Article Text

Abstract
Objective Bile acids are regulators of lipid and glucose metabolism, and modulate inflammation in the liver and other tissues. Primary bile acids such as cholic acid and chenodeoxycholic acid (CDCA) are produced in the liver, and converted into secondary bile acids such as deoxycholic acid (DCA) and lithocholic acid by gut microbiota. Here we investigated the possible roles of bile acids in non-alcoholic fatty liver disease (NAFLD) pathogenesis and the impact of the gut microbiome on bile acid signalling in NAFLD.
Design Serum bile acid levels and fibroblast growth factor 19 (FGF19), liver gene expression profiles and gut microbiome compositions were determined in patients with NAFLD, high-fat diet-fed rats and their controls.
Results Serum concentrations of primary and secondary bile acids were increased in patients with NAFLD. In per cent, the farnesoid X receptor (FXR) antagonistic DCA was increased, while the agonistic CDCA was decreased in NAFLD. Increased mRNA expression for cytochrome P450 7A1, Na+-taurocholate cotransporting polypeptide and paraoxonase 1, no change in mRNA expression for small heterodimer partner and bile salt export pump, and reduced serum FGF19 were evidence of impaired FXR and fibroblast growth factor receptor 4 (FGFR4)-mediated signalling in NAFLD. Taurine and glycine metabolising bacteria were increased in the gut of patients with NAFLD, reflecting increased secondary bile acid production. Similar changes in liver gene expression and the gut microbiome were observed in high-fat diet-fed rats.
Conclusions The serum bile acid profile, the hepatic gene expression pattern and the gut microbiome composition consistently support an elevated bile acid production in NAFLD. The increased proportion of FXR antagonistic bile acid explains, at least in part, the suppression of hepatic FXR-mediated and FGFR4-mediated signalling. Our study suggests that future NAFLD intervention may target the components of FXR signalling, including the bile acid converting gut microbiome.
- bile acid
- bile acid metabolism
- nonalcoholic steatohepatitis
- intestinal microbiology
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Farnesoid X receptor (FXR)-mediated bile acid signalling mediates beneficial effects on hepatic lipid and carbohydrate metabolism.
FXR modulates primary bile acid synthesis in liver.
Gut microbiome converts primary bile acids into secondary bile acids.
What are the new findings?
Serum concentrations of primary and secondary bile acids were increased in patients with non-alcoholic fatty liver disease (NAFLD).
In per cent, the FXR antagonistic deoxycholic acid was increased, while the agonistic chenodeoxycholic acid was decreased in NAFLD.
FXR-mediated and fibroblast growth factor receptor 4-mediated signalling was suppressed in NAFLD livers.
Secondary bile acid production was elevated in the gut of NAFLD.
Similar changes in liver gene expression and the gut microbiome were observed in high-fat diet-fed rats.
How might it impact on clinical practice in the foreseeable future?
Our study suggests that future NAFLD intervention may target the components of FXR signalling, including the bile acid converting gut microbiome.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of obesity-associated metabolic syndrome. NAFLD includes a broad spectrum of liver abnormalities, ranging from simple steatosis to non-alcoholic steatohepatitis (NASH) with varying degrees of inflammation and fibrosis, which can progress to cirrhosis. NAFLD has become the most common liver disease, and its prevalence is about 11% in adolescents1 and 20% in adults2 in the USA, with an annual direct medical cost of about $103 billion ($1613 per patient).3
Bile acids, their receptors and transporters are currently being considered as potential therapeutic targets for NAFLD intervention.4 5 Besides assisting fat absorption in the intestine, bile acids are regulators of lipid and glucose metabolism, and modulate inflammation in the liver and other tissues. These regulatory activities are mediated primarily by bile acid receptors farnesoid X receptor (FXR) and G-protein coupled bile acid receptor 1 (GPBAR1 or TGR5). The effect of hepatic FXR agonism on lipid metabolism is likely mediated by small heterodimer partner (SHP)/sterol-regulatory element-binding protein-1 c6 and peroxisome proliferator-activated receptor α.7 In addition, bile acids may activate FXR in enterocytes to induce fibroblast growth factor 15 (FGF15, mouse) or FGF19 (human) expression. Subsequently, FGF15/FGF19 activates hepatic FGF receptor to decrease hepatic lipogenesis and increase fatty acid β-oxidation.4 8 Bile acid/FXR-mediated pathways may also regulate glucose metabolism and promote insulin sensitivity, although opposite observations related to insulin sensitivity have been reported.4
On the other hand, bile acids may exert a beneficial effect on insulin sensitivity by activating GPBAR1 in intestinal L cells.9 Activation of GPBAR1 in the L cells induces the secretion of glucagon-like peptide 1, which may decrease appetite10 and stimulate pancreatic insulin production.11
Gut microbiota convert primary bile acids to secondary bile acids, and therefore has a potent impact on bile acid signalling.12 In humans, the primary bile acids are cholic acid (CA) and chenodeoxycholic acid (CDCA), which are converted to secondary bile acids such as deoxycholic acid (DCA), lithocholic acid (LCA) and ursodeoxycholic acid (UDCA) by the gut microbiota. Bile acids differ in their potency to activate FXR and GPBAR1. When individually tested, CDCA is the strongest FXR agonist compared with other bile acids, and bile acids other than CDCA exhibited negligible activity in recruiting the FXR coactivator steroid receptor coactivator-1.13 Under more physiological conditions, DCA and LCA actually function as FXR antagonists in the presence of CDCA.13 14 As such, it is expected that human gut microbiome will have a negative impact on FXR signalling by producing antagonistic ligands. In contrast, mouse gut microbiome favours FXR signalling by reducing muricholic acid, one major primary bile acid that is FXR antagonistic.15 Conversely, secondary bile acids DCA and LCA are more potent agonists for GPBAR1 than CDCA and CA.16
The multifaceted impacts of bile acid signalling on liver metabolism suggest that the gut microbiome may be a target for the therapeutic intervention of NAFLD. Recently, Mouzaki et al 17 examined the gut microbiome composition in NAFLD. Using PCR-based method, they found that patients with NASH exhibited small but significant changes in faecal microbiome, which is in line with altered faecal bile acid composition. To understand the possible mechanisms by which the gut microbiome modulates bile acid-mediated signalling in NAFLD, we examined serum levels of bile acid and FGF19, hepatic bile acid-related gene expression and the composition of gut microbiome in patients with NAFLD and control subjects. We found that NAFLD is associated with elevated production of both primary and secondary bile acids, altered gut microbiome and impaired hepatic bile acid-mediated signalling. Our data provide a solid foundation for the development of bile acid-related interventions for NAFLD.
