Article Text

Abstract
Objective The transcription factor SOX9 was recently shown to stimulate ductal gene expression in pancreatic acinar-to-ductal metaplasia and to accelerate development of premalignant lesions preceding pancreatic ductal adenocarcinoma (PDAC). Here, we investigate how SOX9 operates in pancreatic tumourigenesis.
Design We analysed genomic and transcriptomic data from surgically resected PDAC and extended the expression analysis to xenografts from PDAC samples and to PDAC cell lines. SOX9 expression was manipulated in human cell lines and mouse models developing PDAC.
Results We found genetic aberrations in the SOX9 gene in about 15% of patient tumours. Most PDAC samples strongly express SOX9 protein, and SOX9 levels are higher in classical PDAC. This tumour subtype is associated with better patient outcome, and cell lines of this subtype respond to therapy targeting epidermal growth factor receptor (EGFR/ERBB1) signalling, a pathway essential for pancreatic tumourigenesis. In human PDAC, high expression of SOX9 correlates with expression of genes belonging to the ERBB pathway. In particular, ERBB2 expression in PDAC cell lines is stimulated by SOX9. Inactivating Sox9 expression in mice confirmed its role in PDAC initiation; it demonstrated that Sox9 stimulates expression of several members of the ERBB pathway and is required for ERBB signalling activity.
Conclusions By integrating data from patient samples and mouse models, we found that SOX9 regulates the ERBB pathway throughout pancreatic tumourigenesis. Our work opens perspectives for therapy targeting tumourigenic mechanisms.
- IMMUNOHISTOCHEMISTRY
- DEVELOPMENT GENES
- EPIDERMAL GROWTH FACTOR
- GENE TARGETING
- PANCREATIC CANCER
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer death. A better understanding of the mechanisms driving the disease will contribute to better treatments.
Mouse models suggest that PDAC can originate from metaplastic acinar cells that evolve into neoplastic lesions called pancreatic intraepithelial neoplasia (PanIN). Activating mutations in the proto-oncogene KRAS is an initiating event in more than 90% of PDAC.
SRY-related human motility group (HMG) box factor 9 (Sox9) is required for acinar-to-ductal metaplasia (ADM) and PanIN formation in a mouse model of PDAC induced by oncogenic Kras. How Sox9 promotes PanIN formation remains an open question.
Epidermal growth factor receptor (EGFR/ERBB1) is also essential for ADM and Kras-induced pancreatic tumourigenesis. How EGFR and its related pathway are regulated in PDAC is still unknown.
What are the new findings?
In humans, pancreatic tumours showing high expression of SOX9 preferentially belong to the classical PDAC subtype. It is known that cells in this subtype respond to EGFR-directed therapy.
In human PDAC samples, high expression of SOX9 correlates with high expression of several members of the EGFR pathway, in particular ERBB2.
Both in humans and in mice, SOX9 regulates the expression of ERBB2, the preferred coreceptor of EGFR.
Analysis of a mouse model of PDAC reveals that Sox9 controls the activity of EGFR pathway. This mechanism explains at least, in part, the role of Sox9 in PanIN formation.
How might it impact on clinical practice in the foreseeable future?
To determine the expression level of SOX9 in a PDAC sample will contribute to identify the molecular subtype of the tumour.
To define the expression signature of SOX9 and other selected genes in PDAC samples could help to predict the response of these tumours to ERBB-directed therapy and survival outcomes.
The present results warrant future explorations of the role of ERBB2 in PDAC initiation and progression. Identifying an important function for this protein in pancreatic cancer will open the available therapeutic arsenal targeting ERBB2 to PDAC treatment, possibly in combination with EGFR-directed therapy.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers. It is characterised by a rising incidence and a poor patient outcome that has remained unchanged since 40 years. Poor prognosis mainly results from resistance to therapy and late diagnosis, thereby reflecting our incomplete understanding of the molecular mechanisms that drive PDAC initiation and progression.1
PDAC is nearly always associated with activating mutations in the proto-oncogene KRAS, as recently confirmed by exome sequencing.2 Analysis of genetically modified mouse models suggests that acinar cells undergo acinar-to-ductal metaplasia (ADM), a process in which the acinar cells are phenotypically converted to duct-like cells.3–5 In the presence of activating Kras mutations, metaplastic lesions give rise to non-invasive ductal lesions called pancreatic intraepithelial neoplasia (PanIN), which on activation of other proto-oncogenes or loss of tumour suppressor genes, eventually evolve into PDAC.3 ,6 ,7 This model of tumourigenesis is further supported by the observation in humans that pancreatitis, an inflammatory disease of the pancreas with pronounced ADM, is associated with increased risk of PDAC.4 ,8 Therefore, it is essential to unravel the mechanisms that underlie the early stages of pancreatic tumourigenesis.
Loss of acinar differentiation, recapitulation of embryonic features typical of ductal differentiation, inflammation and oncogenic Kras expression are intimately linked in the oncogenic process.9–11 Recent observations indicate that the epidermal growth factor receptor (EGFR/ERBB1) pathway contributes to ADM and is essential for Kras-induced pancreatic tumourigenesis.12 ,13 EGFR signalling stimulates RAS, leading to robust extracellular-signal-regulated kinase (ERK) activation, which is required for pancreatic transformation.12
The human motility group (HMG)-box transcription factor SOX9 is a regulator of pancreatic duct cell fate.14 We previously reported that in inflammatory conditions, SOX9 is expressed in metaplastic acinar cells and contributes to ADM.15 Others showed that Sox9 is critical for PDAC initiation, although the precise mechanism by which Sox9 operates in this context is to be elucidated.5
In the present work, we further addressed the function of SOX9 in the pathogenesis of PDAC. Using bioinformatic analysis of gene expression profiles in human PDAC and experimental manipulation of SOX9 levels in cultured cell lines and in vivo mouse models, we found that SOX9 regulates the activity of ERBB signalling. Therefore, our work establishes a functional link between two key players in PDAC tumourigenesis, namely SOX9 and EGFR signalling.
