gastric ulceration is due to impaired mucosal defence rather than increased acid load.172
Non-steroidal anti-inflammatory drugs
NSAIDs tend to produce ulceration of the stomach rather than the duodenum and do this by impairing mucosal defence mechanisms rather than increasing acid secretion. The mechanism by which NSAIDs induce ulcers in humans is complex and not fully understood and is the subject of a recent review.173 The drugs exert a topical effect involving impairment of the hydrophobic property of the mucus barrier allowing back diffusion of hydrogen ions. They also impair mucosal defence by systemic inhibition of cyclooxygenase (COX) activity, resulting in reduced synthesis of mucus and bicarbonate and impairment of mucosal blood flow, which are all important in preventing and neutralising back diffusion of hydrogen ions. It used to be thought that the mucosal damaging effects were due to inhibition of COX-1, but recent evidence indicates that dual suppression of COX-1 and COX-2 is necessary. Work in animals demonstrates that neutrophil adherence to gastric mucosal microcirculation and the resulting obstruction of capillary blood flow plays an important role in impairing mucosal defences. Nitric oxide and hydrogen sulfide increase mucosal blood flow and administration of agents that release these gases reduces NSAID damage in animal models.
NSAID-induced ulcers are more common in the elderly, in those with a history of ulcers and in the presence of comorbidities. NSAID-associated ulcers are usually treated by PPI therapy and stopping the NSAID treatment. If NSAID treatment needs to be reintroduced, then a COX-2 selective inhibitor should be employed together with a PPI, provided low-dose aspirin (LDA) cardioprotection is not required. A recent study focusing on bleeding peptic ulcer disease showed that a range of other drugs, when combined with NSAIDs or aspirin, significantly enhance the risk of bleeding ulceration173 ,174
H. pylori-negative/NSAID-negative ulcers
Other causes of ulcers are rare and should only be considered when H. pylori infection and NSAID use has been excluded.175 In countries where the prevalence of H. pylori infection, and consequently, the prevalence of H. pylori-related ulcers, is falling, the proportion of ulcers that are H. pylori-negative/NSAID-negative will inevitably increase even if the absolute prevalence does not change. The commonest cause of apparent H. pylori-negative/NSAID-negative ulcers is a false negative H. pylori test and unrecognised or surreptitious NSAID use.175 False negative H. pylori tests are more common with recent use of antibiotics, PPI therapy, bismuth containing medications and in the context of acute upper GI bleeding. Other causes include gastroduodenal involvement with Crohn's disease or sarcoidosis, underlying malignancy, cytomegalovirus in immune-compromised patients, Cameron ulcer of the gastric mucosa within a hiatus hernia and ulceration following gastric surgery, which is now mainly seen following bariatric surgery. The ZES is a rare cause of ulcers and due to high levels of tumour-derived gastrin and consequently excessive acid secretion. Rare causes include gastric ischaemia, a variety of medications other than NSAIDs, radiotherapy and severe systemic disease. However, a true causal role for these factors is not fully established. A very small proportion of patients have idiopathic ulcer disease. Idiopathic ulcers may be more difficult to control as acid inhibitory therapy is less effective in the absence of H. pylori infection. Idiopathic ulcers are also more commonly associated with complications, including bleeding and perforation.
Symptoms of gastric and duodenal ulcers
Historically, it was claimed that ulcers produced typical symptoms that even allowed discrimination between duodenal and gastric ulcers. However, recent studies indicate a very weak association between symptoms and the presence of ulcers. The vast majority of patients investigated for epigastric pain have no evidence of gastric or duodenal ulcer and their symptoms are attributed to reflux disease or non-ulcer dyspepsia. Furthermore, a substantial proportion of patients found to have ulcers have no associated symptoms. Also, 43–87% of patients presenting with upper GI bleeding found to be arising from chronic gastric or duodenal ulcers have no history of preceding symptoms irrespective of NSAID use.176–178 Also, in community-based endoscopic studies of H. pylori positive subjects, 13–21% have been found to have underlying ulcers but few, if any, symptoms.179–181 Curing ulcers by eradicating H. pylori infection results in the resolution of symptoms thought to be due to the ulcer in only approximately 50% of cases.182 These observations suggest that a majority of ulcers are asymptomatic.
