Article Text

Abstract
Objectives The tumour stroma/microenvironment not only provides structural support for tumour development, but more importantly it provides cues to cancer stem cells (CSCs) that regulate their self-renewal and metastatic potential. This is certainly true for pancreatic ductal adenocarcinomas (PDAC), where tumour-associated fibroblasts, pancreatic stellate cells and immune cells create an abundant paracrine niche for CSCs via microenvironment-secreted factors. Thus understanding the role that tumour stroma cells play in PDAC development and CSC biology is of utmost importance.
Design Microarray analyses, tumour microarray immunohistochemical assays, in vitro co-culture experiments, recombinant protein treatment approaches and in vivo intervention studies were performed to understand the role that the immunomodulatory cationic antimicrobial peptide 18/LL-37 (hCAP-18/LL-37) plays in PDAC biology.
Results We found that hCAP-18/LL-37 was strongly expressed in the stroma of advanced primary and secondary PDAC tumours and is secreted by immune cells of the stroma (eg, tumour-associated macrophages) in response to tumour growth factor-β1 and particularly CSC-secreted Nodal/ActivinA. Treatment of pancreatic CSCs with recombinant LL-37 increased pluripotency-associated gene expression, self-renewal, invasion and tumourigenicity via formyl peptide receptor 2 (FPR2)- and P2X purinoceptor 7 receptor (P2X7R)-dependent mechanisms, which could be reversed by inhibiting these receptors. Importantly, in a genetically engineered mouse model of K-Ras-driven pancreatic tumourigenesis, we also showed that tumour formation was inhibited by either reconstituting these mice with bone marrow from cathelicidin-related antimicrobial peptide (ie, murine homologue of hCAP-18/LL-37) knockout mice or by pharmacologically inhibiting FPR2 and P2X7R.
Conclusions Thus, hCAP-18/LL-37 represents a previously unrecognised PDAC microenvironment factor that plays a critical role in pancreatic CSC-mediated tumourigenesis.
- ANTIBACTERIAL PEPTIDE
- PANCREATIC CANCER
- STEM CELLS
- MACROPHAGES
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Pancreatic ductal adenocarcinoma (PDAC) is the most lethal cancer with limited therapeutic options.
Pancreatic cancer stem cells (CSCs) are exclusively tumourigenic and highly resistant to chemotherapy.
Tumour-associated macrophages are important for the progression and metastatic spread of many solid tumours.
What are the new findings?
The immunomodulatory cationic antimicrobial peptide 18/leucine leucine-37 (hCAP-18/LL-37) is overexpressed in the stroma of PDAC and acts on CSCs to potentiate their inherent biological properties.
Tumour-associated macrophages secrete hCAP-18/LL-37 in direct response to CSC-secreted NODAL/ACTIVINA/tumour growth factor-β1.
Small molecule targeting of the LL-37 receptors formyl peptide receptor 2 (FPR2) and P2X purinoceptor 7 receptor (P2X7R), present on pancreatic CSCs, negatively impacts tumour growth and circulating tumour cell numbers.
How might it impact on clinical practice in the foreseeable future?
The discovery of the crucial role of hCAP-18/LL-37 in CSC biology represents an important advancement in our understanding of the PDAC tumour microenvironment.
Targeting pancreatic CSCs using inhibitors of the LL-37 receptors FPR2 and P2X7R may represent a specific therapeutic approach to block the tumour promoting cross-talk that exists within the tumour microenvironment.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers largely due to its high resistance to current treatment strategies.1 This can, at least in part, be attributed to a subpopulation of cells known as pancreatic cancer stem cells (CSCs),2–4 which are defined by their cell-intrinsic and unlimited self-renewal, exclusive long-term tumourigenicity, capacity to recapitulate the entire cancer cell heterogeneity and metastatic potential.5–7 In addition, PDAC is characterised by extensive desmoplasia,8 which is made up of heterogeneous cell populations, including pancreatic stellate cells (PSCs)9 ,10 and immune cells.8 ,11–15 This dynamic cellular microenvironment may directly or indirectly promote CSC features,6 ,10 ,16 ,17 but few comprehensive studies have been performed in PDAC to date. Thus, we set out to identify stroma-specific paracrine drivers that potentiate pancreatic CSC features.
Here we show that the human cationic antimicrobial protein 18 (hCAP-18, cathelicidin antimicrobial peptide (CAMP)), the only known human cathelicidin alarmin,18–20 is strongly and exclusively expressed by macrophages present within the PDAC stroma. Cleavage of hCAP-18 at the COOH-terminal end gives rise to the biologically active 37 amino acid hCAP-18 peptide called leucine leucine-37 (LL-37).19 ,20 Intriguingly, secreted LL-37, via the G protein-coupled receptor, formyl peptide receptor 2 (FPR2)21–23 and P2X(7) purinergic receptor (P2X7R),22 significantly potentiated pancreatic CSC features, such as self-renewal, invasion and tumourigenesis. While the factors that mediate LL-37 expression can vary based on the biological context, we also show for the first time that CSC-secreted tumour growth factor-β (TGF-β) family members Nodal and ActivinA induce hCAP-18/LL-37 expression in macrophages. Pharmacological or genetic inhibition of paracrine activation of pancreatic CSCs by LL-37 markedly reduced their tumourigenicity and metastasis in vivo. Thus, our findings not only identify a previously unrecognised tumour microenvironment factor that potentiates pancreatic CSC features, but also highlight the potential therapeutic impact that targeting this peptide may have on PDAC progression and spread.
Methods
Primary human pancreatic cancer cells and macrophages
Tumours were expanded in mice as xenografts (patient-derived xenografts (PDX)), processed and subsequently cultured in vitro as previously detailed.7 Human blood was obtained from healthy donors with informed consent. Monocyte-derived human macrophage cultures were established and polarised to an M1 phenotype with granulocyte-macrophage colony-stimulating factor (GM-CSF) as previously described.24–26
Tissue microarrays
Four human tissue microarrays (TMAs) containing quadruplicate 1 mm cores from a total of 42 tumours were constructed. All immunohistochemically stained sections were assessed and scored by in-house pathologists.
In vivo assays
The K-Ras+/LSL-G12D;Trp53LSL-R172H;PDX1-Cre mouse model (KPC) of advanced pancreatic cancer has been described previously.27 B6.129X1-Camptm1Rlg/J mice (cathelicidin-related antimicrobial peptide (CRAMP−/−)) were purchased from Jackson Laboratories (Bar Harbor, Maine, USA) and have been previously described.28 Mice were housed according to institutional guidelines.
Statistical analyses
Results for continuous variables are presented as means±SEM unless stated otherwise. Treatment groups were compared with the independent samples t test. Pair-wise multiple comparisons were performed with the one-way analysis of variance (two-sided) with Bonferroni adjustment. p Values <0.05 were considered statistically significant. All analyses were performed using SPSS V.22.0 (SPSS, Chicago, Illinois, USA).
More materials and methods can be found as online supplementary information.
