Article Text

Abstract
Background and aims The unique expression pattern makes oncofetal proteins ideal diagnostic biomarkers and therapeutic targets in cancer. However, few oncofetal proteins have been identified and entered clinical practice.
Methods Fetal liver, adult liver and hepatocellular carcinoma (HCC) tissues were employed to assess the expression of hepatic leukaemia factor (HLF). The impact of HLF on HCC onset and progression was investigated both in vivo and in vitro. The association between HLF and patient prognosis was determined in patient cohorts. The correlation between HLF expression and sorafenib benefits in HCC was further evaluated in patient cohorts and patient-derived xenografts (PDXs).
Results HLF is a novel oncofetal protein which is reactivated in HCC by SOX2 and OCT4. Functional studies revealed that HLF transactivates c-Jun to promote tumour initiating cell (TIC) generation and enhances TIC-like properties of hepatoma cells, thus driving HCC initiation and progression. Consistently, our clinical investigations elucidated the association between HLF and patient prognosis and unravelled the close correlation between HLF levels and c-Jun expression in patient HCCs. Importantly, HLF/c-Jun axis determines the responses of hepatoma cells to sorafenib treatment, and interference of HLF abrogated c-Jun activation and enhanced sorafenib response. Analysis of patient cohorts and PDXs further suggests that HLF/c-Jun axis might serve as a biomarker for sorafenib benefits in HCC patients.
Conclusions Our findings uncovered HLF as a novel oncofetal protein and revealed the crucial role of the HLF/c-Jun axis in HCC development and sorafenib response, rendering HLF as an optimal target for the prevention and intervention of HCC.
- hepatocellular carcinoma
- oncogenes
- tumour markers
- carcinogenesis
- drug resistance
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Due to the unique expression patterns, oncofetal genes are probably of great value in cancer diagnosis and treatment. However, very few functional oncofetal proteins have been identified to date.
What are the new findings?
Hepatic leukaemia factor (HLF) is a novel oncofetal protein that drives hepatocellular carcinoma (HCC) onset and progression.
Oncofetal HLF is reactivated by stemness-associated factors OCT4/SOX2.
HLF transactivates c-Jun to promote tumour initiating cell (TIC) generation and enhance TIC-like properties of hepatoma cells.
HLF/c-Jun axis determines sorafenib response in hepatoma cells and correlates with sorafenib benefits in patients.
How might it impact on clinical practice in the foreseeable future?
Our study suggests that HLF could be an optimal target for HCC intervention as well as a biomarker for sorafenib benefits.
Introduction
Hepatocellular carcinoma (HCC) represents the sixth leading type of cancer globally and ranks as the third most common cause of cancer-related death.1 The incidence of this deadly disease is still on the rise and curative treatments, including resection, ablation or transplantation, are only feasible in patients diagnosed at early stages.2 For those patients with advanced HCC, optimal therapeutic options are still lacking. Sorafenib is the first FDA-approved first-line drug for advanced HCC, which provides limited survival benefits.1 Moreover, due to frequent recurrence and metastasis, the prognosis of this refractory disease remains dismal, further emphasising the urgent need to deepen our understanding of the molecular pathogenesis of HCC and to develop novel intervention strategies.
Embryonic/fetal development and carcinogenesis share many similarities in the profile and regulation of gene expression.3 Expression of oncofetal genes is relatively high in both embryonic/fetal tissues and cancers, but it is turned off in adult normal tissues.4 This unique expression pattern makes oncofetal proteins ideal candidate targets for cancer diagnosis and treatment.5 6 In addition, the reactivation of oncofetal genes implies that they probably have important roles in cancer onset and progression. However, studies on the basic mechanisms beneath the reactivation of oncofetal genes and their functions are scarce to date. Moreover, most oncofetal genes discovered cannot enter clinical practice due to various limitations.4 Therefore, it is necessary to identify novel, reliable oncofetal genes and clarify their biological function and clinical significance.
Hepatic leukaemia factor (HLF) is a transcription factor that belongs to the proline and acidic amino acid-rich family, which function as circadian modulators.7 HLF was also reported to be implicated in the regulation of hematopoietic stem cells as well as hematopoietic malignancies.8–11 Initially, in the 1990s, a part of HLF was discovered in E2A-HLF, a fusion gene identified in pre-B-cell acute lymphoblastic leukaemia, and the mRNA of HLF was found to be absent in normal hematopoietic cells but was detected in liver tissues by northern blot analysis, thus it was named.12 Nevertheless, our previous study demonstrated that HLF protein was not detectable in normal liver, but it was expressed in activated hepatic stellate cells and played an essential role in liver fibrosis.13 Nevertheless, the role of HLF in liver carcinogenesis and HCC progression remains obscure.
In present study, we discover that HLF is a novel oncofetal protein and drives the onset and progression of HCC through transactivating c-Jun expression. Patient cohort studies elucidate that the HLF/c-Jun axis determines the responses of hepatoma cells to sorafenib and correlates with sorafenib benefits in patients.
Material and methods
Patients and analysis
Fetal liver samples were obtained from General Hospital of PLA Rocket Force and Changhai Hospital. The HCC and corresponding peritumoral tissues were collected from surgical resections of patients without preoperative treatment at Eastern Hepatobiliary Surgery Hospital (EHBH) from 2006 to 2010. Two cohorts (157 patients in cohort 1 (the test cohort) and 350 patients in cohort 2 (the validation cohort)) were followed for 60 months, and disease-free survival (DFS) and overall survival (OS) analysis were performed using the Kaplan-Meier method. OS was defined as the interval between the dates of surgery and death. The recurrence was defined as the interval between the dates of surgery and recurrence; if recurrence was not diagnosed, patients were censored on the date of death or the last follow-up. Detailed clinicopathological features of the patients in cohorts 1 and 2 are described in online supplementary tables 1 and 2. Cohort 3 consists of patients who have received adjuvant sorafenib therapy after surgery for primary HCC (online supplementary table 3). Cohort 4 consists of patients who have received sorafenib therapy for the postoperative recurrent HCC (online supplementary table 4). In addition, a group of matched recurrent HCC, primary HCC and peritumoral normal liver tissues from 17 patients were used for immunohistochemistry (IHC) analysis of HLF. Another group of 62 HCC specimens were used for analysing the correlation between HLF and c-Jun mRNA expression.
Supplementary file 1
Cell lines and viruses
The immortalised, normal hepatocyte cell line HL7702(L-02), and liver cancer cell lines HCCLM3 (LM3) and Huh7 were obtained from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai Institute of Cell Biology of the Chinese Academy of Sciences). HLF knockdown and control lentiviruses (designated as shHLF and Control) were purchased from Cyagen (Guangzhou, China). The RNAi target sequence was as follows: GCTGGGCAAATGCAAGAACAT. Adenoviruses expressing FLAG-HLF or FLAG (designated as Ad-HLF and Ad-Con) were obtained from Viagen (Shandong, China). The anti-Flag antibody was used to recognise exogenous Flag-tagged HLF protein in Ad-HLF infected cells.
Mice and HCC induction
HLFPB/PB mice were described previously.13 Briefly, The HLFPB/PB mice were constructed by inserting a piggybac (PB) transposon encoding CAG-RFP (a red fluorescent protein sequence under CAG promoter) into the HLF gene. For HCC induction, 15-day-old male mice were intraperitoneally injected with diethylnitrosamine (DEN) (25 mg/kg body weight) once and were sacrificed 7, 8 or 9 months later. The mouse livers and serum were collected for subsequent experiments. All animal procedures were conducted with the approval of the EHBH.
Spheroid assay
The cells were seeded in 96-well ultra-low attachment culture plates (Corning Incorporated Life Sciences) (300 cells per well) and cultured in DMEM/F12 (Gibco) supplemented with 1% FBS, 20 ng/mL bFGF and 20 ng/mL EGF for 7 days. The number of spheroids was counted and representative views were shown.
