Article Text

Abstract
Objective As the current therapeutic strategies for human hepatocellular carcinoma (HCC) have been proven to have limited effectiveness, immunotherapy becomes a compelling way to tackle the disease. We aim to provide humanised mouse (humice) models for the understanding of the interaction between human cancer and immune system, particularly for human-specific drug testing.
Design Patient-derived xenograft tumours are established with type I human leucocyte antigen matched human immune system in NOD-scid Il2rg−/− (NSG) mice. The longitudinal changes of the tumour and immune responses as well as the efficacy of immune checkpoint inhibitors are investigated.
Results Similar to the clinical outcomes, the human immune system in our model is educated by the tumour and exhibits exhaustion phenotypes such as a significant declination of leucocyte numbers, upregulation of exhaustion markers and decreased the production of human proinflammatory cytokines. Notably, cytotoxic immune cells decreased more rapidly compared with other cell types. Tumour infiltrated T cells have much higher expression of exhaustion markers and lower cytokine production compared with peripheral T cells. In addition, tumour-associated macrophages and myeloid-derived suppressor cells are found to be highly enriched in the tumour microenvironment. Interestingly, the tumour also changes gene expression profiles in response to immune responses by upregulating immune checkpoint ligands. Most importantly, in contrast to the NSG model, our model demonstrates both therapeutic and side effects of immune checkpoint inhibitors pembrolizumab and ipilimumab.
Conclusions Our work provides a model for immune-oncology study and a useful parallel-to-human platform for anti-HCC drug testing, especially immunotherapy.
- hepatocellular carcinoma
- immunotherapy
- immunology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Development of novel antihuman hepatocellular carcinoma (anti-HCC) immunotherapy is often plagued by discrepancies between drug efficacy obtained in normal mice studies and outcomes of clinical trials.
Conventional human patient-derived xenograft (PDX) model lacks human immune environment.
In vivo models for the study of human cancer and immune system are important for both basic and translation research.
What are the new findings?
We have developed a new model where human HCC-PDX grows in the presence of a human immune system.
Our results demonstrate that the HCCs educate the human immune system and condition the tumour microenvironment to support immune suppression and their own development.
Our model can reproduce both therapeutic and side effects of some immune checkpoint inhibitors used in the clinic.
How might it impact on clinical practice in the foreseeable future?
This new HCC-PDX humice model provides a human-specific platform for the study of the interactions between HCC and human immune system, as well as the evaluation and prediction of immunotherapy and combination therapies.
Introduction
Human hepatocellular carcinoma (HCC) is a leading cause of cancer-related death globally, and the burden of HCC is expected to increase further in coming years.1–3 Few strategies have been proven effective in human HCC treatment, particularly for those patients not indicated for curative resection or transplantation.4–6 Thus, the improvement of existing treatments and development of new therapy are urgently needed.
In patients with HCC, various dysfunctions of the human immune components are involved in HCC development and progression including immune cells and cytokines, which were related to HCC proliferation, invasion and drug resistance.7–9 Immunotherapy has been developed for human cancer treatment and garnering more attention as a result of encouraging outcomes of new strategies such as immune checkpoint blockade.10–12 However, the development of novel HCC immunotherapy is often plagued by discrepancies among drug efficacies obtained from in vitro culture system, mice studies and outcomes of clinical trials.13–16 Although in vitro culture systems are advantageous for addressing specific experimental questions, they are often vague, fidelity-lacking deductions that largely ignore the heterogeneity of HCC as well as the complexity of HCC microenvironment.13 For mouse models, significant differences have been found between mice and humans in immune system development, activation and response to challenge, in both the innate and adaptive arms.14 15 For clinical trials, the inconsistencies may be attributed to the lack of clinically relevant cancer models used for human immunotherapy drug testing.16 Undoubtedly, establishing and developing new HCC-patient-derived xenograft (PDX) model with human tumour immune microenvironment is critical to basic and translational research.
NOD-scid Il2rg−/− (NSG) mice have been shown to be able to support the engraftment of PDX tumours.17 18 These PDX models present many features of the patient tumour and have been widely used for anticancer drug testing.18 Also, the human immune system can be developed in NSG mice including functional human T cells, nature killer (NK) cells and monocytes, etc by human haematopoietic stem cells (HSC)transplantation (humanised mouse).19 20 In our study, we showed that patient-derived HCC tumours could be engrafted in humanised mice with human immune system. In this model, human immune system showed strong responses to patients with HCC tumour. In addition, immune checkpoint blockade drugs (pembrolizumab and ipilimumab) in this model could suppress the growth and progression of HCC with human immune system.
Materials and methods
Human fetal liver progenitor stem cells
Fetal liver tissues were isolated from aborted fetuses at 15–23 weeks of gestation, with written consent obtained from guardians of donors, and in accordance with the ethical guidelines of KK Women’s and Children’s Hospital, Singapore. The sample was processed as described previously.21 Human CD34+ cells were isolated and purified using EasySep Human CD34-Positive Selection Kit (Stemcell Technologies) under sterile conditions, according to manufacturer’s instructions. The purity of the CD34+ cells was 90%–99% as determined by flow cytometry.