Methods
Patients
This study was approved by the Institutional Review Board of The State University of New York at Buffalo. Prior to sample collection, written consent from parents of patients and assent from children were obtained. Patients included in this study were biopsy-proven patients with NASH fulfilling Kleiner’s criteria.18 For serum bile acid analysis, 16 patients with NASH and 11 healthy controls were enrolled between March 2015 and February 2016 (table 1). Patients in the NASH group and the control group had similar age and gender ratio. Fractionated and total serum bile acid tests were conducted at LabCorp by liquid chromatography tandem-mass spectrometry (LC-MSMS) method. Serum FGF19 levels in controls and in patients with NASH were measured with the Quantikine ELISA Kit (DF1900) from R&D Systems (Minneapolis, Minnesota, USA).
Patient characteristics for serum bile acid analysis
For hepatic gene expression (online supplementary table 1), 27 NASH liver biopsy samples were collected between July 2010 and September 2013. For healthy liver controls, six total liver RNA samples with similar age of the patients with NASH were purchased from Admet Technologies (Durham, North Carolina, USA). These samples were derived from healthy donor livers intended for liver transplantation. Gender was not matched between patients with NASH and healthy controls because of limited availability of healthy controls. Nevertheless, we confirmed that gender does not have any impact on the expression of the target genes in this study (online supplementary table 2).
Supplementary file 1
Supplementary file 2
Animals
The protocol for animal studies was reviewed and approved by the Institutional Animal Care and Use Committee of University at Buffalo. Four-week-old male Sprague-Dawley rats, standard rodent chow (2018s, 18% calories from fat) and high-fat diet (HFD, TD.06414, 60% calories from fat) were purchased from Harlan Laboratories (Indianapolis, Indiana, USA). Animals were maintained at room temperature on a 12 hour:12 hour light–dark cycle in the Laboratory Animal Facility, University at Buffalo. Rats were randomised into two groups (n=6 for each group). Animals in the (1) control group were fed with standard chow and (2) NAFLD group were fed with HFD, for 16 weeks. All rats in the study were fed ad lib and had unlimited access to water. All rats were then sacrificed for tissue collection.
Histopathology
Liver cryosection and Oil Red O staining were performed as described previously.19
Microarray data
A well characterised microarray database20 was used to examine gene expression in NASH livers and controls (Gene Expression Omnibus accession number: GSE24807, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24807).
Quantitative real-time PCR
Custom primers were designed using the National Institute of Health primer tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), with a melting temperature of >60°C, and separated by at least one intron of longer than 1000 nucleotides (online supplementary table 3). Primers were characterised by melting curve analysis, agarose gel electrophoresis and DNA sequencing, and were synthesised at Eurofins MWG Operon (Huntsville, Alabama, USA). Patient biopsies and animal tissues were stored at −80°C after soaking with RNAlater (Qiagen, Valencia, California, USA). Total RNA isolation, cDNA preparation, real-time PCR (RT-PCR) analysis and data process were performed as described previously.21
Supplementary file 3
Gut microbiome data of patients with NASH and healthy controls
Human gut microbiome data for patients with NASH and healthy controls were described previously.22 Briefly, faecal microbiomes from 22 biopsy-proven patients with NASH and 16 volunteer healthy subjects were pyrosequenced on a 454-FLX-Titanium Genome Sequencer (Roche 454 Life Sciences, Branford, Connecticut, USA). Raw 454 sequencing reads and the associated meta-data are archived at MG-Rast (http://metagenomics.anl.gov/linkin.cgi?project=1195). Sequencing reads were processed with the Quantitative Insights into Microbial Ecology (QIIME) V.1.8.1 software.