Xenografted tumour formation and patient-derived xenograft model
For in vivo transformation assay, HL7702 cells infected with Ad-HLF or Ad-Con were dissociated into single cells and mixed with matrigel at a ratio of 1:1. The mixtures were then injected subcutaneously into eight NOD-SCID mice at 1×103 cells per mouse. Xenografted tumour formation was monitored and the mice were sacrificed 10 weeks postinoculation. For in vivo tumour growth assay, HCCLM3 shHLF or control cells were injected subcutaneously into six NOD-SCID mice at 2×106 cells per mouse. Xenografted tumour formation was monitored and the mice were sacrificed 5 weeks postinoculation. For the patient-derived xenograft (PDX) model, primary tumour samples were obtained for xenograft establishment as described previously.14 The mice with xenografts were given sorafenib (60 mg/kg) or vehicle daily orally for 24 days (n=4 for each group). Tumour volumes were measured at the indicated time points. All procedures and protocols were approved by the Institutional Review Board of EHBH.
IHC and immunofluorescent staining
The tissue samples were fixed with neutral buffered formalin and embedded in paraffin, followed by H&E staining or IHC examination as described previously.13 The antibodies used are listed in online supplementary table 5. The stained sections were further quantified and scored blindly by a pathologist. The score was defined as ‘1’ (positivity ≤10%), ‘2’ (10% ˂ positivity ≤20%), ‘3’ (20% ˂ positivity ≤40%), ‘4’ (40% ˂ positivity ≤60%), ‘5’ (60% ˂ positivity ≤80%) or ‘6’ (80% ˂ positivity ≤100%).
Luciferase reporter assay
The DNA sequences containing the promoter region of HLF (+155 to +670) and c-Jun (−1173 to −690) were cloned into pGL6-luc plasmid (designated as HLF-WT and c-Jun-WT). The potential OCT4/SOX2 binding sites within the HLF promoter and the potential HLF binding sites within the c-Jun promoter were further deleted in pGL6-HLF-WT-luc and pGL6-c-Jun-WT-luc plasmid, respectively (designated as HLF-Mut and c-Jun-Mut). The cells were transfected with pGL6-HLF-WT-luc, pGL6-HLF-Mut-luc, pGL6-c-Jun-WT-luc, or pGL6-c-Jun-Mut-luc and pRL-TK-renilla-luc plasmids. Luciferase activity was measured using a Synergy 2 Multidetection Microplate Reader (BioTek Instruments). Data were normalised for transfection efficiency by dividing firefly luciferase activity by Renilla luciferase activity.
Chromatin immunoprecipitation assays
The chromatin immunoprecipitation (ChIP) assay was performed using an EpiTect ChIP qPCR Kit (QIAGEN) according to the manufacturer’s instructions. Briefly, the chromatin was immunoprecipitated with IgG, anti-OCT4, anti-SOX2 or anti-Flag antibodies. Then, the DNA was purified, and qPCR was performed to examine the bound sequences. The primers used for qPCR analysis were listed in online supplementary table 6.
Data analysis
Statistical analysis was performed using SPSS V.16.0. The data are expressed as the mean ±SD The significance of mean values between two groups was analysed by Student’s t-test or Mann-Whitney U test. Pearson’s correlation analysis was performed to determine the correlation between two variables. A p-value less than 0.05 was considered statistically significant.
The remainder of the description of the materials and methods can be found in the supplementary materials.
Results
HLF is a novel oncofetal protein in liver cancer
To explore the physiological and pathological role of HLF in liver, the expression of HLF was determined in fetal liver, adult liver and HCC tissues. As shown in figure 1A,B, HLF transcripts were relatively high in fetal livers but were low in adult livers. Of note, the expression of HLF was reactivated in HCCs from both mice and humans. Consistently, the distinct expression pattern of HLF was further confirmed at the protein level in mice (figure 1C) and humans (figure 1D,E), suggesting that HLF is a novel oncofetal protein. Oncofetal genes are usually turned off in adult tissues, and the mechanisms underlying their reactivation in cancer remain far from clear. Analysis of the genomic status of HLF in HCCs from TCGA data sets revealed that the alteration of HLF DNA is rare (online supplementary figure S1A).15 16 In addition, the reactivation of HLF was not regulated via DNA methylation or histone acetylation in hepatoma cells (online supplementary figure S1B,C). Bioinformatics analysis of HLF promoter revealed a set of potential binding sites of distinct transcription factors, including SOX2, OCT4, SMAD2/3 and STAT3 (figure 1F). Neither the interference of STAT3 or SMAD3 nor the treatment of IL-6 or TGF-β affected HLF expression in hepatoma cells (online supplementary figure S1D-G). Nevertheless, interference of SOX2 or OCT4 reduced the expression of HLF in hepatoma cells, and HLF levels were dramatically suppressed on simultaneous silencing of SOX2 and OCT4 (figure 1G,H). Consistently, the promoter activity of HLF was suppressed by SOX2 or OCT4 interference (figure 1I). ChIP analysis revealed that SOX2 or OCT4 can bind to the promoter of HLF (figure 1J). Furthermore, the mutation of SOX2/OCT4 binding sites within HLF promoter region abrogated the suppression of the HLF promoter activity triggered by SOX2 or OCT4 interference (figure 1K,L). These findings clarify the pivotal role of SOX2/OCT4 in controlling the reactivation of oncofetal gene HLF in HCC, which sheds new light on the underlying mechanism of oncofetal gene reactivation in cancer.
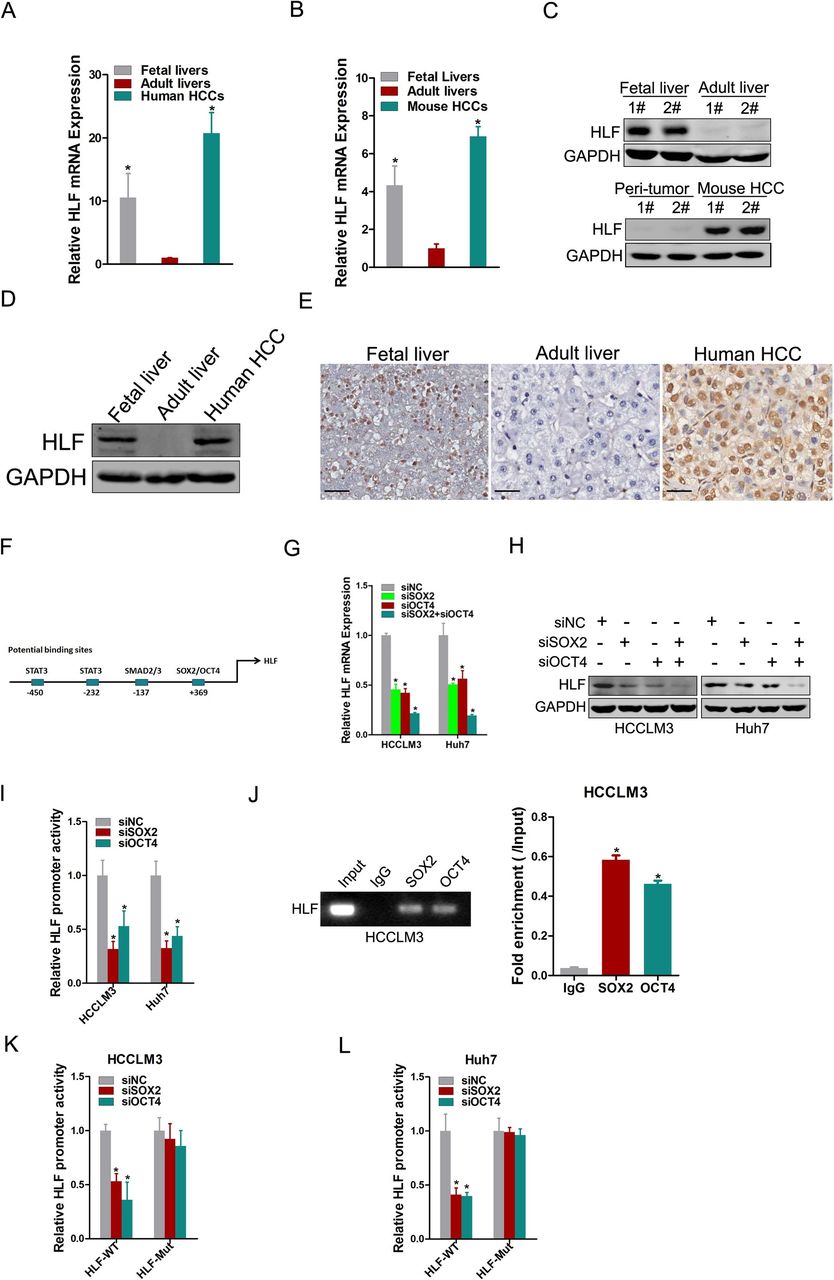
HLF is an oncofetal protein reactivated by OCT4 and SOX2 in HCC. (A&B) The mRNA expression of HLF in human or murine fetal livers (n=5), adult livers (n=5) and HCC tissues (n=5) was detected by real-time PCR, respectively. The mouse HCC tissues were collected from the DEN-induced mouse hepatocarcinogenesis model. (C&D) The protein expression of HLF in fetal livers, adult livers and HCC tissues from mice or humans was examined by western blot. (E) Representative views of IHC staining of HLF in human fetal livers, adult livers and HCC tissues. (F) The potential binding sites within the promoter region of HLF. (G&H) Hepatoma cells were transfected with siSOX2 and/or siOCT4 followed by real-time PCR or western-blot analysis. (I) The hepatoma cells transfected with siSOX2 or siOCT4 were subjected to luciferase analysis of HLF promoter activity. (J) The hepatoma cells were subjected to ChIP assay with anti-SOX2, anti-OCT4 or anti-IgG antibody. (K&L) The luciferase activity of the HLF-WT or HLF-Mut promoter was measured in hepatoma cells transfected with siSOX2 or siOCT4, and the relative activity was presented (relative to siNC), respectively. ChIP, chromatin immunoprecipitation; DEN, diethylnitrosamine; HCC, hepatocellular carcinoma; HLF, hepatic leukaemia factor; IHC, immunohistochemistry.