Results
Macrophages promote PDAC tumourigenesis and produce hCAP-18/LL-37
To appreciate the contributory role of tumour stroma cells in PDAC development, we injected into nude mice 5×105 primary sphere-derived CSC-enriched PDAC cells alone or with equal numbers of immortalised PSCs, primary PSCs or primary human monocyte-derived unpolarised macrophages. No marked differences in early tumour growth were observed between primary PDAC cells injected alone or with PSCs. In contrast, tumour take and growth was significantly accelerated when PDAC cells were co-injected with macrophages (figure 1A). Interestingly, at 1.5 weeks human macrophages were no longer detected in these tumours as determined by a lack of CD68, CD16 and CD163 staining (data not shown), suggesting that the transient presence of human macrophages was sufficient to jumpstart tumour take and promote PDAC cell growth in vivo.

Macrophages promote pancreatic ductal adenocarcinomas (PDAC) tumour take and growth. (A) Summary of in vivo tumour take and growth 3 weeks post-subcutaneous injection of sphere-derived cancer stem cell (CSC)-enriched PDAC cells with or without human monocyte-derived unpolarised macrophages (MØ), immortalised pancreatic stellate cells (iPSCs) or primary PSCs (pPSCs) (n=4 mice/group). (B) Heatmap of top 25 genes upregulated and downregulated (FDR<10−4, |logFC|>2) in primary human monocyte-derived macrophages co-cultured with Panc185 (1), Panc354 (2) or Panc215 (3) in a trans-well assay for 48 h. (C) RT-qPCR analysis of human cationic antimicrobial peptide 18/ leucine leucine 37 (hCAP-18/LL-37) mRNA levels in human monocyte-derived macrophages alone or co-cultured with indicated PDAC cells for 48 h. (D) Immunofluorescence analysis of CD16 (macrophage marker) and LL-37 in human monocyte-derived macrophage-PDAC cell co-cultures. White dashed line marks the perimeter of a PDAC colony. Scale bar=100 µm. (E) RT-qPCR analysis of Oct3/4 and Nanog mRNA levels in Panc215 and Panc185 cultures following 48 h co-cultivation with primary human monocyte-derived macrophages in a trans-well assay.
To begin to understand how macrophages might promote PDAC tumour growth, we co-cultured human monocyte-derived unpolarised macrophages with and without primary PDAC cells in trans-wells (to separate the two cell types) and performed microarray analyses (see online supplementary table S1). Of the top 25 upregulated genes in macrophages (FDR<10−4, |logFC|>2), 19 genes belonged to the family of interferon-stimulated genes (figure 1B), while the six remaining genes (radical S-adenosyl methionine domain containing 2, cytidine monophosphate (UMP-CMP) kinase 2, kertain 19, CAMP, sialic acid binding Ig-like lectin 1 and orosomucoid 1) encoded for proteins with diverse functions. We identified hCAP-18/LL-37 (ie, CAMP) as a gene of interest as it both encodes for a secreted factor and has been shown to be expressed in other solid tumours, including breast,29 ,30 lung31–33 and ovarian cancers,18 ,23 ,34 although in the epithelial compartment. RT-qPCR analysis (figure 1C) and immunofluorescence confocal microscopy (figure 1D) showed upregulation of hCAP-18/LL-37 in monocyte-derived unpolarised macrophages only when co-cultured with primary PDAC cells, validating our microarray results. Moreover, while PDAC cells co-cultured with macrophages did not express hCAP-18/LL-37 (data not shown), we observed upregulation of pluripotency-associated genes, which was blocked by the addition of LL-37 blocking antibodies or inhibitors, suggesting an LL-37-mediated cross-talk between macrophages and CSCs (figure 1E and online supplementary figure S1).
Primary PDAC tumours express hCAP-18/LL-37
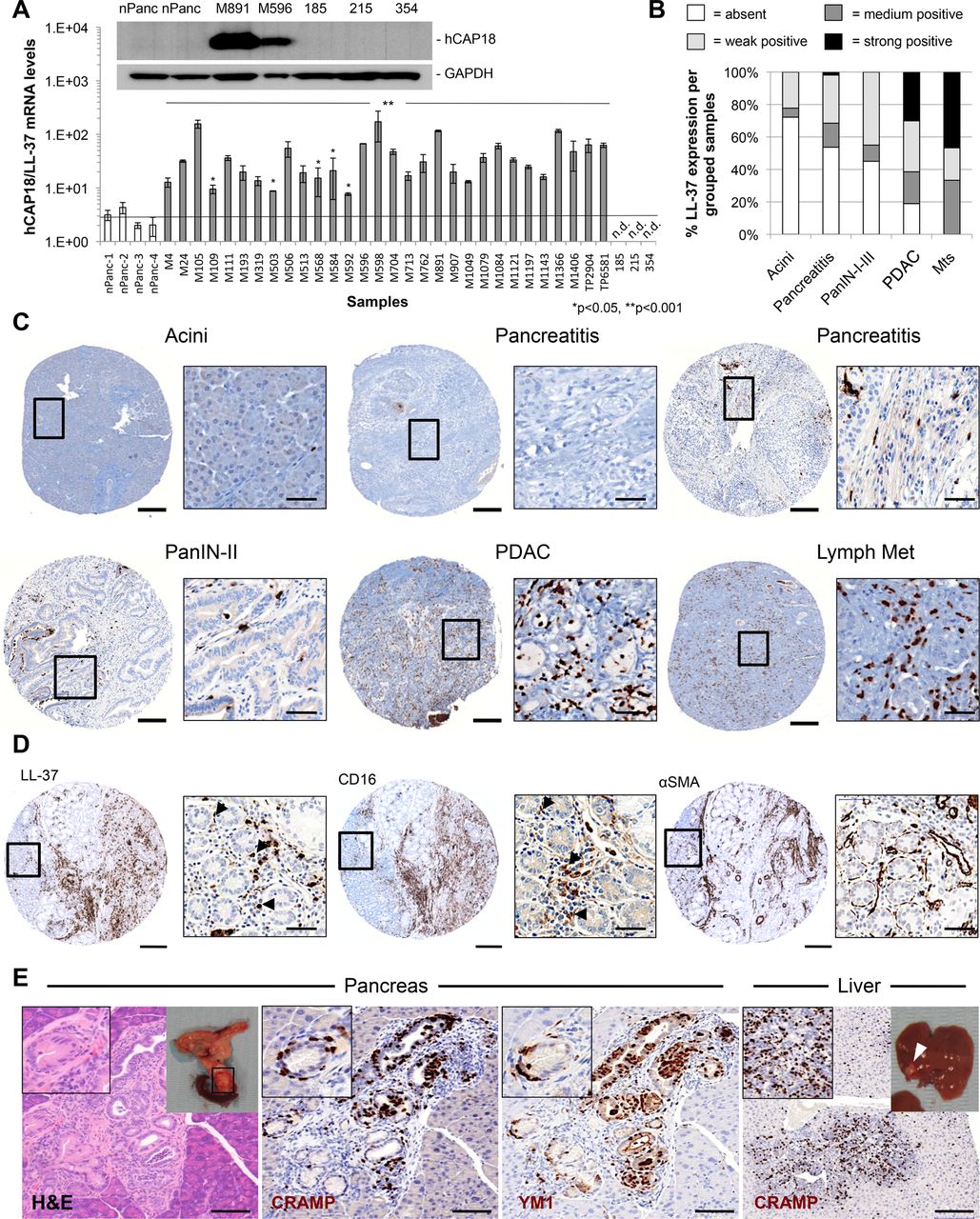
Next, we assessed the expression of hCAP-18/LL-37 by RT-qPCR and immunohistochemical analysis in primary PDAC samples. Compared to normal pancreas, overexpression of hCAP-18/LL-37 mRNA and protein was observed in all PDAC samples (figure 2A and online supplementary figure S2A). Interestingly, only the stroma stained positive for hCAP-18/LL-37 while cancer cells were negative (figure 2A and online supplementary figure S2A–C), which is in contrast to what has been reported for other carcinomas.18 ,29 ,31 ,35

Leucine leucine 37 (LL-37) expression is restricted to the tumour stroma and correlates with advanced neoplastic lesions. (A) RT-qPCR analysis of human cationic antimicrobial peptide 18 (hCAP-18)/LL-37 mRNA levels in a panel of surgically resected human primary pancreatic ductal adenocarcinomas (PDAC) tumours (n=30) and cultures (185, 215 and 354). Western blot analysis of hCAP-18/LL-37 protein expression in a subgroup of samples (inset). (B) hCAP-18/LL-37 expression profile in a tissue microarray (TMA) panel containing normal, pancreatitis, pancreatic intraepithelial neoplasia (PanIN, I–III), PDAC, and metastases (Mts) cores. (C) Representative micrographs of hCAP-18/LL-37-stained TMA cores. (D) Serial sections of a PDAC TMA core stained for hCAP-18/LL-37, the macrophage markers CD16 and the pancreatic stellate cell (PSC) marker αSMA. (E) Immunohistological analysis of cathelicidin-related antimicrobial peptide (CRAMP) (murine ortholog of LL-37) and YM1 (macrophage marker) expression in 26-week-old K-Ras+/LSL-G12D;Trp53LSL-R172H;PDX1-Cre (KPC) mouse pancreas and liver. Scale bars=200 and 50 µm (insets). nPanc, normal pancreas.