More detailed materials and methods can be found in online supplementary material.
Supplemental material
Results
HCC-PDX tumour can grow in human leucocyte antigen type I matched humice
Humice used in this model was constructed by injection of human HSCs. A considerable number of HSC samples had been banked in our stock and human leucocyte antigen (HLA)-typing on HLA-A*, HLA-B* and HLA-DRB1* was performed to define matched pairs between HCC and HSCs. In this study, four HCC-PDX tumours have been established from different donors (HCC#1, HCC#2, HCC#3 and HCC#4). HLA-typing results are shown in online supplementary table S1. The criteria that we applied to pick the matched pairs were minimum two out of four alleles matching on HLA-A* and HLA-B*. Paired HSCs were used to inject NSG pups, and 8–10 weeks later, HCC-PDX was transplanted subcutaneously into humice. NSG mice with PDX transplants were used as a control. HCC-PDX tumours showed similar trend in tumour development and immune profiling but due to the limitation of space, we only describe the characterisation of HCC#1 in the main figures while others in the online supplementary material provided. Interestingly, when comparing the tumour size, HCC-PDX grown in NSG mice without human immune system were significantly smaller than those in humice (figure 1A, B). This suggests that the in vivo immune environment might have been transformed by engrafted HCC tumour to promote tumour growth.
Supplemental material

Establishment of patient-derived xenograft (PDX)-hepatocellular carcinoma (HCC) humice model and the blood immune cell number changes. (A–B) PDX tumours were transplanted subcutaneously to NOD-scid Il2rg−/− (NSG) mice and humice (n=5) aged 8–10 weeks. (A) Representative image of tumours and spleens 8 weeks after transplantation in NSG and humice. (B) The weekly changes in PDX tumour size in NSG and humice after transplantation. Data are presented as fold changes normalised to the size of tumour before PDX transplantation (week 0). *P<0.05, **P<0.01. (C–J) PDX tumours were transplanted subcutaneously to humice aged 8–10 weeks. Blood immune cell frequencies and absolute numbers from humice without tumour (n=5) and humice with tumour (n=5) were analysed biweekly by flow cytometry. Data are presented as fold changes normalised to the cell numbers of specific cell types before PDX transplantation (week 0): human CD45+ (hCD45+) (C), hCD3+ (D), hCD19+ (E), hCD4+ (F), hCD8+ (G), hCD14-HLA-DR-CD56+ (H), hCD14+ (I) and DC (J).
HCC leads to blood leucopenia and reduced production of cytokines in humice
To characterise the responses of human immune system to HCC, we followed the human immune cell profiles in peripheral blood of humice. Human T cells and non-T cells gating panels are shown in online supplementary figure S1A,B respectively. In T cells panel, T cell subtypes and phenotypes include: T helper cells (Th, hCD45+hCD3+hCD4+), type 1 T helper cells (Th1, hCD45+hCD3+hCD4+hCXCR3+hCCR6−), type 2 T helper cells (Th2, hCD45+hCD3+hCD4+hCXCR3-hCCR6−), type 17 T helper cells (Th17, hCD45+hCD3+hCD4+hCXCR3-hCCR6+), regulatory T cells (Treg, hCD45+hCD3+hCD4+hCD25+hCD127low), cytotoxic T cells (Tc, hCD45+hCD3+hCD8+), type 1 cytotoxic T cells (Tc1, hCD45+hCD3+hCD8+hCXCR3+) and type 17 cytotoxic T cells (Tc17, hCD45+hCD3+hCD8+hCXCR3-hCCR4+hCCR6+). We also gated naïve (TN, hCD45RA+hCD197+), effector (TE, hCD45RA+hCD197-), effector memory (TEM, hCD45RA-hCD197-) and central memory (TCM, hCD45RA-hCD197+) in both Th and Tc cells. Non-T cell panel includes: human B cells (hCD45+hCD19+), IgD+ memory B cells (hCD45+hCD19+hCD27+hIgD+), IgD- memory B cells (hCD45+hCD19+hCD27+hIgD-), naïve B cells (hCD45+hCD19+hCD27-), B plasmablast cells (hCD45+hCD19+hCD27-hCD20-hCD38+), NK cells (hCD14-hHLA-DR-hCD56+), cytokine-producing NK cells (hCD14-hHLA-DR-hCD56brighthCD16-/+), cytotoxic NK cells (NKc, hCD14-HLA-DR-hCD56dimhCD16+), macrophage (hCD45+hCD14+), non-classical macrophage (hCD45+hCD14+hCD16+), intermediate macrophage (hCD45+hCD14++hCD16+), classic macrophage (hCD45+hCD14++hCD16-), plasmacytoid dendritic cells (pDCs, hCD45+hCD3-hCD19-hHLA-DR+hCD123+hCD11c-) and myeloid dendritic cells (mDCs, hCD45+hCD3-hCD19-hHLA-DR+hCD123-hCD11c+). After counting the human cell numbers in peripheral blood, we observed that all the major immune cell types including human CD45+ (hCD45+) leucocytes, hCD3+ T cells, hCD19+ B cells, hCD14-HLA-DR-CD56+ NK cells, hCD14+ monocytes and DCs showed significant decrease over the course of tumour growth (figure 1C-1J). In contrast, the number of human blood immune cells in humice without HCC generally showed sign of increase during the tumour progression.