HLF expression correlates with HCC progression and patient prognosis
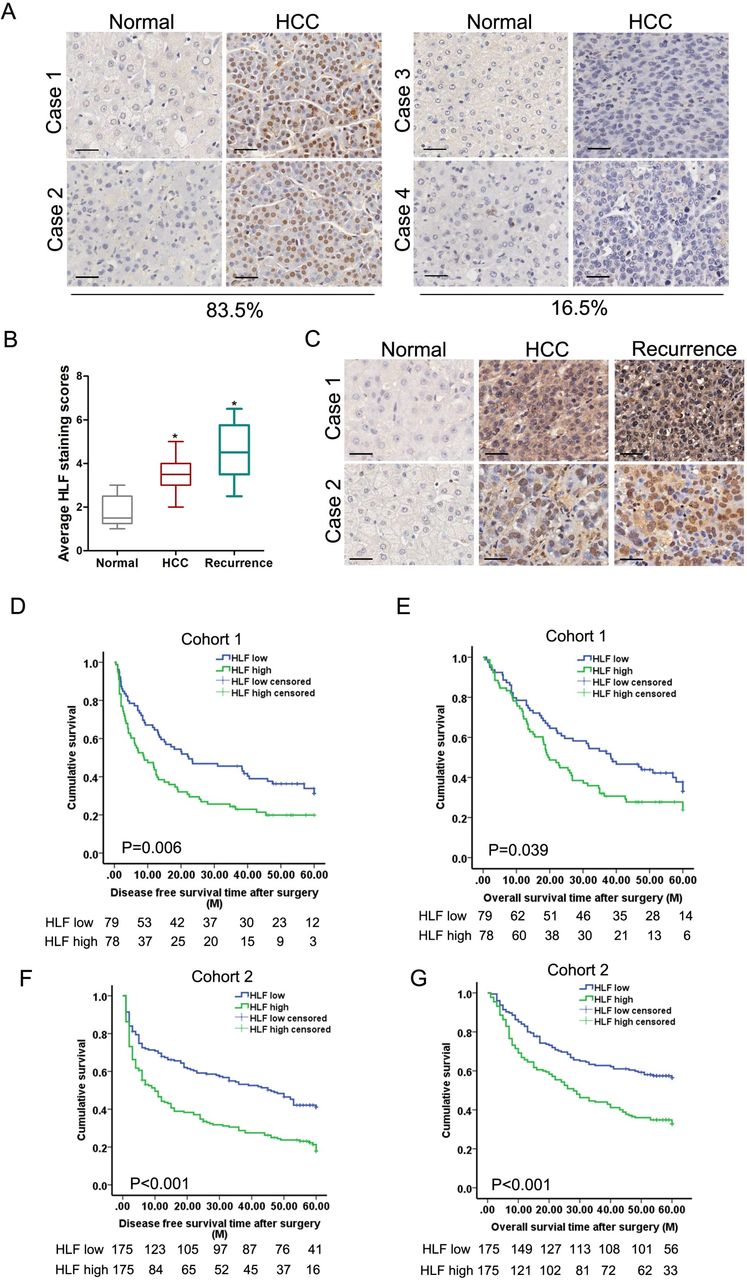
To define the clinical significance of HLF, we further analysed HLF expression in HCCs from patient cohorts. By IHC examination, we observed a significant upregulation of HLF in HCCs from a cohort consisting of 157 patients (cohort 1, the test cohort) (online supplementary figure S2A) where 83.5% of the patients displayed enhanced HLF expression in HCCs (figure 2A). Consistent HLF upregulation in patient HCC compared with peritumoral normal tissues was detected by western-blot assay (online supplementary figure S2B). The levels of HLF were further upregulated in portal vein tumour thrombus tissues compared with that in the corresponding tumours (online supplementary figure S2C). Notably, HLF expression was significantly elevated in recurrent HCCs compared with that in primary HCCs (figure 2B,C), further indicating the close correlation between HLF expression and HCC progression. More importantly, Kaplan-Meier analysis revealed that high HLF levels in HCCs correlated with a worse DFS and OS of patients (figure 2D,E). Consistently, elevated HLF expression in HCCs was confirmed in another independent patient cohort (cohort 2, the validation cohort) (online supplementary figure S2D), and the survival analysis further validated the value of HLF as a biomarker in HCC patients (figure 2F,G).
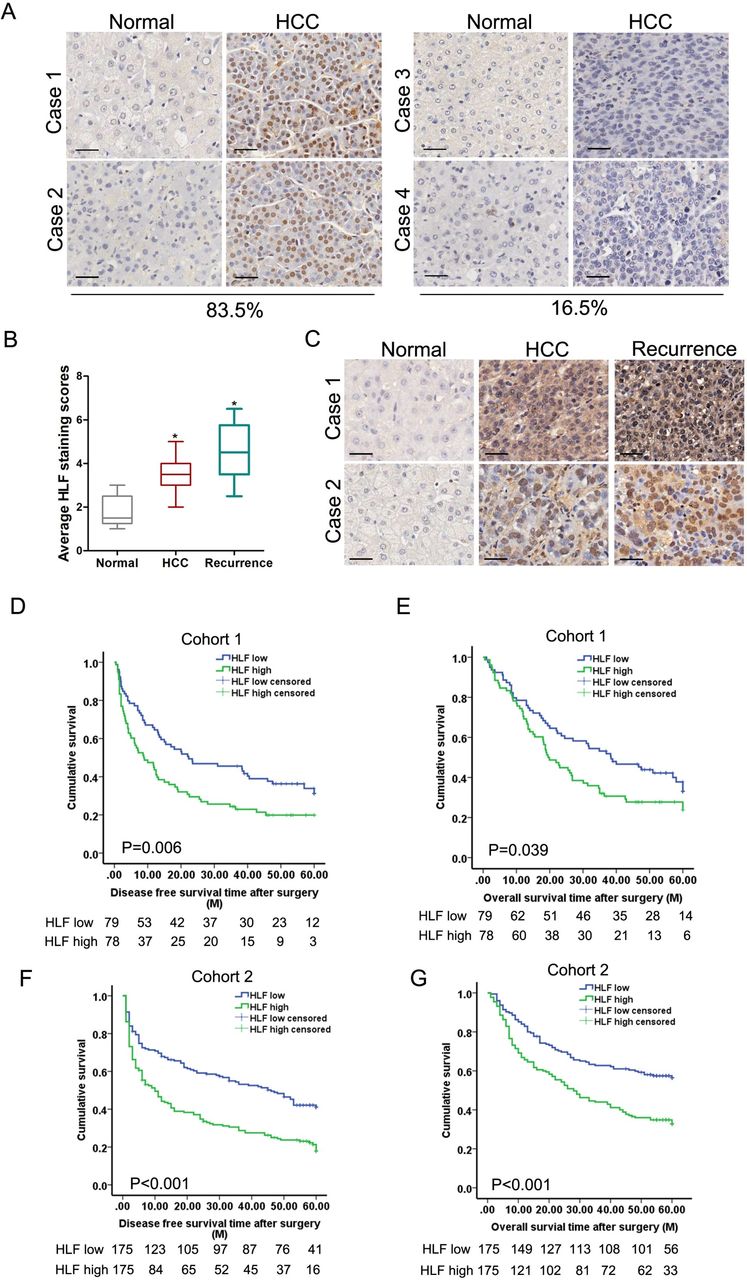
The expression of HLF correlated with HCC progression and patient prognosis. (A) Representative views of IHC staining of HLF in human HCC and peritumoral normal tissues from cohort 1. The vast majority of patients (83.5%) exhibited increased HLF expression in their HCCs. (B) The expression of HLF was analysed in matched human peritumoral normal, HCC and the corresponding relapsing HCC of 17 patients by IHC staining. (C) Representative images of IHC staining of HLF in matched human peritumoral normal, HCC and relapsing HCC tissues. Scale bar=25 µm. (D&E) The overall survival and disease free survival time after surgery of the patients in cohort 1 were compared between the ‘HLF low’ and ‘HLF high’ groups. (F&G) The overall survival and disease free survival time after surgery of the patients in cohort 2 were compared between the ‘HLF low’ and ‘HLF high’ groups. HCC, hepatocellular carcinoma; HLF, hepatic leukaemia factor; IHC, immunohistochemistry.