Analysis of gut microbiome in rats
Colon contents were collected from the rats and stored at −80°C before DNA isolation with the ZR Fecal DNA MicroPrep (Zymo Research, Irvine, California, USA). The V3–V4 hypervariable region was PCR-amplified with primer pair (319F: 5’ACTCCTACGGGAGGCAGCAG 3’; 806R: 3’ACTCCTACGGGAGGCAGCAG 5’). Libraries were multiplexed and paired-end sequenced on an Illumina MiSeq at the Genomics Research Center of University of Rochester, following a dual-indexing protocol.23
Initial Illumina basecall raw data were processed into 2×300 FASTQ paired-end reads using Illumina’s bcl2fastq (v1.8.4) without demultiplexing. The bar codes in each read of the paired sequencing reads were removed and concatenated together for later use. Each pair of reads was then stitched together, with the combined bar codes attached. FASTQ format read files were then converted to FASTA and QUAL files and quality-filtered using QIIME V.1.8.1. These files were then processed to produce an operational taxomonic unit (OTU) table and a phylogenetic tree as described previously.22
Predicting metagenomes by PICRUSt
The 16S rRNA sequencing data were processed with closed-reference OTU picking. The resulting OTU tables were then used for microbial community metagenome prediction with Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt).24 Briefly, OTU tables were normalised for 16S rRNA copy number, followed by metagenome predictions against Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KOs). Then the KO tables were compiled into the KEGG pathways. Differential pathways predicted with the 16S rRNA microbiome data were identified with (linear discriminant analysis effect size (LEfSe), http://huttenhower.sph.harvard.edu/galaxy). Linear discriminant analysis score equal or larger than 2 and p value in Kruskal-Wallis test <0.05 were considered significant. Differential taxa were selected with adjusted p values for multiple comparisons, with False Discovery Rate (FDR) <0.05 considered significantly different.
Additional statistical analysis
Student’s t-tests or Mann-Whitney U tests with two-tailed distribution were performed to examine statistical differences between the two experimental groups. Spearman’s rank correlation coefficient test was performed to examine the correlations between genes of interest. A p value <0.05 was considered significant.
Results
Altered serum bile acid levels in patients with NASH
Serum bile acids were measured in 16 biopsy-proven patients with NASH and compared with those of 11 healthy controls. The characteristics of these age-matched and gender-matched patients with NASH and healthy controls are summarised in table 1. Total serum bile acid levels in patients with NASH were ~3 times that of healthy controls (figure 1A). The composition of the serum bile acid pool also differed between the groups, with patients with NASH having a higher percentage of secondary bile acids (figure 1A,B). Specifically, per cent DCA was ~4-fold elevated in NASH sera, while UDCA was unchanged (figure 1C). Conversely, per cent CDCA, the major primary bile acid in serum, was lower in NASH serum (figure 1C). Per cent representation of CA was not significantly changed (figure 1C). Despite different trends in per cent bile acid distribution between the groups, the absolute concentrations of all bile acid species were increased in NASH serum (figure 1D).

Serum bile acids in patients with NASH and healthy controls. (A) Plotted in the bar graph are total bile acids (mean±SEM) in the serum of patients with NASH and healthy controls. Pie graphs are the mean per cent of the bile acids. (B) Ratio of secondary bile acids (DCA and UDCA) to primary bile acids (CA and CDCA) in the serum of patients with NASH and healthy controls. (C) Per cent of bile acids in the serum of patients with NASH and healthy controls. (D) Absolute serum concentration of bile acids. Values are expressed as mean±SEM. CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; NASH, non-alcoholic steatohepatitis; UDCA, ursodeoxycholic acid.
Hepatic expression of genes related to bile acids
Altered serum bile acid levels prompted us to examine the differential hepatic expression of genes related to bile acid metabolism (figure 2A), using a thoroughly characterised and validated microarray data set from NASH and control livers.19–21 25–28 CYP7A1, the rate-limiting enzyme in bile acid synthesis, demonstrated 9.3-fold increased expression in NASH livers (table 2). Increased expression was also observed for CYP27A1 and CYP8A1, but not for CYP7B1. With a different cohort of NASH subjects and controls (supplementary table 1), we confirmed these findings by quantitative RT-PCR (qRT-PCR). Subjects with NASH demonstrated increased expression of CYP7A1, CYP8B1, CYP27A1, BAAT and BACS, but not CYP7B1 (figure 2B).

Hepatic gene expression in patients with NASH and healthy controls by quantitative real-time PCR. (A) Schematic representation of the two FXR-related pathways. In the enterocyte, bile acid-activated FXR drives the expression of FGF19, which translocates to the hepatocyte via portal circulation and binds to its receptor FGFR4, which suppresses the expression of CYP7A1. FGFR4 activity requires the presence of KLB. On the other hand, bile acids may activate FXR in the hepatocyte to increase expression of BSEP, OATP1B3 and SHP. The nuclear factor SHP suppresses the expression of CYP7A1 and NTCP. Red and green arrows indicate positive and negative effect, respectively. (B) Hepatic expression of genes required for human primary bile acid synthesis in patients with NASH and healthy controls. (C) Hepatic expression of genes required for bile acid transportation in patients with NASH and healthy controls. (D) Hepatic expression of genes that regulate bile acid metabolism. Values are the mean±SEM. *p<0.05, **p<0.01, ***p<0.001. BSEP, bile salt export protein; FGF19, fibroblast growth factor 19; FGFR4, fibroblast growth factor receptor 4; FXR, farnesoid X receptor; KLB, Klotho beta; NASH, non-alcoholic steatohepatitis; NTCP, Na+-taurocholate cotransporting polypeptide; SHP, small heterodimer partner.