Management of patients presenting with symptoms possibly due to ulcer
Over the last 10–15 years, there has been a transformation in the ways in which ulcers are diagnosed and treated. Patients with upper GI symptoms and no sinister symptoms have a non-invasive test for H. pylori infection (urea breath test, faecal test or serology) and if positive, given a course of H. pylori eradication treatment. The great majority of those treated do not have an ulcer-associated H. pylori infection, but the strategy cures the minority with ulcers. This widely applied strategy has resulted in a dramatic reduction in ulcers seen at routine endoscopy.
Ulcer complications and their management
Complications of ulcer disease include bleeding, perforation and rarely pyloric stenosis, which, if seen, may be associated with neoplasia or Crohn's disease. Perforation is mainly associated with acute ulceration and NSAIDs, but bleeding may occur in ulcers associated with H. pylori or NSAIDs especially if patients are also taking aspirin.
In contrast to some earlier recommendations,183–185 recent guidelines recommend that in patients presenting with upper GI bleeding intravenous PPI therapy is not started prior to the diagnosis as they may obscure the presence of underlying vessels.186 ,187 These new guidelines also recommend that endoscopic therapy of visible vessels should include injection of adrenaline plus one other therapeutic modality, such as clipping or heater probe.186 ,187 If further bleeding occurs after the initial endoscopy, the procedure should be repeated and, if bleeding reoccurs, management should be by interventional radiology. Recent guidelines also highlight the risks of over-transfusion as well as under-transfusion of blood.187 Blood transfusion is rarely required if haemoglobin concentration >10 g/dL, and nearly always if <6 g/dL. In patients presenting with upper GI bleeding on LDA for cardiovascular protection, it is recommended that the LDA is recommenced when haemostasis is achieved to reduce the high incidence of cardiovascular events following upper GI bleeding.
Prevention of H. pylori-induced ulcers
Eradication of H. pylori infection prevents the occurrence and reoccurrence of gastric and duodenal ulcers unrelated to NSAIDs. The widespread use of non-invasive H. pylori test and treat strategies for dyspepsia means that a significant proportion of the population are prevented from developing H. pylori-related ulcers or having them cured before they are detected.
Prevention of NSAID-induced ulcers
In patients at increased risk of developing NSAID–induced ulcer and ulcer complication, the risk can be ameliorated by co-prescription of a PPI by using a selective COX-2 inhibitor rather than non-selective NSAID or by combination of these approaches. The regimen employed is based upon the GI risk and whether the patient requires LDA for cardiovascular protection as per recent guidelines.172 ,186 ,187 LDA negates the GI protection afforded by selective COX-2 inhibition.
Eradicating H. pylori infection in patients about to start NSAIDs reduces subsequent ulcer risk and is equivalent to PPI prophylaxis.170 However, eradication of H. pylori alone is not sufficient to prevent ulcer bleeding in NSAID users with increasing GI risk, such as history of ulcer bleeding.171
Gastric cancer
Gastric cancer represents the worst end of the spectrum of an ‘unhealthy’ stomach. Although the age-adjusted mortality due to gastric cancer has continued to decline in most countries of the world, nearly one million people are newly diagnosed with gastric cancer each year, three quarters of whom are at an advanced stage culminating in death; this places it as the third leading cause of cancer death.188 Gastric cancer is a multifaceted disease with different aetiologies, genetic changes and phenotypes. Recent data based on genetic alterations suggested that it could be classified into four subclasses189 (figure 8). However, H. pylori infection is considered as the single most important risk factor leading to gastric cancer,190 through chronic inflammatory changes in the gastric mucosa, followed by pre-neoplastic changes such as atrophy and IM, as in the Correa cascade.111 ,191 As gastric cancer risk increases during these intermediary steps, it is necessary to interrupt the progression by eradicating H. pylori infection that can halt or even regress some of the mucosal changes. Indeed, recent meta-analyses clearly demonstrated the benefit of eradication to reduce gastric cancer.192 ,193 However, the effect on reducing gastric cancer incidence by eradication treatment was small: only about one-third of risk reduction. Furthermore, there are conflicting data on the effect of eradication following endoscopic treatments for early gastric cancer, where most of the background mucosa was in a pre-neoplastic