HLF drives liver oncogenesis via promoting TIC generation
To elucidate the oncogenic role of HLF in liver, we activated HLF in normal hepatocyte cell line and mouse primary hepatocytes (online supplementary figure S3A). Interestingly, ectopic HLF expression strikingly elevated the expression of TIC-associated markers in hepatocytes and facilitated the generation of TICs (figure 3A and online supplementary figure S3B). Moreover, the introduction of HLF induced hepatocyte transformation in vitro (figure 3B) and initiated the tumorigenesis in vivo while the control cells did not, and the formed tumour exhibited the phenotype of HCC with strong alpha fetal protein (AFP) staining (figure 3C,D). Next, the impact of HLF on hepatocarcinogenesis was validated in HLF-intact (WT) and HLF-deficient (HLFPB/PB) mice administrated with DEN. As shown in figure 3E,F, the formation of preneoplastic foci was completely blocked at 7 months and the subsquent tumour onset was also dramatically reduced in HLFPB/PB mice compared with that in WT mice. Furthermore, the tumour load was evidently reduced by HLF disruption, as indicated by a significant decline in tumour numbers, maximal tumour sizes and liver-to-body weight ratios in HLF-deficient mice compared with control mice (figure 3G and online supplementary figure S3C). Ki67 staining also indicated a notable decrease in hepatoma cell proliferation in HLFPB/PB mice compared with that in WT mice (online supplementary figure S3D). There were no differences in the levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) between HLFPB/PB and WT mice, suggesting that the liver injury did not differ between HLFPB/PB and WT mice (online supplementary figure S3E). Importantly, the expression of TIC-associated markers, including CD133, CD24, CD90, EpCAM and OV6, was significantly declined in HCCs from HLF-deficient mice compared with that from WT mice, suggesting that HLF gives rise TICs and drives liver oncogenesis (figure 3H,I).

HLF drives hepatocarcinogenesis via promoting tumour initiation. (A&B) HL7702 cells and mouse primary hepatocytes infected with Ad-HLF or Ad-Con were subjected to spheroid formation and colony formation assays. (C) HL7702 cells infected with Ad-HLF or Ad-Con were injected subcutaneously into NOD-SCID mice at 1×103 cells per mouse. Xenografted tumour growth was monitored, and tumour weight was measured 10 weeks later. (D) The tumour formed in C was subjected to H&E and IHC staining. (E) DEN-induced preneoplastic nodule and tumour formation in HLFPB/PB and WT mice at 7, 8 or 9 months after DEN injection. (F) The preneoplastic foci incidence (7 months) and tumour incidence (8 and 9 months) in HLFPB/PB and WT mice was measured. (G) The tumour numbers, maximal tumour sizes and liver-to-body weight ratios of HLFPB/PB and WT mice at 9 months after DEN injection was measured. (H&I) The mRNA and protein expression of TIC-associated markers in tumours from HLFPB/PB and WT mice at 9 months after DEN injection were examined by real-time PCR and IHC staining analysis. DEN, diethylnitrosamine; HLF, hepatic leukaemia factor; IHC, immunohistochemistry; TIC, tumour initiating cell.
HLF transcriptionally activates c-Jun to promote HCC initiation
We further explored the molecular mechanism underlying the HLF-triggered tumor-initiation. Pathway enrichment analysis of the microarray data (GSE125934) uncovered a set of genes in MAPK signalling pathways were differentially expressed in DEN-induced HCCs between HLF-deficient mice and WT mice (online supplementary figure S4A,B). Expression of these genes was then examined by real-time PCR, and the alteration of c-Jun was found to be the most significant (figure 4A). Western-blot analysis further confirmed the impaired c-Jun activation in HCCs from HLF-deficient mice compared with that from HLF-intact mice (figure 4B). To further investigate the regulation of c-Jun by HLF in hepatocarcinogenesis, HLF was ectopically expressed in normal hepatocytes. As shown in figure 4C,D, the forced HLF expression upregulated the mRNA and protein levels of c-Jun in a dose-dependent manner. Consistent upregulation and nuclear accumulation of c-Jun in HLF-expressing hepatocytes was confirmed by ICC staining (figure 4E). As expected, hyperactivation of c-Jun was detected in HLF-transformed tumours by histological examination (figure 4F). Consistently, c-Jun activation was enhanced by ectopic HLF expression as evident by activator protein one (AP-1) luciferase reporter assay (online supplementary figure S4C). In addition, the enhanced activation of c-Jun promoter by ectopic HLF expression was also observed (online supplementary figure S4D). ChIP assay further detected the significant enrichment of HLF in the promoter region of c-Jun (figure 4G and online supplementary figure S4E). Next, the mutation of HLF binding site within c-Jun promoter region abrogated the enhancement of c-Jun promoter activity triggered by ectopic HLF expression (figure 4H). Moreover, the interference of c-Jun abrogated the enhancement of TIC-associated genes expression, self-renewal, colony formation and tumorigenesis triggered by ectopic HLF expression in normal hepatocytes (figure 4I-L and online supplementary figure S4F), which further demonstrates that HLF promotes tumor-initiation through transactivating c-Jun.