Data were extended using TMAs. The majority (∼74%) of ‘normal’ adjacent non-tumour tissue was negative for hCAP-18/LL-37; however, 81% of PDAC samples stained positive for LL-37 with varying degrees of intensity observed between tissues (figure 2B,C and online supplementary figures S2 and S3). Staining of serial sections for LL-37, CD16, CD163, CD68 and αSMA demonstrated that tumour infiltrating immune cells (ie, macrophages) were the predominant cell type producing hCAP-18/LL-37 (figure 2D and online supplementary figure S2D–E); however, other stromal cells (eg, CD16+ neutrophils) also expressed LL-37.
To allow for a more systematic analysis of pancreatic cancer progression from low-grade to high-grade pancreatic intraepithelial neoplasias (PanINs), PDAC lesions and subsequent metastatic spread, we next studied the K-Ras+/LSL-G12D;Trp53+/LSLR172H;PDX-1-Cre (hereafter referred to as KPC)27 mouse model of PDAC. CRAMP (ie, murine homologue of hCAP-18/LL-37) expression in KPC mice was similarly restricted to the tumour stroma, absent in normal tissue and most prominently expressed in primary PDAC lesions and in all metastatic lesions of secondary organs, such as the liver (figure 2E and online supplementary figure S4A). Staining for the macrophage markers YM1 or F4/80 also revealed a strong correlation between macrophages and CRAMP staining (figure 2E and online supplementary figure S4B, C).
CRAMP promotes tumourigenicity of murine PDAC cells in vivo
To test whether CRAMP expression is necessary for tumour formation in vivo, we eliminated CRAMP from the haematopoietic system of irradiated 5-week-old to 6-week-old KPC mice by syngeneic transplantation of either wild-type or CRAMP knockout (CRAMP−/−) bone marrow. While all mice, regardless of the donor bone marrow received, showed macrophage infiltration in the pancreas (see online supplementary figure S5A), mice transplanted with bone marrow from CRAMP−/− mice did not show any CRAMP expression, which does not only suggest excellent transplantation efficiency but also demonstrates that immune cells and not other stromal cells are the primary source of CRAMP in PDAC tumours (figure 3A). At the tumour level, PDAC tumour tissue and severely altered tissue (ie, acinar-to-ductal metaplasia and inflammation (see online supplementary figure S5B)) were significantly reduced in mice transplanted with CRAMP–/– bone marrow compared with controls at 17 weeks post transplantation (figure 3B and online supplementary figure S5C).

Cathelicidin-related antimicrobial peptide (CRAMP) affects pancreatic ductal adenocarcinomas (PDAC) tumour growth in vivo. (A) Immunohistochemical (IHC) analysis of CRAMP expression in representative formalin-fixed paraffin-embedded sections from pancreata of 22-week-old to 23-week-old K-Ras+/LSL-G12D;Trp53LSL-R172H;PDX1-Cre (KPC) mice transplanted with bone marrow from wild-type control C57Bl/6 mice or CRAMP−/− mice (n=4 mice/group). (B) Quantification of tissue area in mouse pancreata from 22-week-old to 23-week-old KPC mice transplanted with bone marrow from wild-type control C57Bl/6 mice (n=4 mice) or CRAMP−/− mice (n=4 mice/group), categorised as normal acinar tissue, severely altered tissue (acinar-to-ductal metaplasia (ADM) and inflammation) or tumour tissue pancreatic intraepithelial neoplasia (PanINs I–III and PDAC) (see online supplementary figure S5B). Representative images of tumour tissue (right). (C and D) Wild-type and CRAMP−/− mice were subcutaneously injected with the indicated number of primary murine PDAC cells (ie, CHX-BC-RFP-Luc cells) in Matrigel (n=8 injections/group). Tumour take was determined 5 weeks post injection (left) by bioluminescence imaging (BLI) assessment (C). Representative BLI pictures of mice 5 weeks postinjection with indicated numbers of murine PDAC cells (left). Summary of in vivo tumour take and growth (right). Cancer stem cell (CSC) frequencies determined using the extreme limiting dilution analysis algorithm (http://bioinf.wehi.edu.au/software/elda/index.htmL) (right, 95% CI). (D) Tumours from mice injected with 105 murine PDAC cells were excised and CD133 content within the RFP+ population was determined. Quantification of the per cent of cells expressing CD133 is graphed (top) and representative cytometry plots are shown (bottom). (E) Summary of in vivo tumour take and growth of subcutaneously-injected 5×105 sphere-derived CSC-enriched murine PDAC cells with or without murine monocyte-derived unpolarised macrophages (MØ) isolated from wild-type (wt) CRAMP+/+ mice or CRAMP−/− mice (n=8 mice/group). n.s., not significant.