Materials and methods
Acquisition of human PDAC samples and genomic, transcriptomic and survival analysis
Biospecimens were provided by Australian Pancreatic Cancer Genome Initiative (APGI; ethical approval: HREC/11/RPAH/329—The Molecular Pathology of Pancreatic Cancer, incorporating the APGI, approved by Sydney Local Health District—RPA Zone, protocol X11-0220).
Representation of SOX9 mutations was analysed using exome sequencing on tumour–normal pairs from APGI.2 Somatic copy number alterations were detected using Illumina Omni1M Quad single nucleotide polymorphism (SNP) arrays and analysed using GenoCN,16 as in Biankin et al.2
The model of SOX9 functional domains was constructed from refs17 and 18, and the helix domains from UniProt. SOX9 was annotated by naturally occurring variants reported by UniProt and somatic variants from COSMIC. The point mutation described here was previously reported by the authors,2 and the functional impact of this mutation was assessed by Polyphen219 and SIFT.20
Gene expression data are stored at Gene Expression Omnibus (GEO; accession GSE50827), processed as in Biankin et al,2 and include data of 103 primary tumours, 95 of which contain disease-specific survival, and 97 of which contain overall survival annotation. The correlation between SOX9 gene expression and any other gene on the microarray was tested by both Pearson and Spearman's rank correlation. Student t test was applied to test difference of SOX9 expression between two PDAC subtypes defined by Collisson et al.21 KOBAS22 ,23 was used for pathway enrichment analysis. The hypergeometrical test was selected to test statistical enrichment of KEGG and Reactome pathways, and the p values were corrected for multiple comparisons.24
Analysis was also performed on 5 tumours with the lowest and from 5 tumours with the highest SOX9 expression. Genes that were differentially expressed were assessed using LimmaGP (V.19.4; Cowley et al, manuscript in preparation; available at http://pwbc.garvan.unsw.edu.au/gp). LimmaGP produced a preranked file, mapping each gene to its t-statistic as a measure of differential expression; for genes with multiple probes, the value from the probe with the largest absolute t-statistic was reported. Using this preranked file, gene set enrichment analysis (GSEA) was performed using GSEApreRanked (V.3.0; software available at http://pwbc.garvan.unsw.edu.au/gp), which is a GenePattern wrapper around GSEA in preranked mode,25 using 1000 permutations, and the geneset collection comprising the Gene Ontology Biological Process terms defined in the Molecular Signatures Database (V.3.0).26
Median survival was estimated using the Kaplan–Meier method and the difference between survival curves was tested by the log-rank test. Survival analysis was performed using the R/Bioconductor survival package.
Mouse experimentation
Mice received humane care according to the criteria listed by the National Academy of Sciences. Sox9f/f, LSL-KrasG12D and ElaCreERT2 mice have been described.27–29 All strains were maintained in an enriched CD1 background. Six-week-old mice were treated with tamoxifen (Sigma) and 4-hydroxytamoxifen (4OT; Sigma) dissolved in corn oil (Sigma; 30 and 0.3 mg/mL, respectively). At least 10 days after tamoxifen injections, acute pancreatitis was induced by 6 hourly intraperitoneal injections of cerulein (Sigma, 125 µg/kg), every other day, for 5 days. The initial day of treatment was day 1 (D1). Mice were sacrificed on day 7 (D7). To induce chronic pancreatitis, at the end of the acute cerulein treatment, the mice continued to receive a daily injection of cerulein, 5 days/week. Mice were sacrificed on day 65 (D65). It is worth noting that, after tamoxifen injections, and before cerulein treatment, pancreata of all genotypes were histologically normal (see online supplementary figure S12).
Immunohistochemistry and immunofluorescence
Immunohistochemistry (IHC) or immunofluorescence was performed as in Prévot et al.30 Additional details are provided in online supplementary methods a.
Results
SOX9 gene aberrations and altered expression are found in human PDAC
The recently described role of Sox9 in ADM and PanIN development5 ,15 warrants further exploration of Sox9's function in PDAC development. As a first step, we investigated the status of the SOX9 gene in a published cohort of 90 resected sporadic PDAC cases from the APGI.2 Through a combination of automated exome-sequencing data and SNP array analysis on the patient tumours and matched controls, followed by manual review, we uncovered one case with a point mutation in SOX9. In addition, we identified 5 cases with a copy number gain, 4 with a loss of one copy and 4 with copy neutral loss of heterozygosity (LOH) of the SOX9 gene (figure 1A). Expression microarray analysis showed that copy number gain in the PDAC cohort tended to correlate with increased SOX9 expression (see online supplementary figure S1A). The point mutation found was a non-synonymous C to A mutation in the transactivating domain of SOX9 (NM_000346.3: c.1436C>A; NM_000346.3: p.Pro479His; see online supplementary figure S1B), which was verified by resequencing and predicted to affect the protein function as determined by the Polyphen2 score (0.998) and SIFT score (0.0). However, non-mutated and mutated SOX9 shared the same transcriptional activity, indicating that this mutation has no effect on the transactivation function of SOX9 (see online supplementary figure S1C).

Genetic aberrations in the SOX9 gene are frequently found in a cohort of human PDAC and SOX9 is differentially expressed in PDAC subtypes. (A) Frequency (%) of SOX9 somatic mutations and copy number variants in the APGI cohort (n=90). (B) Kaplan–Meier curve for disease specific survival in groups with an IHC score 0–100 (n=12) versus IHC score comprised between 101 and 200 (n=29) and IHC score >200–300 (n=69), respectively the SOX9low, SOX9medium, and SOX9high group; p<0.01. Pie chart represents the proportion of samples within each group. (C) SOX9 mRNA expression levels, obtained from Illumina microarray analysis (total samples, n=89), in the classical and QM PDAC subtypes. SOX9 expression was higher in the classical subtype than in the QM subtype (n=29 and n=26, respectively, *p<0.05). PDAC, pancreatic ductal adenocarcinoma; APGI, Australian Pancreatic Cancer Genome Initiative; IHC, immunohistochemistry; QM, quasi-mesenchymal; LOH, loss of heterozygosity.