In addition, we also analysed the levels of human proinflammatory cytokines and cytolytic proteins in plasma of PDX humice. The major proinflammatory cytokines and cytolytic proteins such as human interferon (IFN)-γ, interleukin (IL)-2, tumour necrosis factor-α, IL-18, granzyme A and granulysin showed inverted-U curve where they showed an increase in the early course of tumour onset (from week 0 to week 4) and an subsequent decreased from week 4 to week 8 (figure 2). These results indicated that human immune system responded to HCC at early stage but was then suppressed by HCC, an observation which is consistent with the features of patients with HCC.22
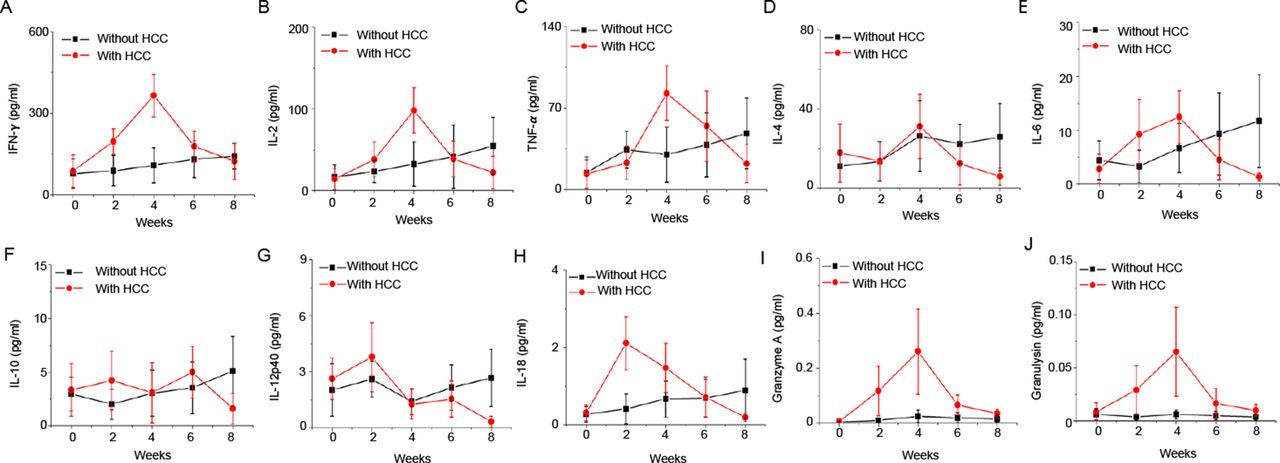
The kinetics of plasma human cytokines and cytolytic proteins after patient-derived xenograft (PDX) tumour engraftment in humice. PDX tumours were transplanted subcutaneously to humice (n=5) aged 8–10 weeks. Humice without tumour (n=5) were used as control. Plasma levels of human cytokines and cytolytic proteins at different time points are shown. (A) Interferon (IFN)-γ; (B) IL-2; (C) tumour necrosis factor (TNF)-α; (D) IL-4; (E) IL-6; (F) IL-10; (G) IL-12p40; (H) IL-18; (I) granzyme A; (J) granulysin. HCC, hepatocellular carcinoma.
We further dissect the subsets of the major immune cell types. The analysis of weekly changes of various immune cell subsets is shown in figure 3A–3G and online supplementary figure S2. All the blood immune cell subsets showed the trend of decreasing cell numbers, at week 4 post-tumour transplantation, including Tc subsets (TcN, TcE, TcEM and TcCM), Th subsets (ThN, ThE, ThEM and ThCM), hCD19+ cells (native, IgD+, IgD- and plasmablast), hCD14+ cells (hCD14+hCD16+, hCD14++hCD16+ and hCD14++hCD16-) and DC (pDC and mDC). Notably, some immune cell subsets such as hCD45RA+hCD197+ ThN, hCD45RA+hCD197+ TcN and hCD56dimhCD16+ NKc seemed to have a more significant drop in cell number compared with others (see online supplementary figure S2A, figure 3A and G). Comparing the proportion of major immune cell types, the percentage of hCD8+ T cells and NK cells consistently had the most dramatic decline in all the HCC humice, while the percentage of hCD4+ T cells increased (figure 3H). Among hCD8+ T cells and NK cells, the most significantly reduced populations were hCD45RA+hCD197+ TcN (figure 3I) and hCD56dimhCD16+ NKc (figure 3J). These results suggested that cytotoxic cells mainly Tc and NKc might be more specifically suppressed than other cell types by cancer in the early stage.