Bile acid-related gene expression in non-alcoholic steatohepatitis (NASH) livers (microarray)*
Proteins encoded by NTCP, OATP1B1 and OATP1B3 are transporters responsible for bile acid uptake at the basolateral side of the hepatocytes. These genes exhibited elevated expression in NASH livers according to both microarray (table 2) and qRT-PCR analyses (figure 2C). The other transporter encoded by BSEP pumps bile acids into the canaliculi. Microarray analysis demonstrated a slight decrease in BSEP gene expression in NASH livers (table 2), which was not confirmed by qRT-PCR (figure 2C).
Further, we examined the mRNA levels of the relevant transcriptional factors. Although a small increase in FXR expression was observed in NASH livers with qRT-PCR (figure 2D), no change was observed for its immediate downstream nuclear factor SHP by either microarray (table 2) or qRT-PCR (figure 2D). Increased expression of fibroblast growth factor receptor 4 (FGFR4) was observed with both microarray and qRT-PCR; however, the expression levels of Klotho beta (KLB), a transmembrane protein required for FGFR4 activity,29 did not correlate with those of FGFR4 (table 2, figure 2D). Consistent with the elevated expression of genes required for bile acid synthesis and transportation, increased HNF4A expression was observed by both microarray (table 2) and qRT-PCR (figure 2D). Expression of HNF4A correlates significantly with the expression levels of CYP7A1, CYP8B1, CYP27A1, BAAT, BACS and NTCP (online supplementary table 4).
Supplementary file 4
Bile acid receptor-mediated signalling regulates the transcription of bile acid metabolising genes and genes related to other important cellular functions, such as PON1, a gene encoding an antioxidative activity paraoxonase 1. PON1 transcription is suppressed by both FXR-mediated and FGFR4-mediated bile acid signalling.30 31 We have previously reported that PON1 expression is elevated in NASH livers.27 Here, with a different cohort of patients and controls, we confirmed a significantly increased PON1 expression in NASH livers (online supplementary figure 1).
Supplementary file 5
To test the hypothesis that diet may contribute to the changes in bile acid-related gene expression, HFD-fed rats were used as an NAFLD model for the study of hepatic gene expression. Compared with rats fed standard chow, the HFD-fed rats exhibited higher body weight starting at week 5 (figure 3A). Oil Red O staining of the liver cryosections revealed drastically increased incidence of hepatic fat deposition, recapitulating a key component of NAFLD pathology (figure 3B).

Hepatic gene expression in HFD-fed NAFLD rat. (A) The body weight (g) of HFD-fed rats and rats fed standard chow (control). (B) Oil Red O staining of the liver cryosections. Representative images were shown. Original amplification: 200×. (C) Quantitative real-time PCR analysis of the hepatic expression of genes that are related to bile acid synthesis, transportation and transcriptional regulation. Plotted values are the mean±SEM. *p<0.05, **p<0.01. FGFR4, fibroblast growth factor receptor 4; FXR, farnesoid X receptor; HFD, high-fat diet; NAFLD, non-alcoholic fatty liver disease; SHP, small heterodimer partner.
Similar to patients with NASH, the HFD-fed rats exhibited elevated expression of CYP7A1, CYP27A1, FXR and HNF4A according to qRT-PCR analysis (figure 3C). Elevated CYP7A1 is expected since HFD leads to increased bile acid production in rats32 and humans.33 Similar to patients with NASH, the HFD-fed rats also exhibited no change in BSEP expression (figure 3C), indicating suppressed FXR signalling. However, in contrast to patients with NASH, hepatic expression of CYP8B1, NTCP and FGFR4 was not increased, while SHP was elevated in HFD-fed rats (figure 3C).