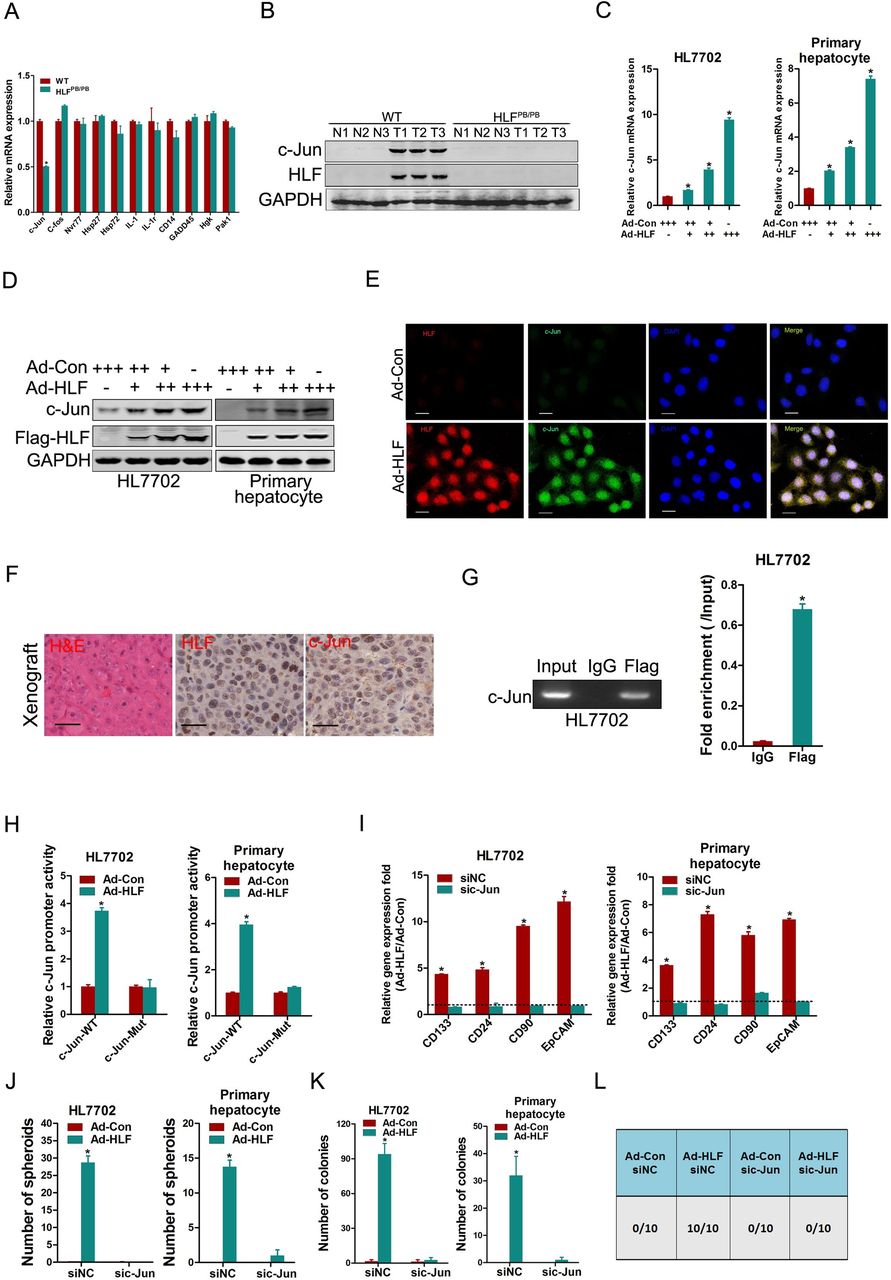
c-Jun is required for HLF-mediated tumour initiation. (A) Real-time PCR analysis of tumours from HLFPB/PB and WT mice at 9 months after DEN injection. (B) Western-blot analysis of tumours and peritumoral normal tissues in the livers from HLFPB/PB and WT mice at 9 months after DEN injection. (C&D) HL7702 cells and mouse primary hepatocytes infected with Ad-HLF or Ad-Con as indicated were subjected to real-time PCR and western-blot analysis. (E) HL7702 cells infected with Ad-HLF or Ad-Con were subjected to dual immunofluorescence staining. Representative images are shown. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Scale bar=20 µm. (F) Representative images of IHC staining of HLF and c-Jun in xenografted tumours formed by Ad-HLF-infected HL7702 cells from figure 3D. Scale bar=25 µm. (G) HL7702 cells infected with Ad-HLF were subjected to ChIP assay with anti-Flag or anti-IgG antibody. (H) The luciferase reporter activity of c-Jun promoter (c-Jun-WT) or the mutant of c-Jun promoter (c-Jun-Mut) was measured in normal hepatocytes infected with Ad-HLF or Ad-Con. (I) The normal hepatocytes infected with Ad-HLF or Ad-Con were transfected with sic-Jun or siNC followed by real-time PCR analysis. (J) The normal hepatocytes infected with Ad-HLF or Ad-Con were transfected with sic-Jun or siNC and were then subjected to spheroid formation assay. (K) The normal hepatocytes infected with Ad-HLF or Ad-Con were transfected with sic-Jun or siNC followed by colony formation assay. (L) HL7702 cells infected with Ad-HLF or Ad-Con were transfected with sic-Jun or siNC and were then injected subcutaneously into NOD-SCID mice at 1×103 cells per mouse. Xenografted tumour formation was monitored 10 weeks later. DEN, diethylnitrosamine; HLF, hepatic leukaemia factor; IHC, immunohistochemistry.
HLF enhances TIC-like properties and facilitates HCC progression
Previous studies showed that HLF was implicated in the functional regulation of hematopoietic stem cells as well as the hematopoietic malignancies.8–11 In this study, we found that HLF, an oncofetal protein, was involved in HCC initiation. However, the role of HLF in modulating the TIC- or cancer stem cell-like properties of hepatoma cells in HCC progression is still unknown. Thus, we first examined the expression of HLF in TICs enriched from hepatoma cells. The results showed that the expression of HLF was elevated in self-renewing spheroids compared with that in attached cells (figure 5A). As expected, HLF expression could return to normal during reattachment in parallel with the differentiation (figure 5B). In addition, increased HLF expression was detected in CD24+ or OV6+ hepatoma cells in comparison with their control cells (figure 5C), further suggesting that HLF expression is enhanced in liver TICs. Next, we found that HLF interference dramatically decreased the expression of TIC markers and stemness-related transcription factors in hepatoma cells (online supplementary figure S5A-C), while HLF ectopic expression increased the expression of TIC markers and stemness-related transcription factors (online supplementary figure S5D-F). Flow cytometry analysis also detected a reduced proportion of EpCAM+ cells in HLF knockdown cells (figure 5D) and an induced proportion of EpCAM+ cells in HLF overexpressing cells (online supplementary figure S5G). Additionally, the self-renewal capacity was attenuated in HLF knockdown cells (online supplementary figure S5H) and was enhanced in HLF overexpressing cells (online supplementary figure S5I). Furthermore, in vitro limited dilution assays revealed that ectopic HLF expression enhanced the TIC-like properties and facilitated TIC expansion in hepatoma cells (online supplementary figure S5J). Next, we conducted ‘loss of function’ and ‘gain of function’ studies to investigate the role of HLF-enhanced TIC-like properties of hepatoma cells in HCC progression. As expected, the interference of HLF suppressed hepatoma cell proliferation and xenografted tumour growth (figure 5E,F, online supplementary figure S5K,L), while ectopic HLF expression promoted the proliferation and cell cycle transition of hepatoma cells (online supplementary figure S5M-O). HLF depletion inhibited the migration and invasion of hepatoma cells (figure 5G,H), whereas forced HLF expression enhanced hepatoma cell migration and invasion (online supplementary figure S5P,Q). Moreover, the lung metastasis in the nude mice inoculated with HLF-silenced hepatoma cells was much less than that in mice inoculated with control cells (figure 5I). Collectively, these data suggest that HLF enhances the TIC-like properties of hepatoma cells and thus facilitating HCC progression.

HLF drives HCC progression via enhancing TIC-like properties. (A) The expression of HLF in hepatoma spheroids and attached cells was examined by real-time PCR. (B) HLF expression in attached cells, spheroids and reattached hepatoma cells was compared by real-time PCR. (C) The expression of HLF in CD24+ or OV6+ HCCLM3 cells and their control cells was examined by real-time PCR. (D) Flow cytometry analysis of the EpCAM+ cell population in shHLF and control hepatoma cells. (E) The proliferation of shHLF or Control hepatoma cells was measured by CCK8 assay. (F) HCCLM3 shHLF or control cells (2×106) were injected subcutaneously into NOD-SCID mice (n=6). Xenografted tumour growth was monitored and tumour weight was measured 5 weeks later. (G&H) The migration and invasion of hepatoma cells were examined with the transwell assay and invasion chamber assay, respectively. Representative images are shown. The cell counts were expressed as the mean number of cells per field of view. (I) Representative images of lung metastatic foci indicated by H&E staining from nude mice inoculated with HCCLM3 shHLF or control cells via tail vein for 12 weeks. The number of lung metastatic foci in each group (n=6) was also calculated. HCC, hepatocellular carcinoma; HLF, hepatic leukaemia factor; TIC, tumour initiating cell.