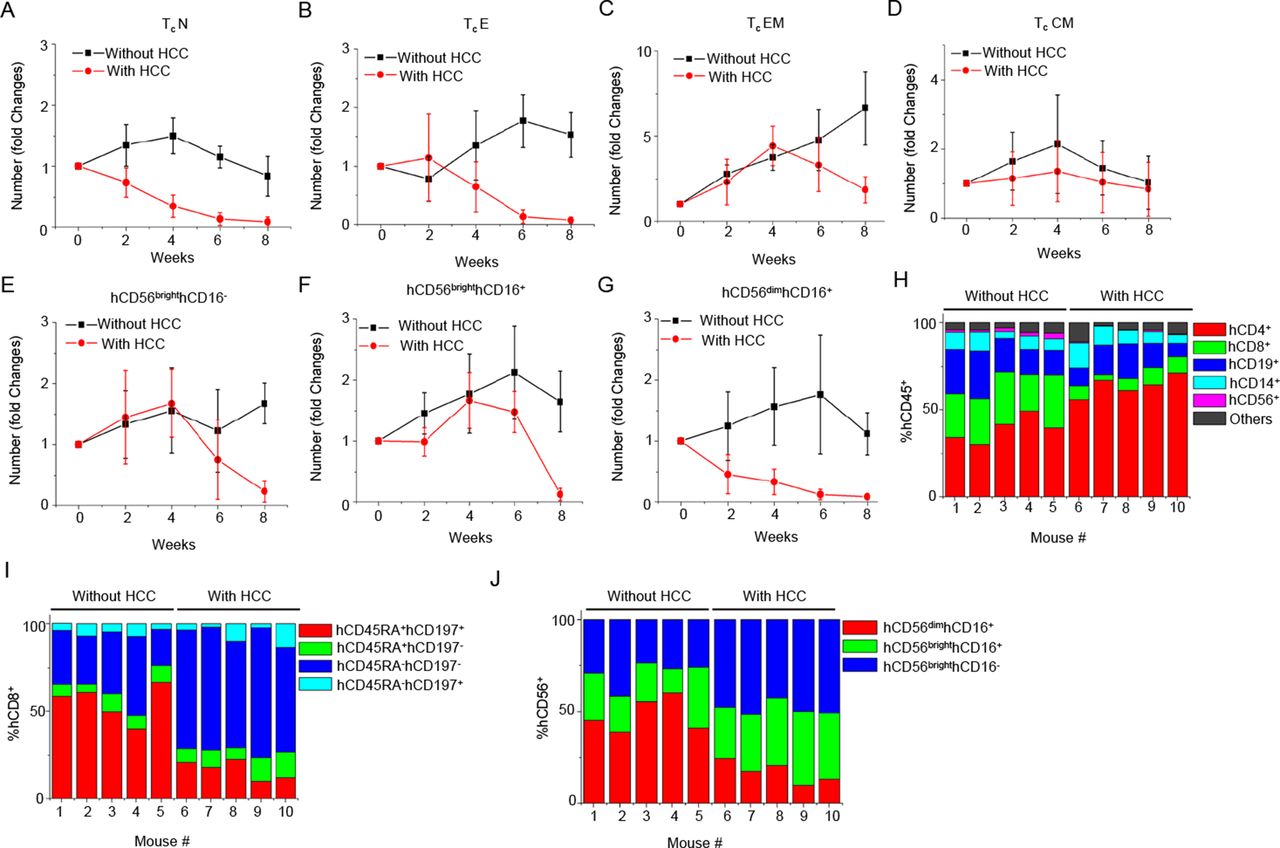
The kinetics of changes in blood cytotoxic cell subtypes responding to hepatocellular carcinoma (HCC)-patient-derived xenograft (PDX) tumour engraftment. PDX tumours were transplanted subcutaneously to humice (n=5) aged 8–10 weeks. Humice without tumour (n=5) were used as control. The numbers of blood immune cell subtypes at various time points were counted and plotted as fold changes normalised to time point week 0. (A) Native cytotoxic T cells (TcN). (B) Effector cytotoxic T cells (TcE). (C) Effector memory cytotoxic T cells (TcEM). (D) Central memory cytotoxic T cells (TcCM). (E) hCD56brighthCD16- NK. (F) hCD56brighthCD16+ NK. (G) hCD56dimhCD16+ NK. (H) The proportion of major human immune cell types in blood hCD45+ cells at 4 weeks after engraftment. (I) The proportion of Tc subtypes in blood hCD8+ Tc cells at 4 weeks after engraftment. (J) The proportion of NK subtypes in blood NK cells at 4 weeks after engraftment.