stage.194–197 Several factors should be considered to explain this paradox. First, we must consider that the subjects registered and screened for gastric cancer prevention trials may have already harboured minute cancer foci that evaded endoscopic detection. Second, early gastric cancer may not be reliably classified by pathologists. In the process of formulating the Vienna classification,198 the diagnostic performance of gastric cancer by pathologists with limited diagnostic experience was shown to be unsatisfactory. Third, it is plausible that pre-neoplastic conditions continue to evolve into true cancerous lesions after eradication treatment as these lesions contain a number of genetic and epigenetic changes, predisposing them to acquire further genetic changes and transforming to cancer.199 Fourth, there might be other aetiological factors involved in the progression to cancer. In the hypochlorhydric stomach, overgrowth of microbes has been well documented, some of which may be responsible for residual inflammation and also the production of carcinogenic substances such as nitrosamines.200 It is also possible that the Epstein–Barr virus may be the major factor for a subtype of H. pylori-negative gastric cancer.199 ,201

Classification of gastric cancer based on gene expression patterns. Adapted from ref. 189. CIMP, CpG Island Methylator Phenotype; EBV, Epstein–Barr virus.
These potential factors may limit the preventive effect of eradication therapy, and so it is highly recommended that eradication should be implemented before advanced atrophy takes place. The recent global conference held in Kyoto came to a consensus that recommended early eradication of the H. pylori infection to enhance the cancer prevention.150 Indeed, after the Japanese government decision to subsidise the cost for eradication of H. pylori gastritis in 2013, a massive surge in the number of people receiving eradication therapy (more than a million per year) has occurred in Japan.202 Thus, we can speculate that most infections will be eliminated by the nationwide implementation of eradication strategies, with the expectation of a sharp decline in gastric cancer incidence. In Taiwan, a community screening programme for H. pylori infection, coupled with an eradication programme, showed promising results for reducing pre-neoplastic lesions,203 although reduction of gastric cancer mortality was not seen, probably due to the short period of observation.
However, not all the gastric cancers, particularly cancer in the cardia in Western countries, are related to H. pylori infection and no plausible preventive measures have yet been proposed for this form of gastric cancer. Early detection during routine endoscopic examination still plays an important role for secondary prevention. Indeed, most of the gastric cancer cases (>90%) are detected by routine clinical examination in Japan.204 To facilitate early detection, high-resolution endoscopy equipped with image-enhanced modalities such as narrow band imaging (NBI), flexible spectral imaging colour enhancement and blue laser imaging (BLI) have recently been introduced into clinical practice in Japan.205–207 As better performance of image-enhanced endoscopy (IEE) has been documented, not only for detecting early gastric cancer, but also for Barrett's lesions, adoption of IEE for routine upper GI endoscopic examination is now highly recommended. This facilitates early detection of gastric cancer, which can be curable in the early stages (figure 9) with minimally invasive therapy. The combination of more efficient primary and secondary preventive measures promises a dramatic decrease in the incidence and mortality of gastric cancer.

(A and B) A small gastric cancer detected by magnification endoscopy equipped with image-enhanced modalities. A small flat lesion with abnormal surface mucosal patters that is sharply demarcated by normal mucosa showing regular pit pattern. Inside the lesion, irregular, tortuous cork-screw like vessels can be identified. Alteration of mucosal surface pattern together with the presence of irregular vessels suggests early gastric cancer. Histology of the endoscopically resected specimen verified the diagnosis of gastric cancer. NBI, narrow band imaging; FICE, flexible spectral imaging colour enhancement.
Detecting the unhealthy stomach
The wide prevalence of dyspeptic symptoms and gastric disease explains why assessment of the stomach is a common procedure frequently assessed for pathology. Assessment of the stomach may occur for screening either because of a hereditary cancer risk or a population risk. On most occasions, however, the stomach is assessed because of symptoms. These include a range of upper abdominal dyspeptic symptoms, as well as others such as stigmata of bleeding. Assessment can occur by various methods.