Reduced serum FGF19 in patients with NASH
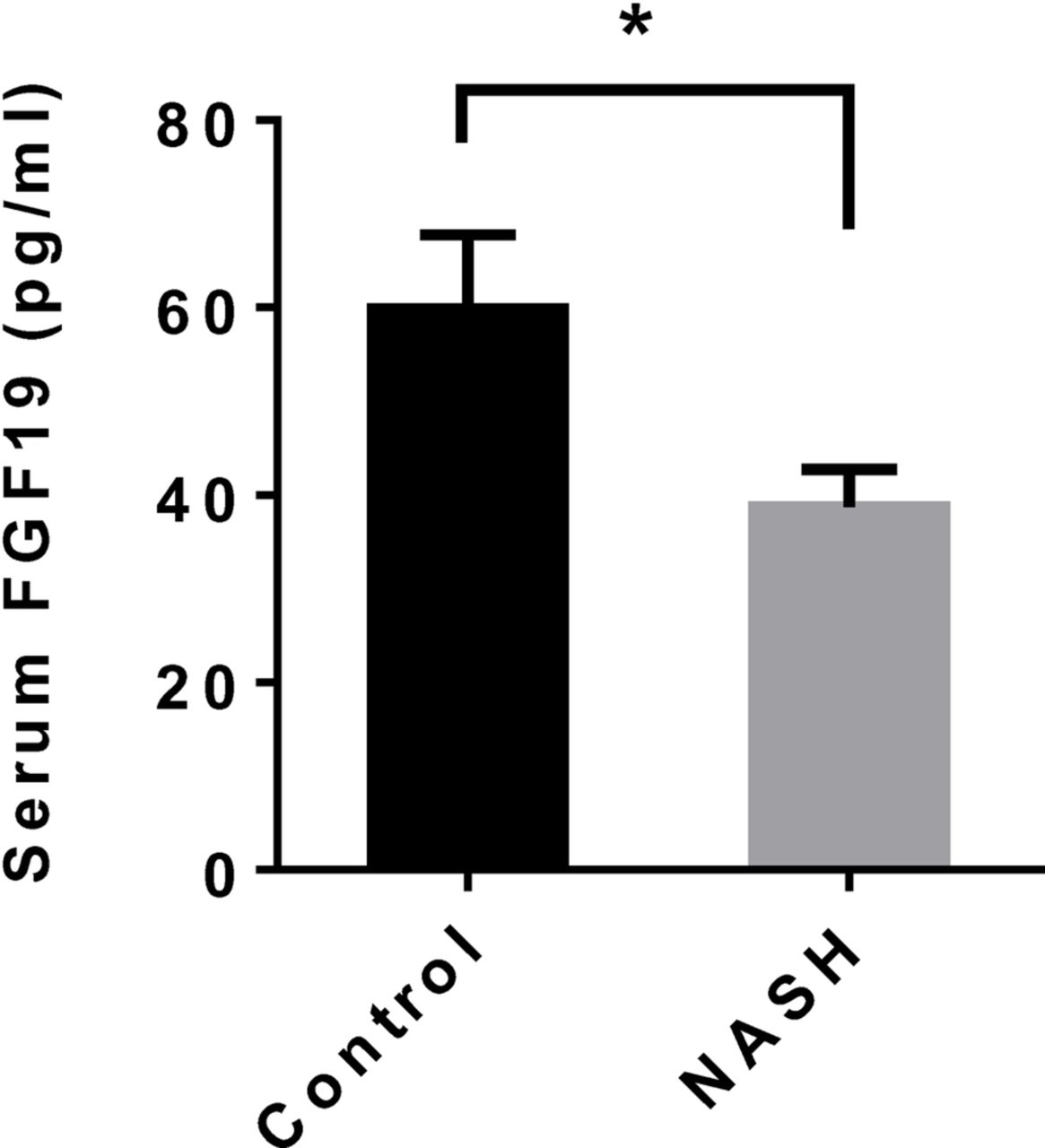
FGF19, a hormone produced in enterocytes, regulates hepatic bile acid, lipid and carbohydrate metabolism. Serum FGF19 was determined in patients with NASH and controls to evaluate its possible impact on hepatic gene expression and NASH pathology. We observed a significantly reduced serum FGF19 in NASH (figure 4).

Serum FGF19 in patients with NASH and controls. Values are the mean±SEM. *p<0.05. FGF19, fibroblast growth factor 19; NASH, non-alcoholic steatohepatitis.
Gut microbiome and secondary bile acid production
The composition of the gut microbiome determines the production of the secondary bile acids, and consequently influences FXR-mediated signalling in the intestine and the liver. We have determined the gut microbiome composition of patients with NASH by 16S rRNA pyrosequencing.22 We revisited the database and identified dozens of pathways that were differentially represented in the gut microbiome between patients with NASH and healthy subjects using PICRUSt24 (online supplementary table 6). The pathway ‘Secondary bile acid biosynthesis’ was not differentially represented between the NASH and the control groups. However, two bile acid-related pathways ‘Glycine, serine and threonine metabolism’ and ‘Taurine and hypotaurine metabolism’ were elevated in NASH samples.
Supplementary file 6
We then examined the bacterial taxa that conduct the key steps of secondary bile acid biosynthesis. Primary bile acids arriving at the intestines are glycine-conjugated or taurine-conjugated. Deconjugation by bile salt hydrolase (BSH) is a prerequisite for downstream modifications by 7-alpha-dehydroxylase to produce DCA and LCA, or by 7-alpha-hydroxysteroid dehydrogenase (HSDH) to produce UDCA (figure 5A). Human gut bacteria that express BSH include species in genera Bacteroides, Clostridium, Bifidobacterium and Lactobacillus.12 Collectively, 25.4% of the gut bacteria belong to these genera in NASH, and no difference in the abundance of these genera was observed between the NASH and the control groups (figure 5B). Similarly, no difference was observed between the study groups in bacteria genera known to express HSDH (figure 5C), 7-alpha HSDH (figure 5D) or 7-alpha-dyhydroxylase (figure 5E). In contrast, gut microbiome in NASH exhibited a 7.2-fold increased abundance in bacteria that metabolise taurine, which was mainly explained by increased abundance in Escherichia and Bilophila (figure 5F).