Human leucocytes infiltrated into HCC-PDX tumour and developed exhaustion phenotypes
Previous studies have shown that tumour-infiltrating leucocytes (TILs) constitute an essential part of HCC microenvironment.23–25 In our model, the infiltration of various human immune cell types was found in HCC tumour environment (figure 4A), which was not present in tumour from NSG mice. TILs were isolated from HCC-PDX tumour and analysed for phenotypes and compositions (see online supplementary figure S3A,B) in comparison with blood. Consistent with the clinical observation, the major component of TILs in this model was hCD3+ T cells (80%~95%) while hCD19+ B cells occupied <5% and the others occupied 5~15% (figure 4B). The frequency of hCD8+ Tc cells (5%~10%) in TILs was significantly lower than blood (12%~18%). In both Th and Tc cells, the proportion of different subtypes was %TEM>%TCM>%TE>%TN (figure 4C). Higher frequency of Treg was also observed in TILs (9%~15%) compared with blood (2%~6%) (figure 4B). Although we could detect CD56+ NK cells in TILs, the majority of these cells were CD56bright cytokine-producing NKs (75%~90% of total NK) (see online supplementary figure S3B). The frequency of CD56dimCD16+ NKc (10%~25% of total NK) was lower compared with blood NKc (30%~55% of total NK). As for macrophages, unlike blood we did not find non-classical macrophages and intermediate macrophages in TILs (figure 4D and online supplementary figure S3B). Instead, the majority of human macrophages in TILs were M∅1 (hCD68+hHLA-DR+hCD86+hCD163-) and M∅2 (hCD68+hHLA-DR-hCD86-hCD163+) macrophages (see online supplementary figure S4A-E). Notably, M∅2 macrophages could only be seen in TILs but not blood, spleen and bone marrow (see online supplementary figure S4A-E). In patients with HCC, myeloid-derived suppressor cell (MDSC) is a unique cell type induced by tumour to suppress immune cell function.24 26–28 MDSC has been grouped into different subtypes.27 29 30 In this model, we found M-MDSC (hCD11b+ hCD14+hHLA-DRlow/- hCD15-), PMN-MDSC (hCD14-hCD11b+hCD15+) and e-MDSC (Lin-(hCD3/19/56) hCD14-hCD15-hHLA-DR-hCD33+) in TILs, and particularly M-MDSC and e-MDSC were highly enriched in tumour than other organs (see online supplementary figure S4F-J). These results demonstrate that the immune cell composition inside tumour microenvironment is very different from peripheral organs.

Infiltration of human immune cells in tumour. Tumours were harvested at 8 weeks postengraftment and analysed for human immune cell infiltration (n=5). (A) In situ stain of various human immune cell types infiltrated in hepatocellular carcinoma (HCC) tumours from NOD-scid Il2rg−/− (NSG) and humice. (B) The frequencies of major human immune cells from blood and HCC-patient-derived xenograft (PDX) tumour analysed by flow cytometry. (C) The proportions of T cell subtypes within T helper and cytotoxic T cells from blood and HCC-PDX tumour. (D) The proportions of myeloid subsets from blood and HCC-PDX tumour. *P<0.05, **P<0.01. TIL, tumour-infiltrating leucocytes.
One of the most important features in immuno-oncology is that HCC can induce the exhaustion of Tc, which results in suppressed cytotoxic function and cytokine production ability.31–33 In our model, the expression of immune checkpoint receptors on Tc cells from blood, spleen and TILs were analysed. Notably, the expression of all the immune checkpoint receptors including PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, KLRG-1, CD244 and BTLA on different subsets of Tc cells particularly TcE and TcEM from TILs was much higher than Tc cells from blood and spleen (figure 5A). Interestingly, the expression of immune checkpoint receptors was mostly found on TIL CD8+ Tc cells instead of TIL CD4+ Th cells while their blood counterparts both only carried low expression levels (see online supplementary figure S5A,B).

Phenotyping and functional assays for cytotoxic T cells (Tc) from blood, spleen and tumour-infiltrating leucocytes (TILs). Leucocytes were isolated from blood, spleen and tumour 8 weeks after tumour inoculation (n=5). (A) Expression of immune checkpoint receptors on leucocytes. (B–D) Intracellular staining of human cytokines and cytolytic proteins. (B) Human interleukin (IL)-2, (C) human interferon (IFN)-γ, (D) cytolytic proteins including perforin, granulysin, granzyme A and B. (E) CD3+ T cells were isolated from spleens from humice inoculated with or without hepatocellular carcinoma (HCC) and TILs. Human IFN-γ levels in culture supernatants were analysed by ELISA secretion after stimulation ex vivo (n=4). *P<0.05, **P<0.01.
To characterise the Tc functions in our model, we compared the cytokine-producing activities among Tc cells from blood, spleen and TILs. The production of IFN-γ and IL-2 was significantly lower in Tc cells from TILs than blood and spleen (figure 5B and C). Similarly, the secretion of cytolytic proteins including Perforin, granulysin and granzyme B by Tc from TILs was also the lowest (figure 5D). When stimulated ex vivo, T cells purified from TILs showed less human IFN-γ production than T cells from spleen (figure 5E). Together, all the above data prove that our model can reproduce the composition, phenotype and function of immune microenvironment in patients with HCC. Moreover, the tumour environment contains exhausted Tc cells and many unique tumour-associated cell types and features compared with other organs.