Non-invasive stomach assessment
Several methods are now available for non-invasive assessment of the stomach. They can be used alone or in combination. These, in particular, include serum gastrin, pepsinogen I and II, and markers for H. pylori infection (serum IgG antibodies, faecal antigens and urease activity determined by urea breath testing) and a breath test for assessing gastric emptying. Each of these markers is indicative, although none confirmatory, of a diseased stomach. The diagnostic methods for H. pylori infection are reliable in daily clinical practice, and if positive, indicate the presence of chronic active gastritis, a condition that virtually always accompanies H. pylori colonisation.208 The level of each marker, however, has no relation to the severity of gastritis nor the presence of further pathology such as ulcer disease or premalignant changes. For the latter purpose, serum gastrin and pepsinogen levels are valuable markers. Serum gastrin is a marker for gastric acid output. A decrease in acid output, either as a result of inflammation or gland loss generally, is associated with an increase in serum gastrin levels. Pepsinogens are aspartic proteinases. They can be discerned by electrophoresis into two groups; pepsinogen I or A, and pepsinogen II or C. Pepsinogen I can be further characterised into several isozymogens. The chief cells in the gastric fundus and corpus produce pepsinogen I. Chief cells in the corpus and mucous neck cells throughout the stomach produce pepsinogen II. Pepsinogens are secreted into the gastric lumen, where they are converted into pepsin as active protease. Pepsinogens can also be detected in serum. Serum levels of pepsinogen I and II increase as a result of gastric inflammation, particularly H. pylori gastritis. Gland loss due to long-standing gastritis eventually leads to a decrease of pepsinogens, particularly pepsinogen I. These phenomena can be used for non-invasive assessment of the condition of the gastric mucosa.209 Positive H. pylori serology with increased serum pepsinogen I and II levels confirms H. pylori gastritis. Positive H. pylori serology with decreased pepsinogen I level and a decreased pepsinogen I/II ratio is indicative of long-standing H. pylori gastritis that has led to atrophic changes. This is further supported by an increased serum fasting gastrin level, which reflects reduced acid output as a result of loss of specialised glands. Finally, the pattern of a markedly decreased pepsinogen I and I/II ratio, with increased gastrin and negative H. pylori serology, can be found in patients with marked atrophic gastritis either as a result of previous H. pylori infection or autoimmune gastritis. Marked atrophic gastritis provides unfavourable conditions for persistent H. pylori colonisation and can thus eventually lead to seroconversion. Autoimmune gastritis can be further detected by the presence of antiparietal cell antibodies.
Serum levels of gastrin and pepsinogens are rarely used in clinical practice with the exception of serum gastrin in patients suspected of ZES. They are, however, frequently applied for screening of populations at high risk for gastric cancer.210 For example, Japanese investigators followed 6983 individuals for a mean 4.7 years after baseline screening with pepsinogens. In subjects with low serum pepsinogens, the incidence of gastric cancer during follow-up was sixfold to eightfold higher than in those with normal pepsinogens at baseline.209 Other serum parameters, such as leptin and ghrelin, have no additional value for assessment of the health of the gastric mucosa.211
Invasive stomach assessment
Endoscopy is the established gold standard for detection of gastric pathology. This includes ulcer disease, mucosal atrophy and metaplasia, and gastric neoplasia, as well as other pathology such as portal hypertensive gastropathy and vascular malformations. For motility disorders, assessment is primarily undertaken with other methods (see previous section), with a limited role for endoscopy other than exclusion of anatomic abnormalities and confirmation of food retention in the stomach.
The accuracy of gastroscopy for diagnosis of macroscopic lesions is high; however, lesions may be missed. In a Japanese population screening study that included 17 522 subjects, 26 (1.5%) patients were diagnosed with gastric cancer 2 years after a negative gastroscopy. In six (23%) of them, the previous endoscopic images in retrospect revealed signs of neoplasia.212 A similar study from Denmark reported a miss rate of 52 (10%) out of 513 oesophageal and gastric cancers in 27 853 patients undergoing a first diagnostic gastroscopy.213 This was the equivalent of missing one cancerous lesion per 536 gastroscopies. These studies confirm that the awareness of the endoscopist for the detection of (early) gastric cancer has to improve.