We then analysed SOX9 protein expression in PanIN lesions (see online supplementary figure S2A) and PDAC tumours from APGI using tissue microarrays (see online supplementary figure S2B). The PanIN lesions showed a heterogeneous expression pattern, ranging from strongly positive to negative, with occasionally both situations found within the same lesion. Heterogeneity persisted in the PDAC samples, with around 60% showing strong nuclear SOX9 expression (SOX9high) and 10% scoring low (SOX9low; figure 1B). These observations extend the recent results from an independent study5 and comprehensively describe the SOX9 gene and protein expression landscape in human early stage I and II PDAC.
PDAC samples with differential expression levels of SOX9 classify into subtypes with different outcomes and therapeutic opportunities
To correlate SOX9 expression with PDAC tumour type and prognosis, we investigated whether SOX9high, SOX9medium and SOX9low tumours presented differential characteristics. SOX9 protein expression did not correlate with tumour size, tumour grade, N-stage or T-stage or vascular space invasion (data not shown). However, we observed that patients with SOX9high and SOX9medium tumours have better disease-specific survival (figure 1B) and overall survival (data not shown), which is also reflected when examining SOX9 mRNA expression (see online supplementary figure S3).
Collisson et al classified PDAC into different subtypes based on gene expression signatures. The classical and the quasi-mesenchymal (QM) subtypes discriminate for patient survival outcomes and potential therapeutic responsiveness; patients with the classical subtype have improved survival and growth of cell lines with the classical signature is more sensitive to inhibition of the EGFR pathway.21 We applied this classification and found that SOX9 mRNA expression is significantly higher in the classical subtype (figure 1C). In addition, the SOX9high tumours (of which 36/69 have gene expression data) were enriched for the classical type (15/36) compared with QM (7/36). This was not apparent in the SOX9medium group (of which 18/29 have gene expression data; 4/18 classical and 6/18 QM) or in the SOX9low group (of which 6/12 have gene expression data; 1/6 classical and 2/6 QM).
We conclude that SOX9high tumours preferentially belong to the classical subtype that has improved survival outcomes and may better respond to EGFR-targeted therapy.
SOX9 is an upstream regulator of ERBB2 expression in human PDAC
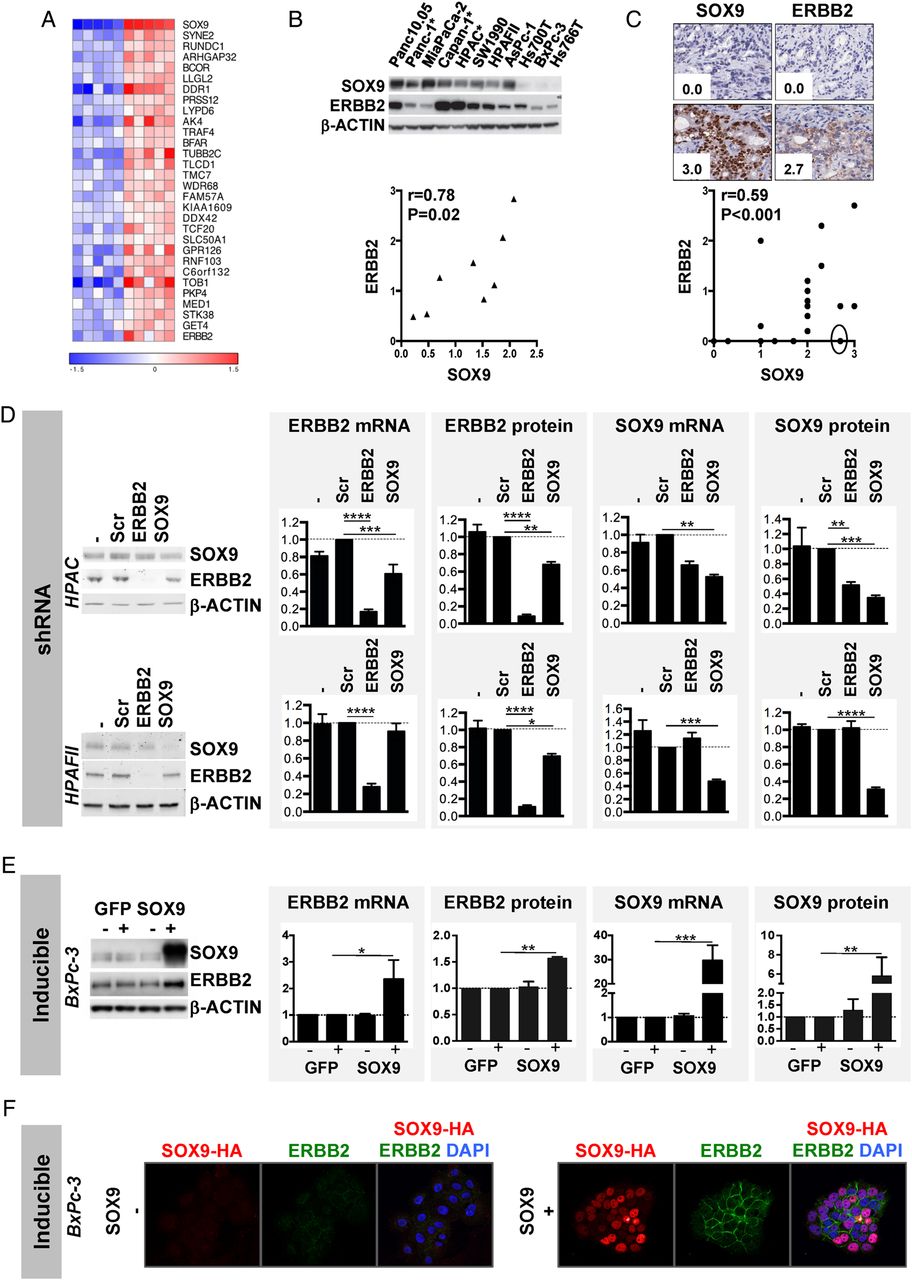
To gain mechanistic insight, we analysed the expression microarray data of the PDAC cohort and identified the genes whose expression correlated with SOX9 (online supplementary table S1). Among these, we found HNF1β, another typical pancreatic ductal transcription factor, HES1, an upstream regulator of SOX9 in adult pancreas and GATA6 for which a functional role unique for the classical subtype of PDAC has been described.14 ,21 Importantly, KOBAS pathway analysis showed enrichment for several mechanisms, including ERBB signalling (online supplementary table S2). Several ERBB family members showed high correlation with SOX9 expression, e.g. ERBB3 and ERBB2.