Tumour alters gene expression profile responding to immune responses
HCC tumour rapidly overpowered and conditioned the immune system in our model. We wonder if the tumour also changes when interacting with immune cells and their responses. Since a considerable level of immune checkpoint receptors was found in Tc cells inside the tumour, we looked into the expression of the immune checkpoint ligands in the HCC-PDX tumour. The mRNA levels of PDL1 (ligand of PD-1), PDL2 (ligand of PD-1), CD80 (ligand of CTLA-4), CD86 (ligand of CTLA-4), CD48 (ligand of 2B4), CD155 (ligand of TIGIT), CEACAM-1 (ligand of TIM-3), HVEM (ligand of BTLA and CD160), GAL9 (ligand of TIM-3) and E-CADHERIN (ligand of CD103) were compared between HCC-PDX tumours from humice and NSG mice. Distinctively, the results showed that tumours grown in the presence of human immune system expressed dramatically higher levels of immune checkpoint ligands than those in NSG (figure 6A). The in situ stain confirmed the protein expression of PDL1 was only found in the HCC-PDX tumour from humice but not NSG (figure 6B). IFN-γ is the most abundant human cytokine found in this model and known to induce immune checkpoint ligand expression in various cell types.34–36 Hep3B cell line was then applied to verify the effects of IFN-γ. As shown in figure 6C, the levels of immune checkpoint ligands were significantly upregulated in Hep3B after adding human recombinant IFN-γ into the culture system. To identify the source of IFN-γ, blood mononuclear cells from humice 4 weeks post-HCC-PDX inoculation were further analysed by intracellular staining. Our results showed that hCD8+ Tc cells expressed the highest level of IFN-γ and among Tc cells, effector Tc cell types including effector and effector memory were the main producers (figure 6D and E). These results suggest that IFN-γ produced mainly by Tc cells is one of the key factors that drive the tumour to evolve further to escape immune surveillance by upregulating immune checkpoint ligands.

Expression of immune checkpoint ligands in hepatocellular carcinoma (HCC)-patient-derived xenograft (PDX) humice and NOD-scid Il2rg−/− (NSG). (A) Four weeks after tumour inoculation, tumours were harvested from NSG and humice and analysed for immune checkpoint ligand expressions (n=7). Shown are relative levels of mRNA expression of immune checkpoint ligands detected by RT-PCR. The mRNA levels of individual genes in tumours from NSG were normalised as 1. (B) Immunohistochemistry staining of human PDL1 in tumour tissues from NSG and humice. (C) Hep3B cell lines were stimulated with human interferon (IFN)-γ and analysed for immune checkpoint ligands. Shown are relative levels of mRNA expression of immune checkpoint ligands in Hep3B cells treated with phosphate-buffered saline (PBS) or IFN-γ. The mRNA levels of individual genes in Hep3B cells with PBS treatment were normalised as 1. The experiment was repeated twice. (D and E) Four weeks after tumour inoculation in humice, blood leucocytes were harvested for intercellular staining to analyse the expression of IFN-γ in T cells (n=6). Shown are the histogram plots (D) and statistical analysis results (E). *P<0.05, **P<0.01.
Immune checkpoint inhibitors suppress HCC growth by re-activating Tc cells and altering the tumour immune microenvironment
To validate this new model, clinically approved human immune checkpoint inhibitors anti-PD1 antibody (pembrolizumab) and anti-CTLA4 antibody (ipilimumab) were applied separately to test the effect of immunotherapy. However, among all the four HCC-PDX tumours, HCC#1 and HCC#4 could respond to immune checkpoint inhibitor treatment, whereas HCC#2 and HCC#3 did not show significant effects. The HCC#1 treatment data are shown in figure 7, online supplementary figure S6 and S7, while HCC#4 treatment data are shown in online supplementary figure S8. After 4 weeks treatment, both groups of pembrolizumab (intravenous 5 mg/kg/week) and ipilimumab (intravenous 5 mg/kg/week) treatment showed obvious suppression in tumour size (figure 7A and B, online supplementary figure S8A,B) in humice but not in NSG, which confirmed that human immune system was essential for immunotherapy. However, the tumour-suppressing effect of pembrolizumab was more significant than ipilimumab. In addition, we observed that the mice treated with ipilimumab lost weight significantly and showed cachexia while the group with pembrolizumab was healthy. It has been reported that ipilimumab has toxicity and causes side effects in multiple organs in clinical setting.37 38 To assess for possible side effects of the immune checkpoint drugs, the major organs liver, lung and kidney were sampled for histological examination. Pathological analysis confirmed that the group treated with ipilimumab developed massive cell infiltration and damage in liver, lung and kidney while the saline-treated and pembrolizumab-treated groups remained normal (see online supplementary figure S7). These results suggest that humice can predict both therapeutic and side effects of the drugs.

The effects of immune checkpoint inhibitors (pembrolizumab and ipilimumab) treatment in hepatocellular carcinoma (HCC)-patient-derived xenograft (PDX) humice. Four weeks after tumour inoculations, humice and NOD-scid Il2rg−/− (NSG) were treated with saline, pembrolizumab or ipilimumab for another 4 weeks before tumour samples and plasma were collected for analysis (n=4). (A) Shown are the representative images of HCC-PDX tumours and spleens from different drug treatment groups in humice. (B) The statistical analysis of HCC-PDX tumour size in NSG and humice with different treatments. (C) The absolute number counts of blood cytotoxic cells after treatments. The data are presented as fold changes normalised to counts from humice without HCC. (D) Blood leucocytes were harvested for intercellular staining to analyse the expression of cytolytic proteins in cytotoxic T cells (Tc) subsets. (E) The proportion changes of myeloid-derived suppressor cell (MDSC) in tumour-infiltrating leucocytes after drug treatments. Each bar presents one mouse. (F) The changes of the ratio of M∅1/M∅2 after drug treatments. *P<0.05, **P<0.01.