To rigorously test the effects of CRAMP on the in vivo CSC compartment, which are exclusively capable of tumour initiation and progression,36 we next performed limiting dilution cell transplantation assays. Primary syngeneic murine PDAC sphere-derived cells expressing a luciferase reporter were transplanted into recipient wild-type and CRAMP−/− mice, and tumour formation was determined 5 weeks post injection (figure 3C, left). While tumours efficiently formed in wild-type mice at dilutions of 104 (4/8) and 105 (8/8) cells, only one tumour was detected in CRAMP−/− mice injected with 105 (1/8) cells, indicating that the capacity for CSC-initiated tumourigenesis was significantly reduced in CRAMP–/– mice (figure 3C, right). In addition, we found a clear reduction in the percentage of CD133+ cells in the tumour formed in the CRAMP−/− mouse (figure 3D). To further dissect these phenotypes at the macrophage level, we injected 5×105 primary sphere-derived CSC-enriched murine PDAC cells alone or with equal numbers of monocyte-derived unpolarised macrophages isolated from wild-type or CRAMP−/− mice. After 4 weeks, tumour take and growth was significantly accelerated when murine PDAC cells were co-injected with wild-type macrophages (similar to what was observed when human macrophages were co-injected with human PDAC cells (figure 1A)), but not with CRAMP–/– macrophages (figure 3E). In line with these observations, murine PDAC sphere formation was consistently enhanced in the presence of wild-type macrophages versus CRAMP−/− macrophages (see online supplementary figure S5D), suggesting that CRAMP enhances/promotes PDAC tumour take and progression by potentiating CSC in vivo self-renewal and tumourigenesis.
hCAP-18/LL-37 has inherent pro-CSC properties
Stimulated by these proof-of-concept studies, we next aimed to further dissect the mechanisms of action for LL-37 on human pancreatic cancer (stem) cells. Using the established CSC cell surface marker CD1332 and a novel biomarker for CSC, autofluorescence,5 we found that treatment of primary xenograft-derived PDAC cultures with recombinant LL-37 peptide (rLL-37) resulted in a consistent approximately twofold enrichment in the CD133+ (figure 4A and online supplementary figure S6A) and autofluorescent populations (figure 4B and online supplementary figure S6A), suggesting that LL-37 increases the CSC pool. Indeed, colony-forming (figure 4C) and sphere-forming efficiency (figure 4D) were both significantly enhanced by rLL-37; the latter even more pronounced during serial passaging, which further enriches for CSCs (figure 4D). Treatment also resulted in overexpression of KLF4, SOX2, OCT3/4 and NANOG in rLL-37-treated spheres (figure 4E and online supplementary figure S6B, C). Of note, recombinant CRAMP functioned similarly to rLL-37 in CSCs derived from KPC mouse pancreatic tumours (see online supplementary figure S7). Lastly and in line with a pro-CSC effect, rLL-37 also increased the inherent chemoresistant potential of CSCs, as measured by increased CD133+ cells following treatment with gemcitabine or abraxane (figure 4F and online supplementary figure S8).

Leucine leucine 37 (LL-37) expands the cancer stem cell (CSC) pool. (A and B) Primary sphere-derived pancreatic ductal adenocarcinomas (PDAC) cultures were treated with recombinant leucine leucine 37 (rLL-37) and the percentage of (A) CD133+ cells and (B) autofluorescent cells was measured by flow cytometry. (C and D) Effects of rLL-37 on CSC (C) colony formation and (D) serial sphere formation. (E) RT-qPCR analysis of pluripotency-associated genes in sphere-derived PDAC cells after stimulation with rLL-37. (F) Percentage of CD133+ cells in control versus gemcitabine (GEM)-treated (0.1 µg/mL) or gemcitabine and rLL-37-treated (5 µg/mL) primary PDAC cells (top), as determined by flow cytometric analysis (bottom).
Importantly, the most defining feature of CSCs lies in their ability to form tumours in vivo. Sphere-derived PDAC cells pretreated with rLL-37 revealed consistently enhanced tumourigenicity, a higher CSC frequency and increased early tumour take as compared to scrambled peptide-treated cells. Notably, this effect was particularly evident when low cell numbers (∼10 cells) were injected (figure 5A, B). In line with our hypothesis that the effects of LL-37 are preferentially affecting CSCs, when we injected adherent cells, which contain few CSCs,6 the differences in tumourigenicity between non-treated and rLL-37-treated cells were not as striking (sphere-derived cells: 10.2-fold (CI 10.1 to 10.4) increase in CSC frequency; adherent cells: 1.4-fold (CI 1.3 to 1.5) increase in CSC frequency) (see online supplementary figure S9). Lastly and to more rigorously test the hypothesis that LL-37 more specifically promotes CSCs, we assessed the sphere-forming capacity and cell cycle profile of CD133+ CSC and their CD133− counterparts in the absence or presence of rLL-37. Indeed, our data confirm that LL-37 preferentially targets and potentiates CSC self-renewal and proliferation (figure 5C, D).

Leucine leucine 37 (LL-37) enhances functional cancer stem cell (CSC) properties. (A and B) Mice were subcutaneously injected with the indicated number of scrambled or recombinant leucine leucine 37 (rLL-37) peptide (10 µg/mL) pretreated CSCs resuspended in Matrigel (n=4 mice/dilution/group). (A) Tumour take was determined 10 weeks post injection (left). Images of resected tumours and CSC frequencies determined using the extreme limiting dilution analysis algorithm (http://bioinf.wehi.edu.au/software/elda/index.htmL) (right, 95% CI). (B) Summary of in vivo tumour take and growth. (C) Primary pancreatic ductal adenocarcinomas (PDAC) cells were sorted for the CSC marker CD133, treated with rLL-37 or a scrambled peptide control, cultured as spheres in anchorage independent conditions and sphere numbers determined 7 days later (n.s., not significant). (D) Proliferation, as detected by BrdU staining, in CD133+ and CD133− PDAC cells after stimulation with rLL-37.
LL-37 promotes CSC invasiveness
We found the highest expression of LL-37 and CRAMP in primary PDAC tumours and metastatic lesions of human and mouse origins (figure 2), respectively, suggesting that LL-37 is likely important late during PDAC progression and may be involved in epithelial to mesenchymal transition (EMT). Supporting this hypothesis, rLL-37 treatment increased the C-X-C chemokine receptor type 4 (CXCR4)+ subpopulation (figure 6A) present in CSCs, a subpopulation of CSCs that drives metastasis.2 Analysis of EMT-related genes following rLL-37 treatment revealed downregulation of E-cadherin and upregulation of vimentin and Snail (figure 6B, C), and rLL-37 treatment also increased migration (figure 6D) and invasion of CSCs (figure 6E). Of particular interest, chemoattraction of invading rLL-37-treated sphere-derived PDAC cells by the CXCR4 ligand SDF-1 was evident even at very low concentrations (eg, 1 ng/mL), which had no impact on control cells. To validate this in vivo, we intrasplenically injected rLL-37 pretreated CSCs stably expressing a luciferase reporter and observed increased dissemination (ie, micro-metastases) of cells pretreated with rLL-37 to the liver (figure 6F, G and online supplementary figure S10).