To better capture the differences between ‘extreme SOX9 expressors’, we then compared the 5 samples showing the highest expression of SOX9 and the 5 samples showing the lowest expression in the cohort. Again, we find ERBB2 featuring in the top 30 genes, together with Mediator 1 (MED1), a transcriptional stimulator of ERBB2 (figure 2A).31 Using GSEA, we found that SOX9 was associated with several upregulated pathways involved in cancer biology. The major themes are protein kinase activity, especially mitogen-activated protein kinase and EGFR/ERBB, as well as cell cycle control, and regulation of gene expression (online supplementary table S3). Besides ERBB2, other components of the ERBB pathway, e.g. EGFR and ERBB2IP/ERBIN, a protein that interacts with ERBB2 and is important for its activity,32 ,33 also showed core enrichment (data not shown).

SOX9 regulates ERBB2 expression. (A) Heat map of the 30 most upregulated genes in the five highest (red) SOX9-expressing tumours versus the five lowest (blue) SOX9-expressing tumours (p<4.37×10−4) based on microarray data from the Australian Pancreatic Cancer Genome Initiative cohort. (B) Correlation plot of SOX9 and ERBB2 band densities (n=8, r=0.78, p=0.02), quantified in western blot analysis, in a panel of ATCC cell lines (cell lines with a star have epidermal growth factor receptor (EGFR) activating mutations). Consequently, they were not included in the quantification). (C) Correlation plot of SOX9 and ERBB2 immunohistochemistry (IHC) scores in a panel of patient-derived xenografts (n=19, r=0.59, p<0.001). The full circle indicates a tumour with loss of heterozygosity of ERBB2. (D) Western blot analysis and quantification of ERBB2 and SOX9 mRNA and protein expression in HPAC and HPAFII cells stably infected with no shRNA (−), or shRNA against a scrambled sequence (Scr), ERBB2 or SOX9, analysed 5 days after selection (n=3). (E) Similar analysis performed on BxPc-3 cells transfected with a doxycycline-inducible eGFP expression plasmid, as a control, or with a doxycycline-inducible SOX9 expression plasmid. Results were analysed 72 h after transfection (n=3). (F) Confocal microscopy for ERBB2 and SOX9 in BxPc-3 cells without (−) or with (+) doxycycline-inducible haemagglutinin (HA)-tagged SOX9. 4’,6-diamidino-2-phenylindole (DAPI) was added to visualise the nuclei. In all graphs, *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Reduced ERBB2 expression impaired cell growth, as indicated by our experiments with pancreatic HPAFII and PanIN cells stably transduced with ERBB2 shRNA (see online supplementary figure S4).6 Therefore, we decided to explore the connection between SOX9 and the ERBB pathway, by focusing on the regulation of ERBB2 by SOX9, as expression of both genes is associated in KOBAS and GSEA. Analysis of ERBB2 and SOX9 protein in western blots of a panel of PDAC cell lines confirmed a correlation between SOX9 and ERBB2 protein expression (figure 2B). Also, IHC scoring for SOX9 and ERBB2 protein in xenografts of primary tumours confirmed a positive correlation of SOX9 and ERBB2 protein expression (figure 2C). Of note, the data point in the full circle in figure 2C, which showed the absence of ERBB2 despite a high SOX9 expression score, comes from a particular tumour that has an LOH of ERBB2 (data not shown). Together these data support that SOX9 and ERBB2 expression is well correlated in PDAC.
To further investigate the relation between SOX9 and ERBB2, we manipulated both in PDAC cell lines. ShRNA-mediated downregulation of SOX9 reduced the level of ERBB2 protein in HPAC and HPAFII cells, but only decreased ERBB2 mRNA levels in HPAC cells (figure 2D). Using siRNA for SOX9, we also observed reduced expression of ERBB2 protein in Panc10.05 and Panc-1 cells; in this experiment, ERBB2 mRNA was not reduced in both cell lines (see online supplementary figure S5). Doxycycline-induced overexpression of SOX9 in BxPc-3 cells upregulated ERBB2 mRNA and protein (figure 2E); in fact, confocal microscopy allowed to reveal that, without doxycycline, barely detectable expression of ERBB2 was seen as dots mostly detected in the cytoplasm, whereas, in the presence of doxycycline, abundant expression of ERBB2 was easily detected at the cell membrane (figure 2F).
These results suggest that SOX9 controls ERBB2 expression at the transcriptional and post-transcriptional level. To better understand this transcriptional regulation, we searched for SOX9-binding sites in the human and mouse ERBB2 promoters and found a potential dimeric site matching the SOX9 binding consensus in these regions (see online supplementary figure S6A). However, the transcriptional activity of reporter plasmids containing the human or mouse promoter was not modified in the presence of SOX9 (see online supplementary figure S6B), indicating that SOX9 could not bind to these sequences or was not sufficient to activate transcription from these regulatory regions.
All together, these observations strongly suggest that SOX9 promotes expression of ERBB2, a stimulator of pancreatic cell growth.
Sox9 controls the expression of members of the ErbB signalling pathway during mouse pancreatic tumourigenesis
To pursue our exploration of the role and mechanism of Sox9 in pancreatic cancer development, we turned to a mouse model of pancreatic tumourigenesis. Cre-recombinase-mediated induction of oncogenic K-Ras (KrasG12D) in acinar cells (ElaCER KrasG12D) is known to induce metaplasia and PanIN when associated with cerulein-driven pancreatic inflammation.10 ,28 ,29 When Sox9 is inactivated in this context (ElaCER KrasG12D Sox9f/f), metaplasia and neoplasia are no longer observed.5 ,27 We confirmed and extended these data using acute and chronic inflammation set-ups (see online supplementary figures S7–10).
We further found that the absence of Sox9 was also associated with reduced inflammatory response in cerulein-induced pancreatitis. Oedema and the number of macrophages and T lymphocytes were reduced in ElaCER KrasG12D Sox9f/f pancreata, compared with ElaCER KrasG12D pancreata (online supplementary figure S11). Importantly, this finding is reminiscent of earlier data indicating that pancreata lacking ErbB1/Egfr, in the same oncogenic Kras background, also present with reduced inflammation and resistance to PanIN development.12 ,13 Together with our observations on human samples, this again suggests that ErbB signalling and Sox9 function are connected.