c-Jun mediates the effects of HLF on HCC progression
Since that HLF could transactivate c-Jun to promote tumor-initiation, we speculated that c-Jun could also be implicated in HLF-induced HCC progression. As expected, the interference of HLF expression suppressed the mRNA and protein expression as well as the nuclear accumulation of c-Jun in hepatoma cells (figure 6A-C). The phosphorylation and transactivation activities of c-Jun were also attenuated by HLF interference (figure 6B,D). Accordingly, ectopic HLF expression increased the mRNA and protein levels of c-Jun in a dose-dependent manner (online supplementary figure S6A,B) and enhanced the activation of c-Jun in hepatoma cells (online supplementary figure S6C). Consistently, decreased c-Jun expression was detected in xenografts formed by HLF-interfered cells compared with that in xenografts formed by control cells (figure 6E), and a close correlation between HLF levels and c-Jun expression was observed in patient HCCs (figure 6F). In further study, we found that the activation of c-Jun promoter was suppressed by HLF knockdown and enhanced by HLF ectopic expression (figure 6G and online supplementary figure S6D). Significant enrichment of HLF in the promoter region of c-Jun was detected by ChIP assay (online supplementary figure S6E). In addition, the mutation of HLF binding site within c-Jun promoter region diminished the distinct activation of c-Jun promoter between HLF knockdown (online supplementary figure S6F) or HLF overexpressing cells (online supplementary figure S6G) and control cells. More importantly, the enhanced expression of TIC- or stemness-related genes triggered by ectopic HLF expression was abolished by c-Jun depletion (online supplementary figure S6H,I). Consistently, the interference of c-Jun attenuated the increase of EpCAM+ cell populations induced by ectopic HLF expression (figure 6H,I). The enhanced self-renewal of HLF overexpressing cells was abrogated by c-Jun depletion (online supplementary figure S6J). Moreover, the interference of c-Jun eliminated the discrepancy of proliferation and invasion between HLF overexpressing cells and control cells (figure 6J-L). Taken together, these data clarify that c-Jun is required for HLF-enhanced TIC-like properties and HCC progression.

HLF activates c-Jun to promote HCC progression. (A) Real-time PCR analysis of the mRNA expression of HLF and c-Jun in shHLF or Control hepatoma cells. (B) Western-blot analysis of c-Jun activation in shHLF or Control hepatoma cells. (C) Representative images of dual immunofluorescence staining of HLF and c-Jun in siHLF or Control hepatoma cells. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Scale bar=20 µm. (D) The luciferase reporter activity of AP-1 was measured in shHLF or Control hepatoma cells. (E) Representative images of H & E and IHC staining of HLF and c-Jun in xenografted tumours formed by HCCLM3 shHLF or Control hepatoma cells. Scale bar=25 µm. (F) The correlation between HLF levels and c-Jun expression in hepatomas was evaluated in a group of 62 HCC specimens using Pearson’s correlation analysis. (G) The luciferase reporter activity of c-Jun promoter was measured in shHLF or Control hepatoma cells. The hepatoma cells infected with Ad-HLF or Ad-Con were transfected with sic-Jun or siNC (H–L). The EpCAM+ populations were analysed in HCCLM3 cells and Huh7 cells, respectively, by flow cytometry (H&I). Cell proliferation was measured by the CCK8 assay (J). Invasion was examined with the Invasion chamber assay (K&L). HCC, hepatocellular carcinoma; HLF, hepatic leukaemia factor; IHC, immunohistochemistry.
HLF-c-Jun axis determines sorafenib response in HCC
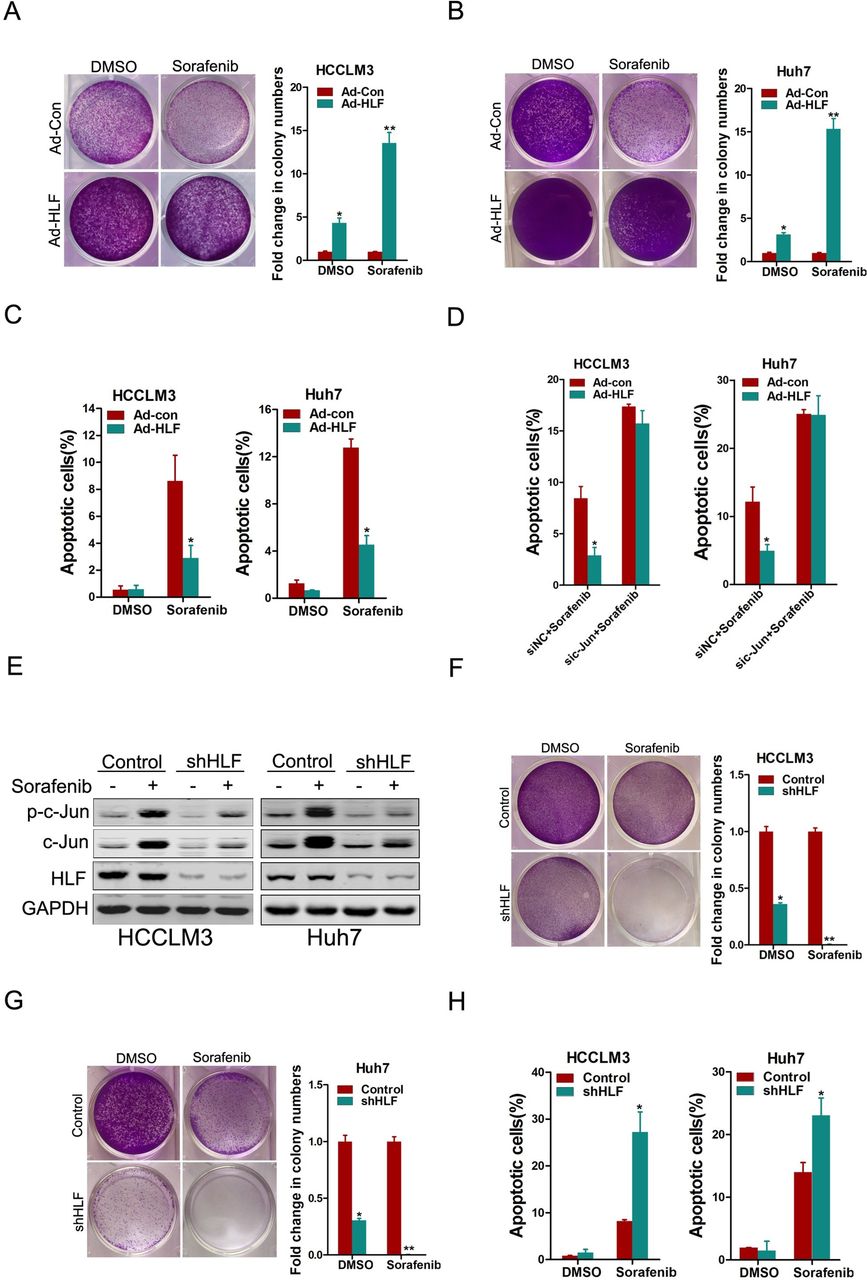
The TIC properties closely correlate with the resistance of cancer to pharmacotherapy.17 Considering that HLF can promote the TIC-like properties of hepatoma cells, we next investigated the correlation between HLF expression and sorafenib response in hepatoma cells. As shown in figure 7A-C, HLF overexpression led to the resistance of hepatoma cells to sorafenib-induced growth inhibition and cell apoptosis. As expected, phosphorylation of ERK was notably inhibited by sorafenib in hepatoma cells (online supplementary figure S7A). Interestingly, our data revealed the notable activation of c-Jun in sorafenib-treated hepatoma cells (online supplementary figure S7A), suggesting that c-Jun activation might be involved in sorafenib resistance. Next, the expression of c-Jun was interfered in hepatoma cells, and the results showed that knockdown of c-Jun remarkably increased the sensitivity of hepatoma cells to sorafenib-induced apoptosis (online supplementary figure S7B,C). As expected, the interference of c-Jun abolished sorafenib resistance in HLF overexpressing hepatoma cells (figure 7D). Moreover, the interference of HLF attenuated sorafenib-induced c-Jun activation (figure 7E) and sensitised hepatoma cells to sorafenib-induced growth inhibition and cell apoptosis (figure 7F-H). Together, these data suggest that HLF-mediated c-Jun activation is required for sorafenib resistance and imply that HLF-c-Jun axis might determine sorafenib response in hepatoma cells.