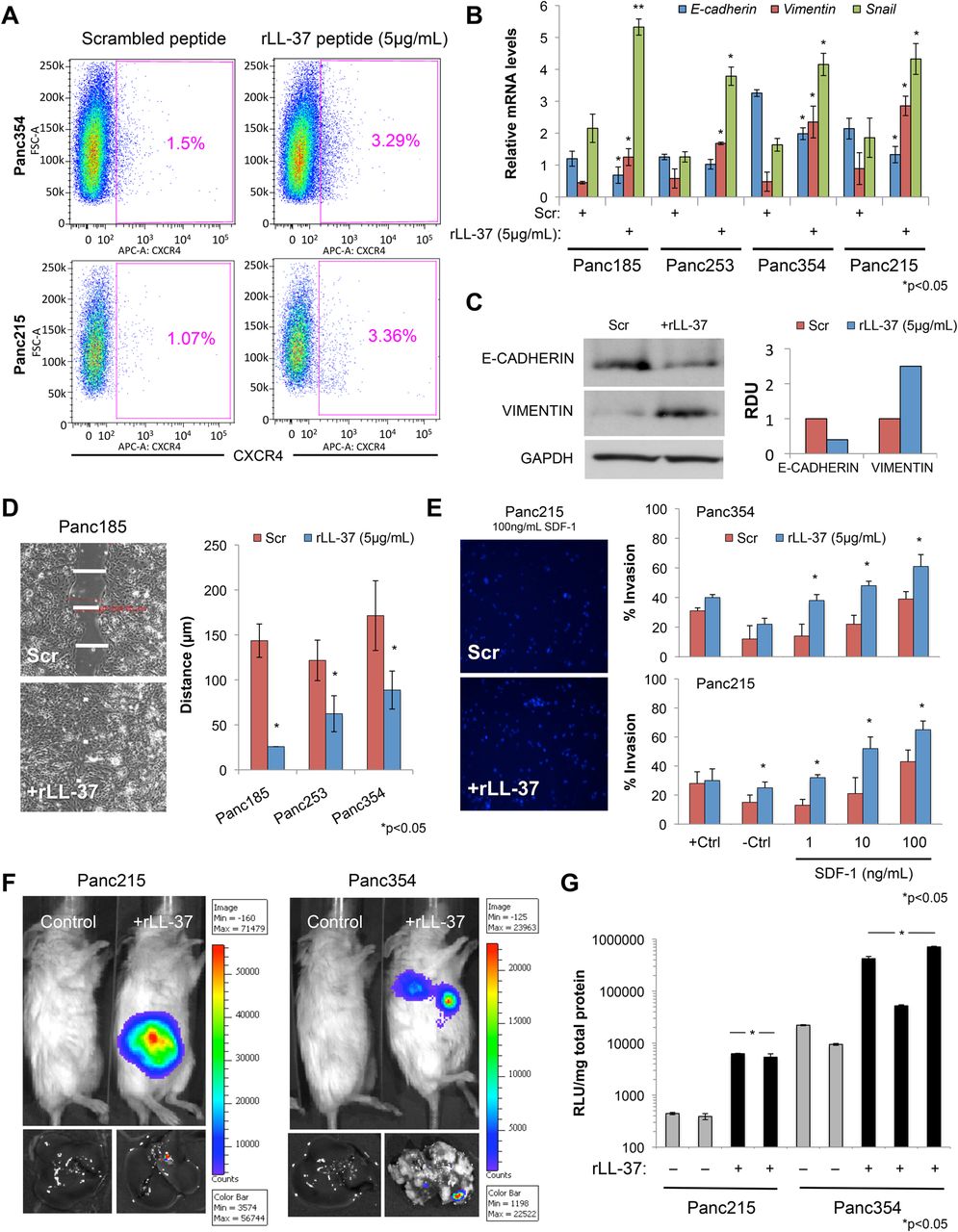
Leucine leucine 37 (LL-37) promotes epithelial to mesenchymal transition (EMT) and cancer stem cell (CSC) invasiveness. (A) Flow cytometry analysis of C-X-C chemokine receptor type 4 (CXCR4) cell surface expression in two primary pancreatic ductal adenocarcinomas (PDAC) cultures treated with recombinant leucine leucine 37 (rLL-37). (B) RT-qPCR analysis of EMT genes in sphere-derived PDAC cells after stimulation with rLL-37. (C) Western blot analysis of E-cadherin, vimentin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in PDAC cells after stimulation with rLL-37 (left) and densitometric analysis of blots (right). RDU, relative density units. (D) Scratch wound assay of PDAC cells after stimulation with rLL-37. Representative micrographs (left) and quantification of wound size 12 h after wound induction (right). (E) Representative images of invaded cells (left) and quantification of invaded rLL-37-treated PDAC cells through Matrigel following stimulation with 20% fetal bovine serum (+Ctl), media alone (−Ctl) or increasing concentration of SDF-1 (right). (F) PDAC cells dissemination in vivo, assessed by non-invasive bioluminescence imaging (BLI) at 10 weeks after intrasplenic injection of luciferase expressing rLL-37-treated (10 µg/mL) PDAC cells in NOD scid interleukin (IL)-2 receptor γ chain knockout (NSG) mice (tops) and BLI assessment of PDAC cell dissemination to the liver (bottoms) (n=5 mice/group). (G) Dissemination of PDAC-luciferase cells to the liver was further verified by measuring luciferase activity in explanted and homogenised tissues, expressed as relative light units (RLUs)/mg of total protein.
Macrophages express hCAP-18/LL-37 in response to CSC-secreted TGF-β1 family members
Macrophages (ie, tumour-associated macrophage (TAMs)), as one of the primary sources of LL-37 in vivo, may respond to cues from the tumour (eg, CSC) to drive the expression of LL-37. To further dissect this putative CSC-TAM cross-talk, we modelled the tumour in vivo microenvironment by first polarising monocyte-derived human macrophages towards an ‘M1’ phenotype with GM-CSF24 prior to exposing them to CSC-conditioned media (CM). Intriguingly, ‘M1’ macrophages exposed to CSC CM upregulated LL-37 (figure 7A, B) and underwent a morphological change from a classic circular morphology to a more elongated shape (figure 7C). This change was accompanied by induction of alternatively-activated/‘M2’ genes, such as oncostatin M (OSM) and vascular endothelial growth factor (VEGF, figure 7D) as well as fluctuations in the expression of the cell surface M2 macrophage markers CD163, CD204 and CD20637–39 (see online supplementary figure S11).

Macrophages express human cationic antimicrobial peptide 18/leucine leucine 37 (hCAP-18/LL-37) in response to cancer stem cell (CSC)-secreted tumour growth factor-β1 (TGF-β1), Nodal and ActivinA. (A) RT-qPCR analysis of hCAP-18/LL-37 mRNA levels in human monocyte-derived macrophages cultured with control media or CSC conditioned-media (CM). (B) Western blot analysis of hCAP-18/LL-37 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in human monocyte-derived macrophages cultured with control media or CSC CM (left) and corresponding densitometric analysis (right). (C) Representative micrographs of primary monocyte-derived macrophages cultured with control media or CSC CM. (D) RT-qPCR analysis of hCAP-18/LL-37, oncostatin M (OSM) and vascular endothelial growth factor (VEGF) mRNA expression levels in human monocyte-derived macrophages cultured with control media or with CSC CM from three primary pancreatic ductal adenocarcinomas (PDAC) cultures. (E–G) Primary human monocyte-derived macrophages were cultured with control media or with CSC CM, TGF-β1 (1 ng/mL), Nodal (300 ng/mL) and ActivinA (100 ng/mL) or a combination of all three. (E) RT-qPCR analysis of hCAP-18/LL-37 mRNA levels, (F) western blot analysis of hCAP-18/LL-37, pSmad2, total Smad2 and GAPDH, (G) RT-qPCR analysis of OSM and VEGF mRNA levels. (H) RT-qPCR analysis of hCAP-18/LL-37, OSM and VEGF mRNA levels in primary human monocyte-derived macrophages pretreated for 1 h with a diluent control (Ctl), SB431542 (20 µM) or SB505124 (20 µM) and subsequently cultured for 48 h with control media, CSC CM, TGF-β1 (1 ng/mL) or Nodal (300 ng/mL) and Activin (100 ng/mL). n.s., not significant.