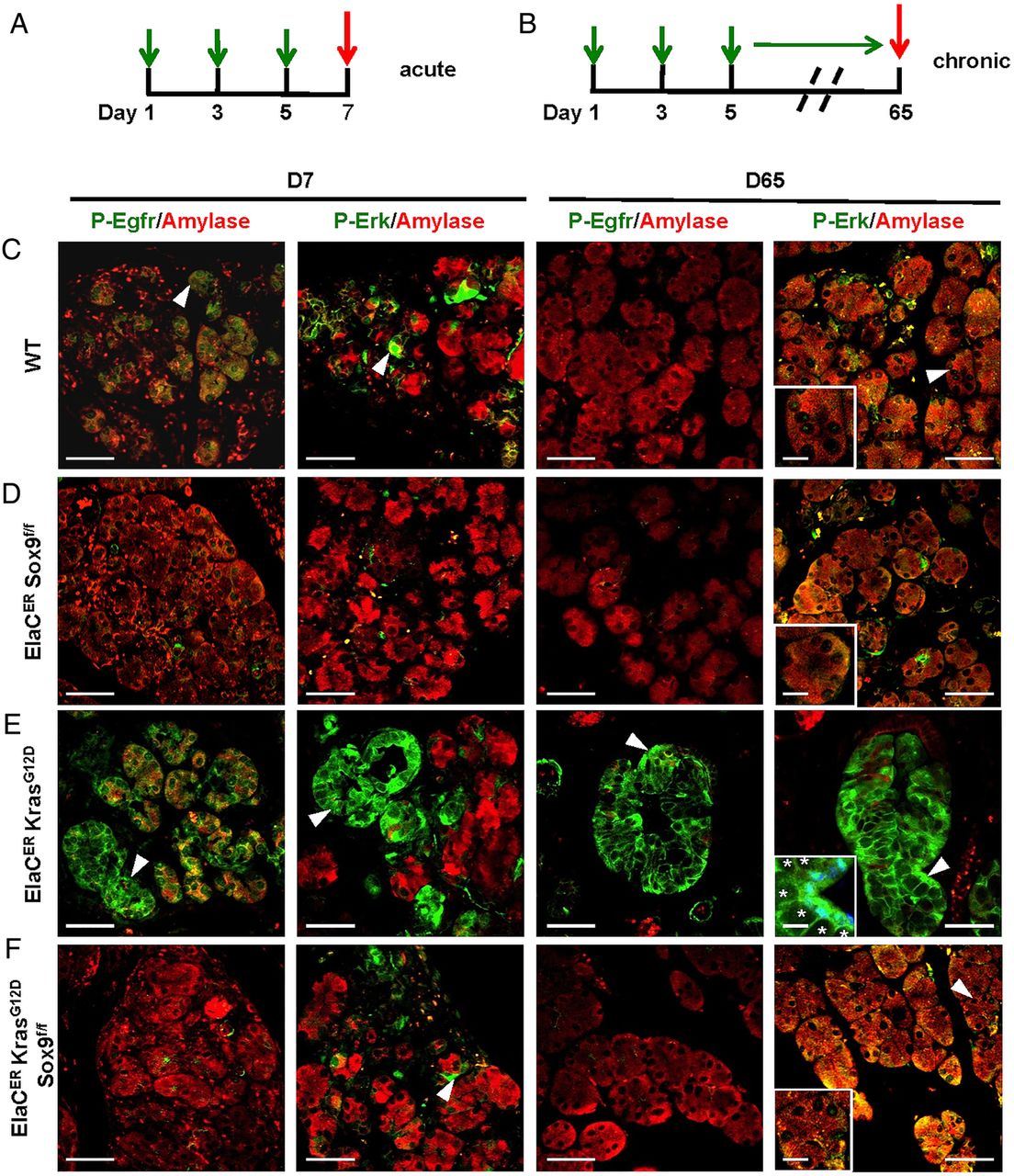
To further corroborate this, we tested the expression of Egfr, ErbB2 and Erbin in the mouse models by immunofluorescent labelling. The experiments also included labelling of Erk, a downstream target of ErbB important for PanIN development,12 and also a potential target of Sox9.34 In normal pancreas, these proteins were not detectable in acinar cells, whereas they were barely expressed in duct and centroacinar cells (online supplementary figure S12). At D7 of pancreatitis (figure 3A), some wild type (WT) metaplastic acini expressed low levels of ErbB2 and Erk, whereas expression of Erbin and Egfr was seen in a larger proportion of metaplastic acini (figure 3B). Strong induction of ErbB2, Erbin, Egfr and Erk was seen in ElaCER KrasG12D mice; typically, their expression was detected in metaplastic acini showing decreased expression of amylase and in tubular complexes (figure 3D). Importantly, ErbB2 labelling complexes distinguished between duct and acinar-derived tubular complex, because amylase was absent in the former and low in the latter (see online supplementary figure S13). In ElaCER Sox9f/f and ElaCER KrasG12D Sox9f/f pancreas, only weak ductal expression of ErbB2 and Egfr was seen and weak acinar expression of Erbin and Erk was detected (figures 3C,E). At D65 in WT mice, expression of ErbB2, Erbin, Egfr and Erk was mainly restricted to the ductal compartment. In a KrasG12D background (ElaCER KrasG12D), these proteins were expressed at high levels in developing PanIN, except when Sox9 was inactivated (ElaCER KrasG12D Sox9f/f; online supplementary figure S14). We conclude that Sox9 is required to stimulate expression of ERBB signalling components during pancreatic tumourigenesis.