The HLF/c-Jun axis regulates sorafenib response in hepatoma cells. (A&B) The hepatoma cells infected with Ad-HLF or Ad-Con were treated with sorafenib (2 µM) for 7 days and their colony growth was examined. (C) The hepatoma cells infected with Ad-HLF or Ad-Con were treated with sorafenib (10 µM) for 48 hours and their apoptosis was examined by flow cytometry. (D) The hepatoma cells infected with Ad-HLF or Ad-Con were transfected with sic-Jun or siNC. The cells were then treated with sorafenib (10 µM) for 48 hours and their apoptosis was examined by flow cytometry. (E) shHLF and Control hepatoma cells treated with sorafenib (10 µM) for 48 hours were subjected to western-blot analysis. (F&G) shHLF and Control hepatoma cells treated with sorafenib (2 µM) for 7 days were subjected to the colony growth assay. (H) shHLF and Control hepatoma cells treated with sorafenib (10 µM) for 48 hours were subjected to flow cytometry analysis. HLF, hepatic leukaemia factor.
The combination of HLF and c-Jun is associated with sorafenib benefit in patients
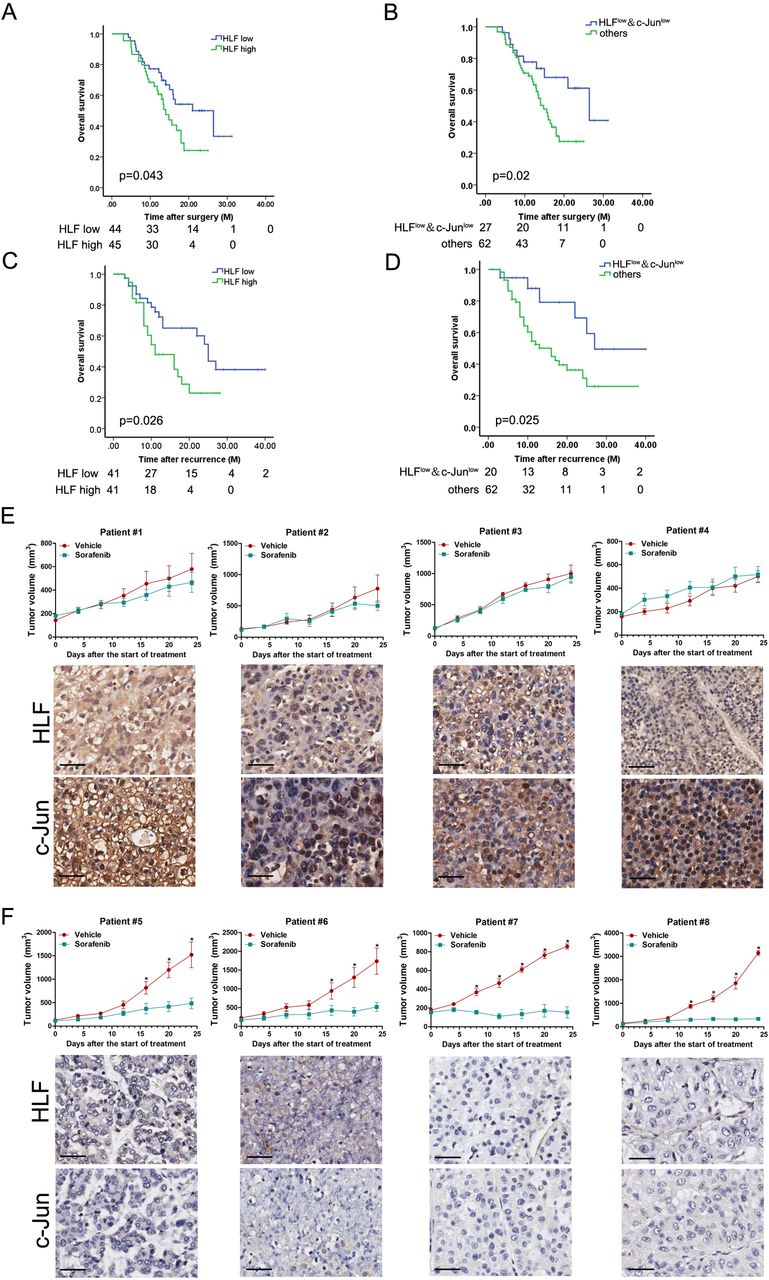
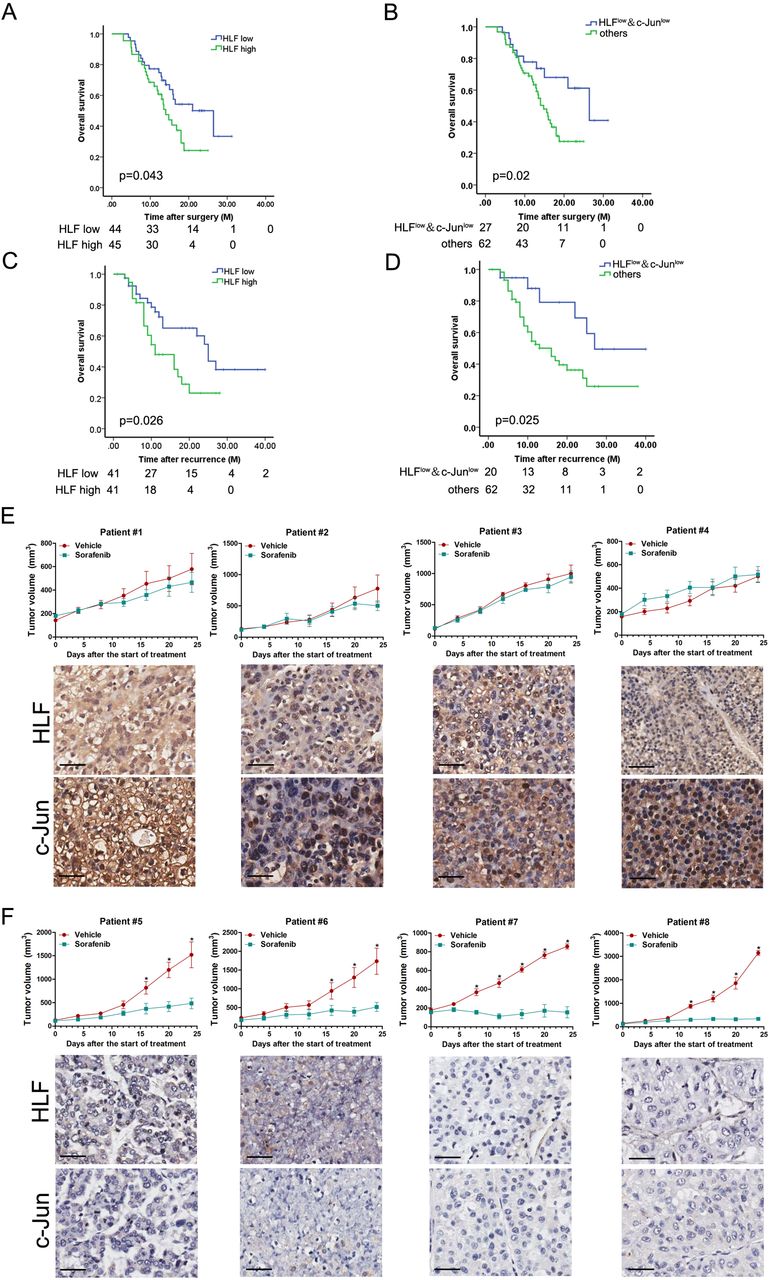
To assess the clinical significance of HLF/c-Jun axis in sorafenib therapy, we examined the expression of HLF and c-Jun in HCCs from a cohort consisting of 89 postoperative patients who had received adjuvant sorafenib treatment (cohort 3). The expression of HLF and c-Jun was then quantified (online supplementary table 3). Kaplan-Meier analysis revealed the survival benefits in adjuvant sorafenib-treated HCC patients with low HLF or low c-Jun levels (figure 8A and online supplementary figure S8A). Of note, the combination of low HLF and low c-Jun correlated with a better OS of patients who have received sorafenib therapy (figure 8B). We also investigated the expression of HLF and c-Jun in the primary tumours from another cohort of patients who had received sorafenib after HCC relapse (cohort 4, online supplementary table 4). Kaplan-Meier analysis indicated that low HLF or low c-Jun levels in the primary HCCs were significantly associated with prolonged OS in patients received sorafenib to treat their recurrent tumours (figure 8C and online supplementary figure S8B). Moreover, a better OS was further observed in recurrent patients with both low HLF and low c-Jun levels in their primary tumours (figure 8D). Next, by virtue of PDXs, we found that the PDXs derived from tumours with high HLF and high c-Jun levels were resistant to sorafenib treatment (figure 8E). In contrast, compared with the vehicle control, sorafenib almost blocked the growth of PDXs derived from tumours with low HLF and low c-Jun levels (figure 8F), indicating that combination of HLF and c-Jun in patient HCCs might serve as a biomarker for sorafenib response.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The association of HLF/c-Jun axis and sorafenib benefits in patients. (A) IHC staining and scoring of HLF expression was performed in HCC samples from 89 patients who had received adjuvant sorafenib therapy after surgery (cohort 3). The overall survival (OS) of patients between HLF-high or HLF-low groups was evaluated by Kaplan-Meier analysis. (B) OS was compared between the patients exhibiting low HLF and low c-Jun levels with the other patients in (A) using Kaplan-Meier analysis. (C) IHC staining and scoring of HLF expression was performed in the primary HCC samples from 82 patients who received sorafenib for recurrent tumours (cohort 4). OS of patients between HLF-high or HLF-low groups was evaluated by Kaplan-Meier analysis. (D) OS was compared between the patients exhibiting both low HLF and low c-Jun levels and the other patients in (C) using Kaplan-Meier analysis. (E) PDXs with both high HLF and high c-Jun levels in their primary tumours indicated by IHC staining were treated with sorafenib (60 mg/kg body weight) or vehicle for 24 days (n=4 for each group). The xenograft growth was monitored. The data are expressed as the mean ±SEM. (F) PDXs with both low HLF and low c-Jun levels in their primary tumours indicated by IHC staining were treated with sorafenib (60 mg/kg body weight) or vehicle for 24 days (n=4 for each group). The xenograft growth was monitored. The data are expressed as the mean ±SEM. HCC, hepatocellular carcinoma; HLF, hepatic leukaemia factor; IHC, immunohistochemistry; PDXs, patient-derived xenografts.