TGF-β1 can polarise ‘M1’ macrophages to an ‘M2’/alternatively activated phenotype40 and cancer cells, including PDAC cells do secrete large amounts of TGF-β1 (see online supplementary figure S12A).41 As many advanced PDAC tumours harbour inactivating mutations in the TGF-β signalling pathway rendering them unresponsive,42 we reasoned that TGF-β1 secreted by PDAC cells may instead primarily act on macrophages inducing LL-37 expression. In addition, we recently showed that PDAC CSC overexpress other members of the TGF-β superfamily, namely Nodal/ActivinA,6 which may also be implicated in the macrophage-CSC-cross-talk. Indeed, pretreatment of M1-polarised GM-CSF-treated monocyte-derived human macrophages with the TGF-β family members Nodal/ActivinA or TGF-β1 significantly increased LL-37 levels (figure 7E, F) as well as alternatively activated/‘M2’ genes, such as OSM and VEGF (figure 7G) and phospho-p38, nuclear factor (NF)-κB phospho-p50 and the transcription factor vitamin D receptor (VDR) (see online supplementary figure S12B,C), the latter three of which have been shown to regulate hCAP-18/LL-37 expression. Blocking Nodal/ActivinA/TGF-β1 signalling in CSC CM-treated macrophages with SB431542 and SB505124, both of which are inhibitors of the respective receptors Activin-like 4 (Alk4) and Alk5, abolished the enhanced expression of LL-37 and additional target genes (figure 7H).
Targeting LL-37 signalling impairs CSC tumourigenesis
hCAP-18/LL-37 is believed to exert its effects through the receptors FPR221 ,22 and/or P2X7R22 ,43 (figure 8A), both of which were detectable at the mRNA level and by flow cytometry in several primary PDAC cultures (figure 8B, C and online supplementary figure S13A). Receptor expression was restricted to a small subpopulation of adherent cells (<1%), which increased when cells were cultured as spheres (2–5%) and treated with rLL-37 (4–8%). More importantly, the expression of both receptors was primarily (>60%) restricted to CD133+ cells (figure 8C and online supplementary figure S13A). The latter was particularly evident in a freshly digested PDAC patient tumour (see online supplementary figure S13B). These data together suggest an enrichment of these receptors in CSCs. Using inhibitors specific for both FPR2 and P2X7R (WR-W4 and KN-62, respectively), LL-37-induced colony formation (figure 8D), invasion (figure 8E) and CD133+ cell expansion (figure 8F) were reduced with each inhibitor alone or in combination, confirming that the effects of LL-37 are indeed mediated through these receptors.
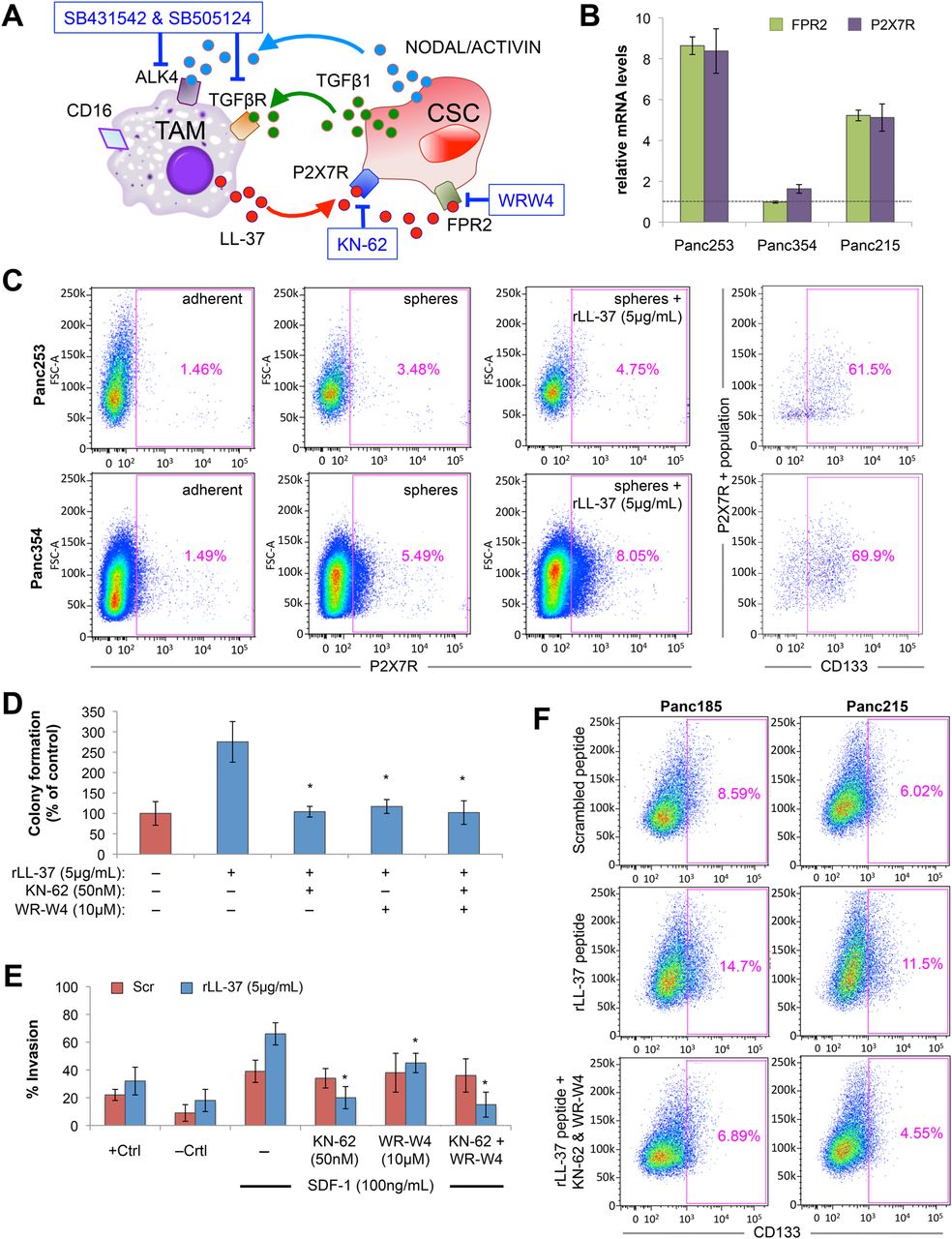
Targeting leucine leucine 37 (LL-37) receptors inhibit cancer stem cell (CSC) phenotypes. (A) Diagram of the cross-talk between macrophages and CSCs, receptors and secreted factors involved, and specific inhibitors of these pathways. (B) RT-qPCR analysis of formyl peptide receptor 2 (FPR2) and P2X purinoceptor 7 receptor (P2X7R) mRNA levels in three primary pancreatic ductal adenocarcinomas (PDAC) cultures. (C) Flow cytometry analysis of P2X7R cell surface expression in two primary PDAC lines cultured as adherent or sphere cultures and treated with recombinant leucine leucine 37 (rLL-37). Percentage of CD133-positive cells within the P2X7R-positive subpopulation is shown in the right panels. (D and E) LL-37-treated sphere-derived PDAC cells were pretreated with diluent control or WR-W4, KN-62 or a combination of both and (D) colony formation on Matrigel, (E) invasion through Matrigel following stimulation with SDF-1 (+Ctrl=20% fetal bovine serum; −Ctrl=media alone) or (F) the percentage of CD133-positive cells measured by flow cytometry was determined.