Sox9 controls expression of members of the epidermal growth factor receptor (EGFR) pathway during pancreatitis. (A) Scheme of cerulein treatment. (B-E) Immunofluorescent labelling of amylase, ErbB2, Erbin, Egfr and extracellular-signal-regulated kinase (Erk) in wild type (WT) (B), ElaCER Sox9f/f (C), ElaCER KrasG12D (D) and ElaCER KrasG12D Sox9f/f (E) mice at day 7 (D7) of acute pancreatitis. The expression of ErbB2, Erbin, Egfr and Erk is strongly induced in metaplastic acini and tubular complexes in the ElaCER KrasG12D pancreas (arrowhead, D), whereas it is only weakly induced in the WT metaplastic acinar cells (arrowhead, B). In ElaCER Sox9f/f (C) and ElaCER KrasG12D Sox9f/f (E) pancreas, this expression is restricted to the apical pole of the metaplastic acinar cells for Erbin (C and E) or to the ducts, for ErbB2 (C and E) and Egfr (C and E). In both genotypes, Erk expression is weakly observed in metaplastic acinar cells (arrowhead, E). Magnifications of WT and ElaCER KrasG12D metaplastic cells are shown in the insets (B and D). Scale bars=50 µm.
Sox9 controls ErbB signalling during mouse pancreatic tumourigenesis
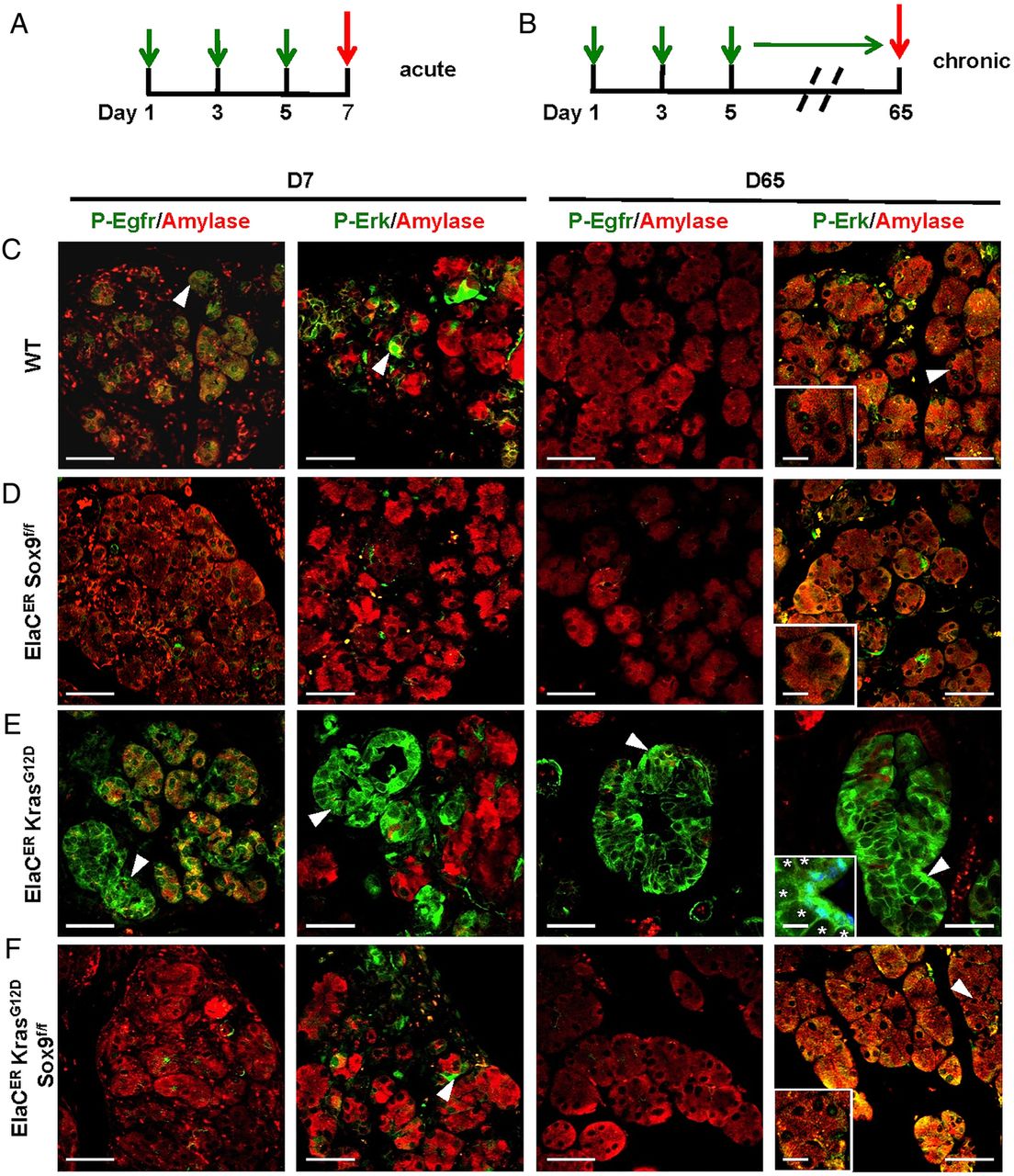
The reduced expression of ErbB signalling components in ElaCER KrasG12D Sox9f/f mice suggested that this pathway had little or no activity in these mice. Phosphorylation of Egfr and Erk is associated with activation of the ErbB pathway and was taken for further investigation of pathway activity. In the absence of pancreatitis, staining for P-Egfr and P-Erk was barely detected in the ducts of the tested genotypes (see online supplementary figure S12). At D7 of acute pancreatitis (figure 4A), in WT mice, P-Egfr and P-Erk were detected in a subset of metaplastic acinar cells (figure 4C). These phosphorylated forms were no longer detected in the acinar compartment in the absence of Sox9 (figure 4D). In contrast, in ElaCER KrasG12D mice, strong levels of P-Egfr and P-Erk were found in metaplastic acini and tubular complexes (figure 4E). In ElaCER KrasG12D Sox9f/f mice, no P-Egfr and weak levels of P-Erk were detected in acinar cells (figure 4E). In the chronic setup (figure 4B), at D65, Erk was weakly phosphorylated in WT, ElaCER Sox9f/f and ElaCER KrasG12D Sox9f/f mice, whereas high levels of phosphorylated Erk were detected in PanINs of ElaCER KrasG12D mice. P-Egfr was detected only in PanINs of ElaCER KrasG12D mice (figure 4E). These data indicate that detection of P-Egfr and P-Erk and activation of the ErbB pathway are dependent on Sox9 in the oncogenic Kras background.