Despite these false negative procedures, gastroscopy is the most effective tool for the diagnosis of gastric cancer, and timely diagnosis improves outcome. A recent study from the UK compared 6513 primary care practices for their elective gastroscopy rate per capita.214 These ranged from 4.4 to 12.9/1000 adults per year. Among 22.488 patients diagnosed with oesophageal or gastric cancer, rates of surgery and 12-month survival were 14% lower in those practices with the lowest compared with the highest use of gastroscopy.214
Gastroscopy was for decades considered an inadequate tool for the diagnosis of microscopic lesions. However, IEE has now become an accurate tool for evaluation of the gastric mucosa. Image enhancement can be obtained with chromoendoscopy as well as NBI, both in combination with magnification.140 ,142
For various purposes, endoscopy is often supplemented with other methods of investigation. Gastric histology can confirm and grade H. pylori colonisation, mucosal inflammation, atrophy and metaplasia, as well as neoplasia. Grading of atrophy and metaplasia is relevant for determining the risk of cancer.140 ,142 ,147 The yield of diagnostic gastroscopy with biopsy sampling has greatly increased because image enhancement allows targeted biopsy sampling of visible lesions. Furthermore, H. pylori urease testing and culture can confirm colonisation, and culture may provide further information on antimicrobial resistance. Other supplementary methods for the assessment of gastric pathology include mucosal oxygen saturation, which can corroborate a diagnosis of gastro-duodenal ischaemia.215
Healthy stomach: future approach to gastric diseases
The stomach occupies the central role in orchestrating the digestive process, and this is frequently underestimated. Moreover, gastric acid secretion in the last decades has been seen as a ‘bystander’ with little function but with deleterious potential for itself and adjacent organs, the oesophagus and duodenum. As a consequence, the pharmacological approach has been towards the development of more potent drugs for acid inhibition. Due to the increasing awareness of GI functional disorders, the role of the stomach has been revisited in its role as site of origin for dyspeptic symptoms. More recently, attention has focused on the stomach for its control function in food intake and for contributing to maintenance of metabolic balance (figure 10).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key functions of the stomach and common harmful and noxious agents that affect gastric mucosal, secretory and motor functions.
The future approach to gastric diseases (box 3) is directed to maintaining a healthy stomach, which is free from discomfort, ulceration and the risk of complications and malignancy. The main challenge remains the elimination of H. pylori infection from individual patients and from populations. An estimated 20% of H. pylori-infected people will continue to suffer from overt clinical upper GI symptoms and complications over their lifetime, and some may develop extra-digestive diseases. The individual outcome of anyone infected with H. pylori cannot be predicted. Therefore, a public health approach should be directed towards ‘screen and treat’ strategies that will have to be adapted to the needs of different populations according to the prevalence of H. pylori infection and gastric cancer risk stratification. Gastric disease prevention programmes should be integrated with more comprehensive GI prevention strategies. The combination of H. pylori screening and eradication programmes with colorectal cancer screening is an initiative promoted and coordinated by the Healthy Stomach Initiative (HSI) (http://www.hsinitiative.org).
Future approach to gastric diseases
Prevention of gastric cancer by H. pylori screen and treat strategies.
Role of the stomach in functional disorders.
Mechanisms of gastric carcinogenesis for development of molecular targeted therapies.
Role of gastric acid in maintenance of a healthy GI microbiome.
Role of gastric control in food intake, obesity and metabolic disorders.
Create more public awareness to keep a healthy stomach and fight illness.
Gastric cancer is still a major challenge worldwide, and because detection is frequently made only at an advanced stage, mortality has remained high.216 ,217 Gastric cancer prevention programmes by H. pylori eradication have been shown of benefit in high-risk populations.193 The best results from gastric cancer prevention strategies are obtained when H. pylori eradication is performed before advanced atrophic gastritis with pre-neoplastic changes becomes established and thus implementation of H. pylori screening and treatment in early adulthood is required. Secondary prevention by H. pylori eradication following endoscopic resection of early gastric cancer has major limitations.218 With pre-neoplastic conditions such as atrophy and IM already present, carcinogenic pathways are more likely to progress in spite of the eradication of H. pylori infection. Future research will need to focus on unravelling mechanisms involved in progression from pre-neoplastic lesions to cancer.