To rigorously test the therapeutic potential of anti-LL-37 therapy in vivo, we treated KPC or KPCR (KPC mice that additionally express a pancreas-specific RFP reporter) mice with WR-W4 and KN-62 for either 5 weeks (short term) or 16 weeks (long term) (figure 9A). With as little as 5 weeks of treatment, there was a marked reduction in the number of RFP+ circulating tumour cell (CTCs) of KPCR mice (figure 9B). In addition, we observed reduced numbers of low-grade PanINs and significantly fewer high-grade PanINs and PDAC lesions (figure 9C) in treated mice. Of note, no liver metastases were detected in either group at this stage. By extending our treatment to 16 weeks, the number of CTCs (epithelial cell adhesion molecule (EPCAM+)) in treated mice was still reduced (figure 9D). With respect to the pancreas, while no impact on the incidence of low-grade PanINs was observed, we still quantified significantly fewer high-grade PanINs and PDAC lesions (figure 9E) in treated mice. In addition, and in line with our CTC analyses, liver metastases were histologically detected in 75% of untreated mice, while only one treated mouse developed liver metastasis (data not shown). These results, in their entirety, highlight the promising therapeutic potential of targeting LL-37 signalling in PDAC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Targeting LL-37 receptors inhibit pancreatic ductal adenocarcinomas (PDAC) development in vivo. (A) Experimental set-up for in vivo short-term and long-term treatment. (B) Analysis of serum circulating tumour cells (CTCs, RFP+). Representative flow cytometry plots are shown (left) and quantification of the frequency of CTCs/lineage negative cells following 5 weeks of treatment are shown (right) (n=4 mice/group). (C) Representative images of mouse pancreata from control (n=4 mice) versus treated (n=4 mice) KPC mice after 5 weeks of treatment (left). Quantitative analysis of pancreatic intraepithelial neoplasia (PanIN)/PDAC frequency and grading (right). n.s., not significant. (D) Analysis of serum CTCs (epithelial cell adhesion molecule (EPCAM+)). Representative flow cytometry plots are shown (left) and quantification of the frequency of CTCs/lineage negative cells following 16 weeks of treatment are shown (right) (n=4 mice/group). (E) Representative images of mouse pancreata from control (n=4 mice) versus treated (n=4 mice) KPC mice after 16 weeks of treatment (left). Quantitative analysis of PanIN/PDAC frequency and grading (right).
Discussion and conclusion
While the PDAC tumour microenvironment is composed of many different cells, TAMs are likely one of the most dynamic resident cells of the tumour stroma.12–14 ,16 ,17 ,39 ,44 ,45 Not only can they enhance tumour growth when co-injected in vivo, but our microarray analyses additionally showed that monocyte-derived human macrophages are transcriptionally reprogrammed by PDAC cells. For example, macrophages upregulated a large number of genes belonging to the family of interferon-stimulated genes ((2’-5’) oligo A synthetase, interferon-induced GTP-binding protein, interferon stimulated gene 15), which we hypothesise is due to IFN-β produced by PDAC cells,46 similar to what has been shown for other Kras-transformed tumours.47 Macrophages also downregulated the expression of chemokines and proinflammatory genes, such as CXCL1, CXCL2, interleukin (IL)-1 and IL-6, as well as macrophage M1 markers, such as CD68, thus providing a first indication that PDAC cells alter the differentiation/polarisation state of macrophages. Most intriguingly, however, macrophages also upregulated the expression of hCAP-18/LL-37, a peptide previously reported to be either pro-tumourigenic or antitumourigenic in other cancer models,18 ,29 ,48 ,49 but had not yet been studied in the context of pancreatic CSC to date and revealed several interesting and novel findings.
The antimicrobial peptide hCAP-18/LL-37 was detectable across a large set of PDAC tumours at both the mRNA and protein level, and its expression was most prominent in PDAC and secondary metastatic lesions. hCAP-18/LL-37 was originally identified as a host immune antimicrobial defensin molecule and is constitutively secreted by a variety of immune cells (eg, neutrophils and macrophages), but can also be expressed by epithelial cells and fibroblasts.50 Interestingly, human and murine PDAC epithelial cells were univocally negative for hCAP-18/LL-37 or CRAMP, respectively, although previous studies examining breast, lung and ovarian cancers had shown that hCAP-18/LL-37 is expressed by the tumour epithelium.18 ,29 Instead, in PDAC, expression of hCAP-18/LL-37 was clearly restricted to the tumour stroma, revealing a previously unappreciated and important difference between tumour entities with respect to the source of hCAP-18/LL-37 expression. We predict that PDAC cells may be deficient in certain signalling factors that are necessary for hCAP-18/LL-37 expression, such as vitamin D-activated PPARγ signalling,51 NF-κB-mediated CCAAT/enhancer-binding protein α activation via MAPK-mediated phosphorylation,52 or other yet unidentified pathways. Within the tumour stroma, we identified macrophages as the primary source of LL-37; however, polymorphonuclear neutrophils can also secrete LL-3750 and have been shown to be one of the many types of tumour-infiltrating cells detected in pancreatic neoplasias.53 Thus, while we focused on TAMs, we note that other immune-infiltrating cells likely also produce LL-37 in vivo.
LL-37 has been shown to signal through P2X7R and FPR2,21–23 both of which were detectable on the surface of PDAC cells, and their expression was primarily restricted to CSCs (ie, CD133+ cells). As CSCs are the driving subpopulation of cells in PDAC with exclusive tumourigenic potential, and since tumour take and growth was significantly increased when PDAC cells were co-injected with macrophages, we thus reasoned that macrophages potentiate tumour growth via an LL-37-mediated pro-CSC mechanism. We confirmed this hypothesis in vivo by using CRAMP knockout mice as a source of bone marrow for transplantation experiments in irradiated KPC mice, as a syngeneic model to study CSC-mediated tumour take and growth or as a source of monocyte-derived macrophages for co-injection tumourigenicity studies with murine PDAC CSCs. All three approaches conclusively demonstrated that CRAMP is necessary for CSC-mediated PDAC development and progression in vivo. Importantly, these results are in agreement with those published by Li et al,32 ,33 where they show that cathelicidin (ie, CRAMP) expressed from murine myeloid cells promotes cigarette smoke-induced lung tumour growth in vivo.