Discussion
Despite the recent progress in HCC prevention and intervention, increasing prevalence and grim prognosis make this intractable disease a global health challenge, which reminds us of our deficient understanding about the basic nature of this disease.18 Therefore, more effort is required to illuminate the in-depth mechanism driving the genesis and progression of HCC. In this study, for the first time, we clarify that HLF is a novel oncofetal protein that determines the onset and progression of HCC. Our clinical investigations revealed that HLF levels are associated with the outcome of HCC patients and also correlate with sorafenib benefit in patients.
Due to the unique expression patterns, oncofetal genes are probably of great value in cancer diagnosis and treatment, which have attracted considerable attention.5 In terms of HCC, the most known oncofetal gene is AFP, which was identified more than half a century ago.19 AFP has been used as a serum diagnostic marker for HCC since its initial discovery, but the function of AFP is still unclear. In recent years, other oncofetal genes, including GPC3 and SALL4, have been discovered,6 20 but the diagnostic or prognostic values of these genes are yet to be determined. Furthermore, whether these genes drive carcinogenesis and cancer progression, and more importantly, whether they are functionally essential targets remain unclear. In this study, we discovered that HLF is an oncofetal protein and also illustrated the pivotal roles of HLF in HCC initiation and progression, suggesting that HLF is a crucial onco-driver in HCC. Although HLF protein was undetectable in normal liver, the mRNA expression of HLF is low but detectable,13 which is similar with SALL4 and GPC3 (online supplementary figure S9).6 21 The divergence between HLF mRNA and protein in normal liver might be due to the complicated post-transcriptional regulation, which warrants further investigation. In addition, our clinical investigations revealed the close correlation between HLF levels with HCC progression and patient prognosis. Moreover, as an oncofetal protein, HLF is not expressed in normal hepatocytes but is rather required for tumour initiation and progression, thus making it an optimal target for HCC therapy.
Recent advances in genomic research of HCC have brought novel perceptions about hepatocarcinogenesis, and a series of oncogenes with DNA alterations have been revealed.22 However, in this study, we have not observed significant alterations of HLF DNA in HCCs. Likewise, remarkable DNA alterations were neither detected in other oncofetal genes, including AFP, GPC3 and SALL4, which are reactivated in HCCs (online supplementary figure S10).15 16 Recent studies suggest that the reactivation of certain oncofetal genes is regulated epigenetically.4 11 Nonetheless, our data revealed that the reactivation of HLF in HCC is not controlled epigenetically but is regulated transcriptionally by the stemness-associated genes SOX2 and OCT4, which also play essential roles in fetal liver development. Our findings represent a novel paradigm of oncofetal gene reactivation and broaden the knowledge on the mechanism of hepatocarcinogenesis.
c-Jun belongs to the AP-1 transcription factor family, and the homodimer or heterodimer of c-Jun is the predominant active isoform for AP-1. Activation of AP-1 was reported to be regulated by mitogenic signalling,23 but the exact mechanism beneath c-Jun activation in HCC remains vague. Previous studies have elucidated that c-Jun plays essential roles in hepatocyte survival and liver regeneration and also controls tumour initiation in hepatocarcinogenesis.24–28 In addition, c-Jun was reported to be involved in the modulation of TICs or CSCs.29–31 In these studies, the basal level of c-Jun protein was rather low and even absent in normal liver samples. In present study, we also found that c-Jun protein was extremely low or undetectable in normal liver or mouse primary hepatocytes (figure 4B,D and online supplementary figure S4F). Moreover, we revealed that HLF transactivates c-Jun to promote TIC generation and enhance TIC-like properties of hepatoma cells, thus driving the initiation and progression of liver cancer. Consistently, our clinical investigations indicated a close correlation between HLF levels and c-Jun expression in patient HCCs. These discoveries not only provided a novel mechanism of c-Jun activation and also demonstrated the crucial role of HLF/c-Jun axis in HCC. Considering the important role of HLF/c-Jun axis in HCC, we believe that targeting the HLF/c-Jun axis could be a novel therapeutic strategy for HCC.
Sorafenib, a receptor tyrosine kinase inhibitor, is the first-line targeted drug for the patients with advanced HCC, but the patients who have received sorafenib only exhibited limited survival improvement.1 Therefore, increasing studies have concentrated on the quest for biomarkers of sorafenib response and patient outcome.32–34 In the current study, we found that the activation of c-Jun could mediate sorafenib resistance in hepatoma cells. More importantly, our data demonstrated that the interference of HLF abrogated c-Jun activation and enhanced sorafenib response in hepatoma cells, which suggests that the HLF/c-Jun axis determines sorafenib response. Therefore, targeting HLF/c-Jun signalling could be an optimal combinational therapeutic strategy to overcome sorafenib resistance in a subset of HCCs. Sorafenib cohort and PDX studies further demonstrated that low HLF and c-Jun levels are associated with a better response and survival in patients who have received sorafenib treatment. These data suggest that the HLF/c-Jun axis may act as a biomarker for sorafenib benefit in HCC patients, thus facilitating patient selection in the precision therapy of sorafenib, which merits further investigation in biomarker-guided clinical trials.
References
Footnotes
D-MX, WS and TZ contributed equally.
Contributors D-MX and TFZ conducted all experiments and analysed the data. HL, RYW, HYL, GJH and CYH provided clinical samples. G-ZJ provided pathology evaluation and D-MX analysed clinical data. ZC, S-CL, XLC, WQJ and GF provided support with experimental techniques. WS wrote the manuscript and JD contributed to the revision. JD and HYW conceived the project and supervised all experiments.
Funding This work was supported by grants from the State Key Project of China (2017YFA0504503), the National Natural Science Foundation of China 81572897, 81770602 and 81702736, Program of Shanghai Municipal Commission of Health (20174Y0144), Shanghai Rising-Star Program (18QA1405300) and Program of Shanghai Academic Research Leader (18XD1405400).
Competing interests None declared.
Ethics approval The procedure of patient specimen collection was approved by the Ethics Committee of Eastern Hepatobiliary Surgery Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.