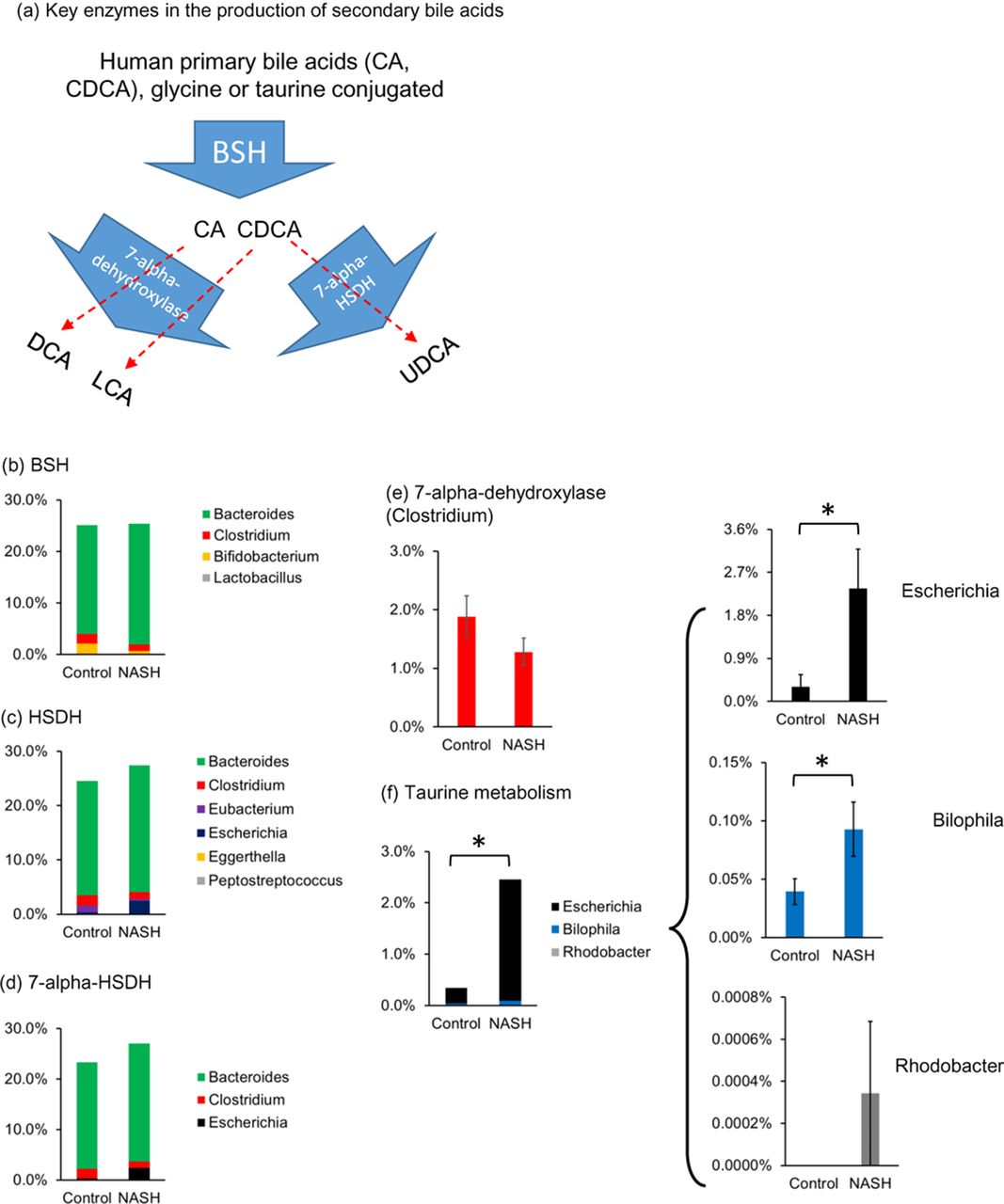
Altered microbiome composition in the gut of patients with non-alcoholic steatohepatitis (NASH). (A) Schematic representation of the bacterial enzymes and their substrates (bile acids). All primary bile acids are deconjugated by bile salt hydrolase (BSH) before further modification. Deconjugated cholic acid (CA) is converted to deoxycholic acid (DCA) by 7-alpha-dehydroxylase; chenodeoxycholic acid (CDCA) is converted to lithocholic acid (LCA) by 7-alpha-dehydroxylase; CDCA is converted to ursodeoxycholic acid (UDCA) by 7-alpha-hydroxysteroid dehydrogenase (HSDH). Average abundance of bacterial genera expressing specific enzymes involved in bile acid synthesis: (B) BSH, (C) HSDH, (D) 7-alpha HSDH, (E) 7-alpha-dehydroxylase (Clostridium) and (F) taurine metabolising bacterial genera in the gut of patients with NASH and healthy controls. Values are the mean±SEM. *p<0.05.