To validate the biological relevance of our in vivo findings in the human setting, we showed that rLL-37 increased the CSC pool using two independent markers for CSCs (ie, CD133 and autofluorescence). This apparent activation/stimulation of the CSC compartment was validated at the functional level and by in vivo limiting dilution tumourigenicity assays, where we demonstrate that rLL-37 treatment increases sphere-derived CSC-mediated tumour formation and fitness. While sphere-derived cells are enriched in CSCs, tumour growth following injection of low numbers of sphere-derived cells is rarely achieved at single-digit numbers. Treatment of cells with rLL-37, however, further expanded the CSCs pool during sphere culture translating into a significantly higher CSC frequency and an apparent increase in viability, such that when injected at low numbers CSCs were able to survive in the recipient host and form tumours. In addition, while it is known that the population of CD133+ CSCs enrich during chemotherapy, we show that rLL-37 treatment further augments this enrichment in the presence of either gemcitabine or abraxane, arguing that LL-37 can enhance the inherent chemoresistance of CSCs. These results parallel those of Acharyya et al,54 wherein they show that the chemokine S100A8/9, secreted by myeloid cells in the breast cancer microenvironment, also enhances cancer cell chemoresistance.
LL-37 treatment also activated an EMT-like profile in CSCs. We observed increased phosphorylation of the kinases ERK1/2, AKT and PKC within 10 min of rLL-37 treatment in CSCs, and the EMT transcriptional mediators vimentin and Snail were additionally upregulated. At the functional level, rLL-37 treatment increased cell motility and invasion and more importantly increased metastasis in vivo. Of particular interest was the observation that rLL-37-treated CSCs were able to invade towards the chemoattractant SDF-1 at concentrations of only 1 ng/mL, while invasion of control cells required 100 ng/mL, a concentration that is hardly relevant in vivo. Wu et al55 reported that rLL-37 enhances the responsiveness of haematopoietic stem progenitor cells to an SDF-1 gradient by CXCR4 cell surface stabilisation. In line with these findings, we observed a threefold increase in CXCR4 cell membrane expression in rLL-37-treated cells, indicating that LL-37 likely primes CSCs for the SDF-1/CXCR4 axis via a similar CXCR4-dependent mechanism.
Members of the TGF-β superfamily, namely bone morphogenic proteins, TGF-β1, and Nodal/ActivinA, exert multiple and sometimes opposing effects on a variety of cell types. We have previously shown that TGF-β1 and Nodal/ActivinA are produced by PDAC CSCs and stromal cells,10 while only Nodal/ActivinA directly promote CSC-mediated tumourigenesis.6 In this study, we demonstrate that these CSC-secreted factors also stimulate macrophages in a paracrine fashion. For example, treatment of ‘M1’-polarised macrophages with CSC-conditioned media rapidly polarised macrophages towards an alternatively activated phenotype at the morphological, transcriptional and cells surface receptor level. More importantly, we also observed a marked increase in hCAP-18/LL-37 mRNA and protein expression following treatment. While Li et al33 also showed that ovarian cancer cells can activate in vitro cultured macrophages to produce hCAP-18/LL-37 via cancer cell secreted versican V1, we did not detect expression of versican V1 in our primary PDAC cultures (data not shown). Instead, we identified TGF-β1, Nodal and ActivinA as the factors responsible for CSC-mediated activation of macrophages, and inhibitors of both the TGF-β1 and Nodal/Activin receptors (Alk5 and Alk4, respectively) could reverse the effects in vitro and in vivo (data not shown). While the transcriptional regulation of hCAP-18/LL-37 expression has not been fully elucidated, the CAMP gene is a direct target of the VDR transcription factor33 ,56 and hCAP/LL-37 expression has also been shown to be regulated by NF-κB phospho-p50 and phospho-p38.52 In macrophages treated with CSC-conditioned media or TGF-β1, Nodal and ActivinA, NF-κB phospho-p50 and phospho-p38 activation was observed as well as transcriptional upregulation of VDR, implicating these factors as common mediators by which TGF-β superfamily members regulate LL-37 expression and thus promote formation of a pro-CSC niche.
Targeting the tumour microenvironment has gained enormous attention over the past decade, particularly the development of agents that can disrupt the cross-talk between cancer cells and the stroma. Having determined that the majority of cells expressing LL-37 receptors are also CD133+, we tested the efficacy of targeting FPR2 and P2X7R in the clinically relevant KPC PDAC mouse model. We observed that in mice treated with WR-W4 and KN-62 high-grade PanIN and PDAC lesions were significantly reduced, and CTCs were essentially eliminated from the blood of treated mice, confirming that LL-37 signalling is indeed important for PDAC tumour progression and dissemination. It is important to note that in the intervention studies performed in KPC mice we cannot rule out the fact that the treatments used may have also targeted TAMs, which also express FPR2 and P2X7R. Nevertheless, our data provide clear proof of principle for targeting these LL-37 receptors, although more studies will be needed to understand how WR-W4 and KN-62 reduced PDAC progression in vivo (ie, at the level of the cancer cell, TAM or both).
Acknowledgments
We are indebted to Sara M Trabulo and Alexandra Aicher for their technical in vivo assistance and to the CNIO Histopathology Core Unit, particularly Raquel Pajares and Maria Lozano.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online tables
Footnotes
Contributors BSJr developed the study concept, obtained funding, acquired, analysed, and interpreted data and wrote the manuscript. SA assisted in the development of the study concept, acquired, analysed and interpreted data. YS-R, MMA, MCi, MT, IM-L, GGL and SG-S acquired and analysed data. MCa and EG analysed and scored histological samples. MH, ME and JK provided extensively characterised PDAC and PSC samples. EG, PS and PCH assisted in the development of the study concept, interpreted the data and edited the manuscript. CH developed the study concept, obtained funding, interpreted the data and wrote the manuscript.
Funding CH: ERC Advanced Investigator Grant (Pa-CSC 233460), European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no 256974 (EPC-TM-NET) and no 602783 (CAM-PaC), the Subdirección General de Evaluación y Fomento de la Investigación, Fondo de Investigación Sanitaria (PS09/02129 and PI12/02643) and the Programa Nacional de Internacionalización de la I+D, Subprogramma: FCCI 2009 (PLE2009-0105; both Ministerio de Economía y Competitividad (es), Spain). BSJr: Rámon y Cajal Merit Award from the Ministerio de Economía y Competitividad, Spain and Clinic and Laboratory Integration Program (CLIP) grant from the Cancer Research Institute, NY, NY. MCi: La Caixa Predoctoral Fellowship.
Competing interests None.
Patient consent Obtained.
Ethics approval The use of human material was approved by the local ethics committee of each respective hospital or university, and the use of human tissue samples for the construction of the TMAs was approved by the Ethics Committee of the Hospital de Madrid Norte Sanchinarro. All experiments were approved by the Animal Experimental Ethics Committee of the Instituto de Salud Carlos III (Madrid, Spain).
Provenance and peer review Not commissioned; externally peer reviewed.