To explore the mechanisms and phenotype changes on drug treatment, blood mononuclear cells, TILs and plasma were further analysed. The number drop of Tc cells particularly TcEM and NKc cells in the blood caused by HCC-PDX tumour could be recovered by pembrolizumab, whereas the effect of ipilimumab was not significant (figure 7C and online supplementary figure S8C). The proportion of hCD8+ Tc cells and hCD56+ NKc cells in both blood and TILs increased more significantly after pembrolizumab treatment than ipilimumab (see online supplementary figure S6A,B and online supplementary figure S8D,E). Consistent with the cell number increase, the proportion analysis on blood and TIL Tc subtypes also revealed that TcEM subset contributed most robustly to the recovery postpembrolizumab treatment (see online supplementary figure S6C,D). NKc cells also occupied higher proportion among NK cells post the inhibitor treatments (see online supplementary figure S6E). Intracellular staining of human IL-2 and IFN-γ showed that pembrolizumab was more potent at inducing cytokine production by TcE and TcEM cells, whereas ipilimumab exerted more effect on TcCM (see online supplementary figure S6F,G and supplementary figure S8F). Correspondingly, the levels of human IFN-γ and IL-2 in plasma were also upregulated after the inhibitor treatments (see online supplementary figure S6H,I). In addition to cytokines, the productions of cytolytic proteins such as granulysin and granzyme B were also recovered in TcE and TcEM particularly after pembrolizumab treatment (figure 7D and online supplementary figure S8G). We also characterised myeloid compartment postdrug treatment in HCC#1 and HCC#4. Pembrolizumab downregulated M-MDSC cells in both HCC#1 and HCC#4 and upregulated the proportion of e-MDSC in HCC#1, which might impair the immunosuppressive function of MDSC (figure 7E and online supplementary figure S8H). The ratio of M∅1/M∅2 was also successfully reversed by upregulating M∅1 and downregulating M∅2 after treatment with immune checkpoint drugs (figure 7F and online supplementary figure S8I). These results prove that tumour immune microenvironment in our model can respond to immune checkpoint inhibitors and it can be used to unravel the underlying mechanisms of therapeutic and side effects of immunotherapeutic drugs.
Although HCC#2 and HCC#3 exhibited the similar trend in tumour growth rate and immune profiles (online supplementary figure S9-S12) to HCC#1 and HCC#4, they did not respond to pembrolizumab and ipilimumab treatment. It indicated that not all the HCC-PDX humice responded to immune checkpoint inhibitors, which were consistent to clinical trial data.39
Altogether, our study provides a new model for the understanding of the interactions between human cancer and immune system (figure 8). At the early stage, the engagement of immune system with cancer triggers anticancer responses from cytotoxic cells such as Tc and NKc cells with the production of proinflammatory cytokines, for example, IFN-γ. In a defensive response, the tumour quickly evolves by upregulating suppressing molecules, for example, immune checkpoint ligands to educate immune cells by inducing immune cell apoptosis, migration and exhaustion. This response results in the reduction of immune cell number, cytokine production and killing functions and the generation of tumour-promoting cell types such as Treg, MDSC and tumour-associated macrophages. This process can be reversed by the immune checkpoint blockade to reactivate anticancer responses.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The model of hepatocellular carcinoma (HCC)-patient-derived xenograft tumour and human immune cell interactions and immune checkpoint drug testing. IFN, interferon; MDSC, myeloid-derived suppressor cell; NK, natural killer.
Discussion
On average, only about 5% of new cancer drug candidates are approved by the Food and Drug Administration, and the majority failed due to the lack of a preclinical model which can accurately recapitulate patient tumour heterogeneity and microenvironment, particularly the human immune system.40 In the recent years, immunotherapy which targets the body’s natural immune defences to fight cancer has grown dramatically, showing great promise in treating cancers.41 42 Hence, a small animal model where the human tumour grows in the presence of human immune system is acutely required.
In this study, we successfully established a new preclinical model to grow human HCC-PDX with a partially HLA-matched human immune system. All the major human immune cell types are found in this model and are able to generate responses to HCC-PDX. A number of interesting observations in this model have helped us to gain new insights into immune-oncology. First, we found that cancer could educate the immune system and suppress immune responses. Human cancer study is now largely limited by the availability of clinical biopsies from different stages, thus the mechanisms of how human cancer educates human immune system over the entire tumour progression are still not well understood. It is believed that the process involves a complex network such as soluble factors secreted by cancer, for example, exosomes, cell-cell interactions through inhibition receptors and the induction of immune cell death/exhaustions at different stages of cancer development.43 44 This new model mimics both early and late periods of cancer and immune cell interactions and will help to address these questions. Second, cancer is found here to respond to immune responses and continue to evolve by changing gene expression profiles to escape immunosurveillance. New mutations may have also been generated in cancer cells over time in the context of inflammation, which contributes to the HCC heterogeneity. This is a major difference to the conventional PDX models, which only grow tumours without an immune system. In this sense, the development of our model will help to improve the prediction of drug responses and the understanding of drug resistance mechanisms. Third, we show the tumour microenvironment is unique and different from other peripheral organs in terms of immune cell composition, phenotypes and functions. Cytotoxic cells are greatly inhibited in tumour compared with peripheral organs. Although mildly increased immune checkpoint receptor expression is found in blood and spleen T cells from HCC humice, these cells can still respond aggressively to stimulations, even more than normal T cells from humice without HCC. Thus, they are more like activated T cells defined by the expression of immune checkpoint receptors. However, T cells in TILs express a much higher level of immune checkpoint receptors and are the true exhausted cells crippled cytokine production. It suggests that the expression levels of immune checkpoint receptors determine the activation or exhaustion status of immune cells. Also, TILs efficiently enrich special immune cell types which are known to promote tumour development such as Treg, tumour-associated macrophages (M∅2) and MDSCs.24 27 45 46 Such a specialised environment also explains why tumour grows faster in humice than NSG.