The absence of Sox9 reduces epidermal growth factor receptor (EGFR) signalling activity during pancreatitis. (A and B) Scheme of cerulein treatments. (C–F) Immunofluorescent labelling for amylase, P-Egfr, and P-extracellular-signal-regulated kinase (Erk) in wild type (WT) (C), ElaCER Sox9f/f(D), ElaCER KrasG12D (E), and ElaCER KrasG12D Sox9f/f (F) mice at day 7 (D7) of acute pancreatitis and D65 of chronic pancreatitis. At D7, the activity of Egfr and Erk is strongly induced in tubular complexes of ElaCER KrasG12D mice (E) whereas this activity is reduced in WT (C), ElaCER Sox9f/f (D) and ElaCER KrasG12D Sox9f/f (F) mice. At D65, the activity of Egfr and Erk is elevated in pancreatic intraepithelial neoplasia (PanIN) (E) found in ElaCER KrasG12D mice, whereas the EGFR pathway is strongly reduced or inactive in WT, ElaCER Sox9f/f and ElaCER KrasG12D Sox9f/f mice (C, D, F). Insets show magnifications revealing P-Erk labelling in nuclei: levels of P-Erk were lower in WT, ElaCER Sox9f/f and ElaCER KrasG12D Sox9f/f metaplastic acinar cells (C, D and F), as compared with PanIN cells of ElaCER KrasG12D mice (labelled in blue by 4’,6-diamidino-2-phenylindole (DAPI) staining) (E). Star: mucin vesicle. Scale bars=50 µm.
To further test the function of the ErbB pathway in the absence of Sox9, pancreatic cell clusters, obtained by enzymatic digestion, were embedded in collagen gel and cultured in the absence or in the presence of epidermal growth factor (EGF). In such conditions, acinar cells undergo a metaplastic change and convert into duct-like cysts in an EGF-dependent process.35 In the absence of EGF, the size of cysts was similar in WT and ElaCER Sox9f/f tissue cultures (figure 5A, D). ElaCER KrasG12D showed significantly larger cysts, confirming the propensity of oncogenic Kras to produce metaplastic structures in these culture conditions (figure 5B, D).36 EGF induced a 2.0-fold and 1.5-fold increase in cyst size in WT and ElaCER Sox9f/f cultures, respectively; this indicates that Sox9 does not significantly affect the function of EGF in basal conditions (figure 5A, D). In ElaCER KrasG12D cultures, a 3.8-fold increase in cyst size was observed in the presence of EGF (figure 5A, D). Importantly, this increase was 1.8-fold lower in the absence of Sox9 (ElaCER KrasG12D Sox9f/f; figure 5C, D). From these tissue culture experiments, we conclude that Sox9 is required for full EGF response in the presence of mutant Kras.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The formation of epidermal growth factor (EGF)-induced duct-like cysts in collagen cultures of ElaCER KrasG12D pancreata is inhibited in the absence of Sox9. (A-C) Digested pancreata are cultured in the absence (−) or in the presence (+) of EGF. Phase contrast microscopy shows cyst formation in acinar cell cultures. Magnifications of cysts are shown in the insets. Scale bars=50 µm. (D) Quantification of cyst area in wild type (WT), ElaCER Sox9f/f, ElaCER KrasG12D and ElaCER KrasG12D Sox9f/f cultures, in the absence (−) or in the presence (+) of EGF (n=3, ***p<0.001, comparing ElaCER KrasG12D vs WT, ElaCER Sox9f/f, and ElaCER KrasG12D Sox9f/f cultures). Scales bars=100 µm.
All together, these experiments indicate that Sox9 stimulates the ErbB pathway in Kras-induced ADM. We suggest that the lack of ADM resulting from the absence of Sox9 and of ErbB2 signalling prevents progression to PanIN.
Discussion
SOX9, a key transcription factor in pancreatic cell differentiation, has recently come in the picture as critical for ADM and initiation of neoplastic lesions in the pancreas.5 ,15 This urged for an in depth study of the mechanism(s) driven by SOX9 in PDAC development and progression.
Our genomics data showed that SOX9 aberrations in human early stage I and II PDAC are seen in approximately 15% of the samples. We found a gain in copy number in about 5% of PDAC and this tends to correlate with higher SOX9 expression. Given the high SOX9 gene expression in the majority of samples, a finding that agrees with another recent study,5 and the equal rate of copy number gains and losses, we discount the increase in copy number as a major mechanism behind the high SOX9 expression in human PDAC samples.
PDAC samples are characterised by a stark heterogeneity among different patients.2 ,7 ,21 We started by studying SOX9 in the perspective of different subsets of tumours as this is possibly informative for deciphering the mechanism by which SOX9 operates. We employed a study where PDAC was categorised into different subtypes based on comprehensive gene expression signature analysis.21 We found that SOX9 expression is higher in classical PDAC and that concurrently, a SOX9high tumour is more likely within a better prognostic subtype. Reassuringly, we also found that GATA6, that has a functional role restricted to PDACs of the classical subtype, correlated well with SOX9 in our patient samples. It can be hypothesised that SOX9high tumours can evolve into a SOX9low state and that the EGFR pathway would then be downregulated in the latter. This is supported by mouse studies, which show that PDAC is initially dependent on Egfr but become independent eventually.12 ,13 This suggests that Sox9 function is always required for the initial phase of PDAC development but may become dispensable later, likely depending on the mutational landscape of the developing tumour.
Our analysis of human PDAC highlights the relation between high expression of SOX9 and high expression of ERBB signalling pathway members, e.g. ERBB2 and EGFR. In a mouse model recapitulating the initial steps of PDAC development, Sox9 is required for EGF-dependent ADM and for development of PanIN, a lesion which also depends on the activity of the EGFR pathway.12 ,13 All together, our mouse and human data suggest that SOX9 is required throughout formation of EGF-responsive pancreatic tumours, namely from the initial metaplastic and PanIN lesions, to the formation of malignant tumour. However, blockage of ADM and PanIN development seen in ElaCER KrasG12D Sox9f/f mice could explain by itself the downregulation of the ERBB pathway observed in these mice, although our results establishing the link between SOX9 and the ERBB pathway do not favour this view.