To test the hypothesis that HFD feeding contributes to the altered secondary bile acid production in the gut, the gut microbiome from HFD-fed rats and the controls were examined by16S rRNA next-generation sequencing. UniFrac-based principal coordinates analysis indicated that the HFD-fed and control groups were distinctly separated (figure 6A), suggesting significant difference between the study groups in beta diversities calculated from phylogenetic distances. Abundant differences were observed at every taxonomic level between the HFD-fed rats and the controls (online supplementary table 6). Similar to patients with NASH, the abundance of the bacteria genera that are known to express BSH and 7-alpha-dehydroxylase in the gut of HFD-fed rats was comparable to those fed with standard chow, while the HSDH and 7-alpha HSDH-expressing bacteria genera were enriched in the gut microbiome of HFD-fed rats (figure 6B–D). Elevated HSDH/7-alpha-HSDH-expressing bacteria were mainly explained by increased abundance of Bacteroides. Taurine metabolising bacteria were 110-fold elevated in the gut of HFD-fed rats, mainly because of increased abundance in Bilophila (figure 6E).
Supplementary file 7

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Altered gut microbiome in the gut of HFD-fed rats. (A) The beta diversity of the gut microbiomes of the HFD-fed rats and rats fed standard chow (control) was evaluated by UniFrac-based principal coordinates analysis. (B) Average abundance of the BSH-expressing bacterial genera in the gut of HFD-fed rats and controls. (C) Average abundance of the HSDH and 7-alpha HSDH-expressing bacterial genera in the gut of HFD-fed rats and controls. (D) Average abundance of the 7-alpha-dehydroxylase expressing Clostridium in the gut of HFD-fed rats and controls. (E) Average abundance of the taurine metabolising bacterial genera in the gut of HFD-fed rats and controls. Plotted values are the mean±SEM. *p<0.05, **p<0.01. BSH, bile salt hydrolase; HFD, high-fat diet; HSDH, hydroxysteroid dehydrogenase.
Discussion
Elevated primary bile acid production
Here we report that all primary and secondary bile acids were elevated in the serum of patients with NASH compared with healthy controls, with DCA disproportionally increased more than other bile acids. Increased hepatic expression of CYP7A1 suggests that the increase in primary bile acids was driven by increased synthesis. Since NAFLD is the hepatic manifestation of metabolic syndrome, our observation of elevated bile acid production in NASH is in harmony with the elevated bile acid production in patients with obesity, diabetes and metabolic syndrome.34–36
Previous studies arrived at contradictory conclusions regarding bile acid production in NAFLD. Elevated serum total bile acids in NASH were first reported by Ferslew et al.37 Using approaches that are complementary to ours, Mouzaki et al 17 concluded that bile acid production is elevated in NAFLD livers, based on the findings that faecal total bile acid and faecal primary bile acid levels are elevated in patients with NAFLD, and that serum marker of bile acid synthesis 7-alpha-hydroxy-4-cholesten-3-one is elevated in NAFLD. In support of increased bile acid production, Min et al 38 also observed elevated hepatic expression of CYP7A1 in NAFLD livers. In contrast, Bechmann et al 39 reported no difference in serum bile acid between NAFLD and healthy controls, while Jahnel et al 40 reported decreased serum bile acids in NAFLD. These inconsistencies may be explained partly by different ages between patients with NAFLD and healthy controls in both studies of Bechmann et al and Jahnel et al, as age is negatively correlated with bile acid production.41
HFD is associated with, and contributes to, the obesity epidemic.42 Since HFD induces higher bile acid production than high protein or high carbohydrate diet in rats32 and humans,33 we hypothesise that HFD contributes to the elevated bile acid production in NAFLD. Our finding that hepatic CYP7A1 expression was elevated in HFD-fed NAFLD rats supports this hypothesis.
Impaired FXR-mediated and FGFR4-mediated signalling in NASH livers
Although all serum bile acid species were increased in NASH, DCA, an antagonistic ligand for FXR,13 was the only bile acid that was increased in per cent. In contrast, per cent CDCA, the most potent agonistic natural ligand for FXR, was decreased in NASH. Thus, increased total bile acid level may not stimulate FXR-mediated signalling in the liver and the intestine of patients with NASH. Indeed, the expression of SHP, a nuclear factor regulated by FXR,43 was not altered in NASH livers. Consistent with the absence of SHP suppression, levels of the downstream effector genes CYP7A1, NTCP and PON1 were increased in NASH livers. Like SHP, BSEP is under positive regulation by FXR.44 The unaltered expression of BSEP is therefore additional evidence for impaired FXR signalling in NASH livers.
Initiated in the intestine, the FGFR4-mediated signalling has a negative impact on CYP7A145 and PON131 expression. Thus, elevated hepatic expression of both CYP7A1 and PON1 is a clear indication for impairment of FGFR4-mediated signalling in NASH. The suppressed FGFR4 signalling in NASH is likely a consequence of reduced serum FGF19, the ligand for FGFR4. Since most of the bile acids are recycled within the enterohepatic circulation, FXR in the enterocytes of patients with NASH is likely exposed to a similar bile acid pool as in liver, with DCA disproportionally increased more than other bile acids. Consequently, FGF19 expression in enterocytes would be suppressed, leading to decreased FGFR4 signalling in hepatocytes. In addition, decreased hepatic expression of KLB, a cofactor required for FGFR4 activity,29 provides additional explanation for impaired hepatic FGFR4 signalling.