The recent definition of H. pylori gastritis as an infectious disease by the Kyoto global consensus conference, January 2014, is expected to raise concern and engender support from regulatory authorities towards the global elimination of H. pylori infection and its serious sequelae.150
Despite these important indications and calls for a widespread approach to the eradication of H. pylori infection, there remain substantial challenges. The first includes achieving the ideal effective therapy without significant side effects and no antibiotic resistance. Such an ideal therapy is not yet available, and therefore, H. pylori eradication therapy, beyond the established specific clinical indications, should embark on selected screen and treat strategies. For the time being, these strategies will have to address populations with a high to moderate incidence of gastric cancer. The search for a ‘golden treatment bullet’ continues to remain one option while the second option is an intensified search for a vaccine.
Second, H. pylori infection may confer some benefits to those who do not have gastroduodenal symptoms, nor present with gastroduodenal disease or complications. The reduced prevalence of atopic diseases, such as asthma in patients infected with H. pylori up to young adulthood, requires intensive investigation to understand the mechanism of this phenomenon. Epidemiological and experimental evidence is still limited and cannot yet offer any conclusions about a causal relationship.93 ,219 ,220 Studies on the relationship of H. pylori gastritis will lead to better understanding of both local and systemic immune responses and their impact on gastric diseases.
In addition to the initiatives and strategies to eradicate H. pylori infection, studies are required to better understand the role of the stomach in food intake, accommodation, pre-digestion and the delivery of nutrient for intestinal digestion.38 Research should focus on ways to modulate gastric functions and their role as ‘weight watcher’ and their integration in the balance of hunger and satiety. Studies will need to address to what extent gastric acid should be inhibited and for how long during the 24 h period in patients who suffer from acid-related diseases. Moreover, it will be important to define just how much acid is required to preserve a ‘healthy’ gut microbiome. The role of the gastric microbiota in the presence and absence of H. pylori infection on the diversity of microbiota in the small bowel will be of enormous relevance in understanding and tackling gastric, hepatic and intestinal diseases.221–223
Last, but by no means least, education with effective presentation of new knowledge to the general public to ensure gastric health and prevent disease is a major task to be accomplished and the creation of the HSI for public awareness is a step in this direction.
References
Footnotes
Correction notice This article has been corrected since it published Online First. The axis have been added to figure 7.
Contributors On invitation to author this review, RHH planned the content together with EME-O, briefed the authors, coordinated, assembled and edited the sections into a single manuscript that was then reviewed, commented on and edited by RHH, EME-O and each of the authors before finalising the text. Although the content was largely brought together, the original submissions were written by RHH (history); PM (gastric hormones); JT (neurogastric physiology); MC (stomach in appetite control and obesity); SEC (immunology of the stomach); JGF (gastric microbiome); MG (gastritis associated with cancer); AS (epidemiology); KELMcC (gastric and duodenal ulceration); KS (gastric cancer); EJK (investigation); and PM (future directions).
Competing interests RHH, consulting: Takeda R&D, Yuhan, Speaking AstraZeneca, Danone; PM, investigator support: Trio Medicines, Ipsen; MC, research support: Rhythm, SK Life Science, Salix, Tsumura, NPS, Vibrant and Advisory Boards with consulting fee to Mayo Clinic: Rhythm, Ironwood, Theravance, Takeda, Shire; JT, research support: Novartis, Shire, Zeria and Consulting or speaker: Alfa Wassermann, Almirall, AstraZeneca, Danone, GlaxoSmithKline, Ironwood, Janssen, Menarini, Novartis, Rhythm, Shire, Sucampo, Takeda,Theravance, Tsumura, Yuhan, Zeria; KELMcC, speaker: Reckitt Benckiser and Astra Zeneca; KS, consulting or speaking: Takeda Pharma, Astellas Pharma and Zeria Pharma, Fuji Film; PM, speaking: Aptalis, Astra Zeneca, Reckitt Benckeiser, Steigerwald and Takeda.
Provenance and peer review Commissioned; externally peer reviewed.