The new model was further validated by testing immunotherapeutic drugs, for example, immune checkpoint inhibitors. The anti-HCC effects of immune checkpoint inhibitors like pembrolizumab and ipilimumab have been shown in clinical trials, although they only showed effects in a small portion of patients.39 The expression of PD-1 and CTLA-4 in immune cells, particularly Tc cells, was consistently reproduced in all the HCC-PDX tumours in humice model; however, the treatment of pembrolizumab and ipilimumab only had significant inhibition effects on the tumour growth in a few samples. We noticed that the responders HCC#1 and HCC#4 both grew slower compared with the other two PDX tumours, which suggested that slow growing tumours might respond better to immunotherapies. The underlying mechanisms for the differential responses to immune checkpoint inhibitors are worth further investigation and this model could serve as a good tool.
In humice, which showed responses to immune checkpoint inhibitors, the therapeutic effect was proven to be associated with the re-activation of cytotoxic cells like Tc and NKc cells. Furthermore, the alteration of tumour microenvironment by immune checkpoint inhibitors also caused chain reactions including the reversal of phenotypes and proportion of M∅1/M∅2 macrophages and MDSC. Further investigation on these cells would lead to the exploration of new potential targets and treatment strategies. Moreover, although pembrolizumab showed a significant effect in suppressing HCC growth, it could not eliminate cancer. This suggests that anti-PD1 or immune checkpoint inhibitor alone is not sufficient to HCC cure. Combination treatments with chemotherapy, targeted-based therapy and other immunotherapy drugs may yield better outcomes and can be tested in humice.
Moreover, we also noticed that ipilimumab exhibited immunotoxicity and caused side effects in our model, which was consistent to clinical data. This toxicity may be due to the reactivation of human immune cells, for example, T cells by CTLA-4 blockade, which lead to autoimmunity-like responses. In mice, the deletion of CTLA-4 could induce autoimmune disease.47 The adverse effect of antibody-based immunotherapy on activation of autoimmune responses has also been observed in clinic and seriously concerned.48 The ability of our model to reproduce such side effects could be valuable for testing both therapeutic and side effects of drugs.
Nonetheless, there is still room for improvement which requires further improvement to this model. The HLA between HCC and the immune system is not fully matched, but there is no sign of rejection because humice immune system is educated by both mouse and human main histocompatibility complex (MHCs).49 Ideally, peripheral blood mononuclear cell (PBMCs), blood or bone marrow stem/progenitor cells from the same patient with cancer should provide a full match. However, the issues of graft-versus-host diseases by PBMCs and the weak long-term repopulating ability of adult stem/progenitor cells make it difficult to generate a humice model with large cohort and robust immune responses. Optimisation with more HLA matches will lead to future improvement.
In general, taking into consideration the advantage of humice technologies, this new model presents an attractive option for studying how a functional human immune system reacts with the tumour, to reproduce the complexity and specificity in humans. This would open new doors to the development of novel therapeutic targets and overcome immunotherapy resistance.
References
Footnotes
Contributors YZ designed and performed experiments, analysed and interpreted data and prepared the manuscript. XYC, SYT and ML performed experiments and analysed data; TWHS, TBT, YF, HY, SGI, GKB, EL, KTEC, TCT, WZ, JKYC, EK-HC, CEC, GHL, YYD, PK-HC, HCT and SGL contributed research tools and reagents and prepared the manuscript. QC conceived the study, designed experiments, supervised the project and prepared the manuscript.
Funding This study was supported by the Industry Alignment Fund Cat 3 (IAF311020), Agency for Science, Technology and Research (A*STAR) Singapore, MOH Industry Alignment Fund Cat 2 (MOHIAFCAT2001), National Medical Research Council Singapore and by the Eradication of HBV TCR Program: NMRC/TCR/014-NUHS/2015 and NMRC/TCR/015-NCC/2016 National Medical Research Council Singapore. QC is also supported by the National Research Foundation Fellowship Singapore NRF-NRFF2017-03.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Singhealth, National Healthcare Group Research Ethics Committees of Singapore and Singapore General Hospital specifically approved this study (CIRB Ref: 2012/064/B, DSRB Reference Number: 2014/00231 and TCR Reference Number: 2016/2626).
Provenance and peer review Not commissioned; externally peer reviewed.