An alternative hypothetical mechanism for reduced serum FGF19 in NASH could be reduced total bile acid availability in enterocytes, causing a similar effect as increased antagonistic bile acids. It was shown that ileal bile acid malabsorption caused by deficiency in ASBT gene leads to repressed FXR activity in enterocytes, and through reduced FGF15 expression, influences hepatic bile acid metabolism.45 Similar effect is observed when bile acid availability is reduced by enhanced bile acid deconjugation and faecal excretion.46 A better understanding of the FXR/FGF signalling pathway in NASH requires further studies on bile acid metabolism in the enterocytes.
In contrast to suppressed FXR activity, we observed elevated hepatic expression of HNF4A, the liver-specific transcriptional factor that drives the expression of CYP7A1. A strong and significant correlation between HNF4A and CYP7A1 suggests that the elevated HNF4A expression may be part of the mechanism for increased bile acid production in NASH.
Gut microbiome, FXR signalling and novel targets for NAFLD intervention
We found that bacteria that produce secondary bile acids were abundant in the gut of patients with NASH, which allows increased production of DCA with an elevated supply of substrate CA. Increased DCA may suppress FXR signalling in the liver and the gut. Because FXR signalling has beneficial effects on lipid47 48 and glucose49 metabolism, our observations identified impaired FXR signalling as a potential target for therapeutic intervention for NAFLD and metabolic syndrome. Since the FXR antagonistic DCA was highly elevated in patients with NAFLD, microbiome intervention aimed at reducing secondary bile acid production could be an effective strategy to boost FXR signalling.
It was a surprise that the bile acid converting gut bacteria were not differently represented between patients with NASH and controls. One explanation suggested by our data is that these bacteria are so abundant that they exceed the demand for elevated bile acid metabolism in NASH. Thus, elevated primary bile acid exposure in NASH may not induce the bile acid metabolising bacteria. On the other hand, the composition of the gut microbiome in patients with NASH does reflect the increased secondary bile acid production. Taurine and glycine metabolising bacteria were elevated in the gut microbiota of patients with NASH compared with healthy controls. Since the dietary proteins and amino acids were not different between patients with NASH and controls,22 bile acids are likely the major source of elevated taurine and glycine metabolism in gut microbiome of patients with NASH.
In HFD-fed rats, the overall abundance of bile acid converting bacteria was similar to that of control rats, although HSDH-expressing bacteria were elevated in HFD-fed rats. Elevated bile acid metabolism in the gut of HFD-fed rats was suggested by highly elevated taurine metabolising bacteria in the gut of HFD-fed rats. The similarity of the gut microbial change in HFD-fed rats and patients with NASH supports that the HFD may initiate the elevated bile acid metabolism in the gut of humans.
Future studies are needed to address several gaps and limitations of this study. For ethical reasons, the current study did not examine bile acid metabolism and FXR signalling in enterocytes. Second, the hepatocytes are exposed to bile acid profiles more similar to those found in portal vein than those in peripheral blood. Examination of the bile acid composition in the portal vein will provide more accurate understanding about liver bile acid metabolism and signalling. Third, examination of the gene and protein expression levels and activities of bile acid converting enzymes in the gut microbiome is a future direction for understanding the production of secondary bile acids in NAFLD. Nevertheless, evidence presented here supports an impaired bile acid/FXR signalling in NAFLD and that gut microbiome contributed to this abnormality.
In summary, the serum bile acid profile, the hepatic gene expression pattern and the gut microbiome composition in patients with NASH consistently support an elevated bile acid production in these patients. The secondary bile acids, mainly FXR antagonistic DCA, were disproportionally increased more than the primary bile acids. The increased DCA level explains, at least in part, the suppression of FXR-mediated and FGFR4-mediated signalling. Our study suggests that future NAFLD intervention may target the components of FXR signalling, including the bile acid converting gut microbiome.
References
Footnotes
Contributors LZ, SSB and RZ conceived and designed the study. NJ, SSB, AC-R, WL, CAN and MT performed the experiments. LZ, SSB, RZ, NJ, AC-R, WL, MJB, RDB, RJG and LM analysed the data. LZ, SSB, RZ, NJ and AC-R wrote the draft of manuscript. WL, CAN, MT, LM, MJB, RDB and RJG critically revised the manuscript.
Funding This work was supported by National Natural Science Foundation of China (No 31200986 and No 41530105, to RZ), Natural Science Foundation of Shanghai (No 16ZR1449800, to RZ), the University at Buffalo Departmental StartUp Fund (to LZ), GEM Community of Excellence, the University at Buffalo (to LZ) and the Peter and Tommy Fund, Buffalo, NY (to SSB and LZ).
Competing interests None declared.
Patient consent Parental/guardian consent obtained.
Ethics approval Children and Youth Institutional Review Board of The State University of New York at Buffalo.
Provenance and peer review Not commissioned; externally peer reviewed.