The role of ERBB2 in PDAC development is less well documented than that of EGFR.12 ,13 However, ERBB2 is the preferred dimerisation partner of EGFR, and the EGFR/ERBB2 heterodimer binds EGF with a seven-fold higher affinity than the EGFR homodimer.37 Moreover, ErbB2 is invariably and intensely expressed in PanIN lesions, in contrast to Egfr which shows heterogeneity; also, P-Erk levels correlated very well with ErbB2 expression, but much less with v,6 suggesting that ErbB2 has a functional role in the PDAC development. This is supported by our observations on growth of PanIN and HPAFII cells treated with ERBB2 shRNA. Consequently, ERBB2 might be considered as a therapeutic target.6 Interestingly, recent work characterising the clinical features of pancreatic cancer with ERBB2 amplification dismissed the conclusions of earlier work proposing that targeting ERBB2 was inefficient to treat PDAC.38–40 Instead, promising effects of novel dual targeting therapy to ERBB2 and EGFR in pancreatic cancer was observed.41 These observations and our present results warrant further explorations of the role of ERBB2 in early stage PDAC.
Manipulation of SOX9 expression resulted in altered ERBB2 expression, suggesting that SOX9 is an upstream regulator of ERBB2-mediated signalling. The nature of this regulation seems complex: whereas SOX9-dependent changes in ERBB2 protein levels are seen in all the cell lines tested, there is a variable correlation between SOX9 and ERBB2 mRNA in these cell lines. This suggests that ERBB2 is controlled by SOX9 at the transcriptional and post-transcriptional levels. Our results show that activity of the ERBB2 proximal promoter is not dependent on SOX9, raising the possibility that the transcriptional control exerted by SOX9 is indirect. MED1 could play a role in this control.31 Interestingly, our microarray data show that high expression of SOX9 correlates with high level of MED1 in human PDAC samples and a recent publication has revealed by chromatin immunoprecipitation that SOX9 binds to the MED1 promoter.42 ERBIN, which is known to stabilise ERBB2 protein and to restrict its localisation to the cell membrane,32 ,33 likely plays a role in the post-transcriptional control. Thus, high levels of SOX9 are associated with high level of ERBIN in human PDAC samples and downregulation of ERBIN expression is seen in ElaCER KrasG12D Sox9f/f mice. Interestingly, when SOX9 was overexpressed in BxPC3 cells, increased localisation of ERBB2 at the cell membrane was associated with an upregulation of ERBIN expression (data not shown).
Intriguingly, a number of immune-related pathways were downregulated in SOX9 high-expressing tumours, including immune response, response to virus, adaptive immune response and cytokine biosynthesis and secretion, primarily driven by changes in FOXP3, CD40L, SFTPD, TLR7 and 8, as well as a number of chemokines, cytokines and interleukins. This further underscores the functional differences between SOX9high and SOX9low tumours.
In conclusion, our results uncover the regulation of EGFR/ERBB2 by SOX9 in PDAC development, a finding that opens perspective for further exploration of therapeutic intervention targeting EGFR and ERBB2 during pancreatic cancer development.
Acknowledgments
We thank M. Sander and M. Hebrok for sharing results prior to publication, S. Konieczny for advice, Rolf Kemler and the Developmental Studies Hybridoma Bank (University of Iowa) for CK19 antibody, L. Mei for Erbin antibody, B. de Crombrugghe for p89 plasmid, P.P. Prévot for discussions, and D. Stoffers, D. Tuveson and G. Scherer for providing the ElaCreER, LSL KrasG12D and Sox9f/f mice, respectively. We thank B. Pastorelli, B. Pirlot and J. Pettitt for expert technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 10 - Online figure 9
- Data supplement 11 - Online figure 10
- Data supplement 12 - Online figure 11
- Data supplement 13 - Online figure 12
- Data supplement 14 - Online figure 13
- Data supplement 15 - Online figure 14
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
- Data supplement 5 - Online figure 4
- Data supplement 6 - Online figure 5
- Data supplement 7 - Online figure 6
- Data supplement 8 - Online figure 7
- Data supplement 9 - Online figure 8
Footnotes
IR and PJ contributed equally.
Contributors AG: study concept and design, acquisition of data, analysis and interpretation of data, statistical analysis and drafting of manuscript. AVP, MJC, AM and JW: acquisition of data, analysis and interpretation of data and statistical analysis. CA, MG-L, GVdS and NW: acquisition of data, analysis and interpretation of data. MP: technical or material support. CS: analysis and interpretation of data. SMG and AVB: obtaining funding, technical or material support. FPL, IR and PJ: study concept and design, analysis and interpretation of data, drafting of manuscript and obtaining funding.
Funding This work was supported by grants from the Cancer Institute NSW; National Health and Medical Research Council of Australia; Australian Government: Department of Innovation, Industry, Science, Research and Tertiary Education; Australian Cancer Research Foundation; Queensland Government; University of Queensland; Cancer Council NSW; Garvan Institute of Medical Research; Avner Nahmani Pancreatic Cancer Research Foundation; R.T. Hall Trust; Petre Foundation; Jane Hemstritch in memory of Philip Hemstritch; Gastroenterological Society of Australia; American Association for Cancer Research Landon Foundation—INNOVATOR Award; Royal Australasian College of Surgeons; Royal Australasian College of Physicians; Royal College of Pathologists of Australasia; HGSC-BCM: NHGRI U54 HG003273; CPRIT grant RP101353-P7 (Tumour Banking for Genomic Research and Clinical Translation Site 1); Fondation contre le Cancer (Belgium); Fonds de la Recherche Scientifique-FNRS (Belgium); Université catholique de Louvain. AG was supported by a grant from EU FP7 (Marie Curie Initial Training Network BOLD) and a Télévie fellowship. AVP and MJC are supported by early career fellowships from Cancer Institute NSW. CA holds a Télévie fellowship, IR is a Future Research Fellow of the Cancer Institute NSW (10/FRL2-03) and PJ is a Research Associate of the Fonds de la Recherche Scientifique-FNRS.
Competing interests None.
Ethics approval Sydney Local Health District—RPA Zone, protocol X11-0220.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Unpublished data are available upon e-mail request to the corresponding authors.