Article Text

Abstract
Chronic hepatitis B virus (HBV) infection remains a major health burden and the main risk factor for the development of hepatocellular carcinoma worldwide. However, HBV is not directly cytopathic and liver injury appears to be mostly caused by repeated attempts of the host's immune responses to control the infection. Recent studies have shown that the unique replication strategy adopted by HBV enables it to survive within the infected hepatocyte while complex virus–host interplays ensure the virus is able to fulfil its replication requirements yet is still able to evade important host antiviral innate immune responses. Clearer understanding of the host and viral mechanisms affecting HBV replication and persistence is necessary to design more effective therapeutic strategies aimed at improving the management of patients with chronic HBV infection to eventually achieve viral eradication. This article focuses on summarising the current knowledge of factors influencing the course of HBV infection, giving emphasis on the use of novel assays and quantitative serological and intrahepatic biomarkers as tools for predicting treatment response and disease progression.
- Hepatitis D
- hepatocyte
- interferon α
- immunology in hepatology
- hepatitis B
- infectious disease
- immunology in hepatology
- hepatitis B
- hepatitis C
Statistics from Altmetric.com
- Hepatitis D
- hepatocyte
- interferon α
- immunology in hepatology
- hepatitis B
- infectious disease
- immunology in hepatology
- hepatitis B
- hepatitis C
Introduction
The human hepatitis B virus (HBV) is a small enveloped DNA virus causing acute and chronic hepatitis. Despite the availability of an effective vaccine, more than 360 million people are chronically infected worldwide and about 1 million people die per year due to HBV-associated liver pathologies.1 Although HBV replication is not considered directly cytopathic, HBV infection leads to a wide spectrum of liver disease ranging from acute to chronic viral hepatitis, which often progresses to liver cirrhosis and development of hepatocellular carcinoma (HCC). Even though numerous epidemiological studies indicated that chronic HBV (CHB) infection represents the main risk factor for liver cancer development,2–4 the molecular mechanisms determining HBV persistence and pathogenesis are still poorly defined. HBV displays a unique genomic organisation and replication strategy which confers on the virus the ability to persist in infected hepatocytes. A hallmark of HBV infection is the formation in hepatocyte nuclei of a stable HBV-DNA minichromosome, the so-called covalently closed circular DNA (cccDNA), which serves as template to generate all RNAs necessary for protein production and viral replication. Even if it is known that failure of viral clearance and relapse of viral activity after cessation of antiviral therapy with polymerase inhibitors are mostly due to the persistence of the cccDNA in chronically infected individuals, the virological and immunological mechanisms that prevent virus eradication leading to the development of chronic infection are still poorly understood. The development of innovative experimental infection models and quantitative serological and intrahepatic biomarkers is starting to provide new insight into the mechanisms of HBV persistence and pathogenesis.
HBV virology
HBV is a member of the Hepadnaviridae family, which are the smallest DNA-containing, enveloped animal viruses known so far. Characteristic of HBV is its high tissue and species specificity, and a unique genomic organisation and replication mechanism. Despite decades of research, essential steps of the viral life cycle, such as viral entry and organisation of the cccDNA minichromosome, are still poorly understood.5 Only recently, innovative infection models and molecular techniques have opened new possibilities to investigate specific steps of the lifecycle and virus–host interactions influencing viral activity in infected hepatocytes.6
Infectious HBV has a spherical structure of 42–44 nm and the hepatitis B surface antigen (HBsAg) envelops the viral nucleocapsid, which is formed by the core protein (HBcAg). The encapsidated viral genome is typically organised as a relaxed circular partially double-stranded DNA (rcDNA) of around 3200 bp, covalently linked to the terminal protein of the viral polymerase (figure 1). The HBV genome displays four overlapping open reading frames (ORFs)1: the preS/S encoding the three viral surface proteins; the precore (PC)/core encoding the core protein and the non structural PC protein, also known as secreted e-antigen (HBeAg); the pol ORF encoding the viral polymerase, which possesses reverse transcriptase, DNA polymerase and RNase H activities, and the terminal protein for priming; and the X ORF, encoding the small regulatory X protein, which is essential in vivo for viral replication.7 8 All four ORFs use a single common polyadenylation signal motif; hence, the HBV-RNA transcripts are polyadenylated and capped.9

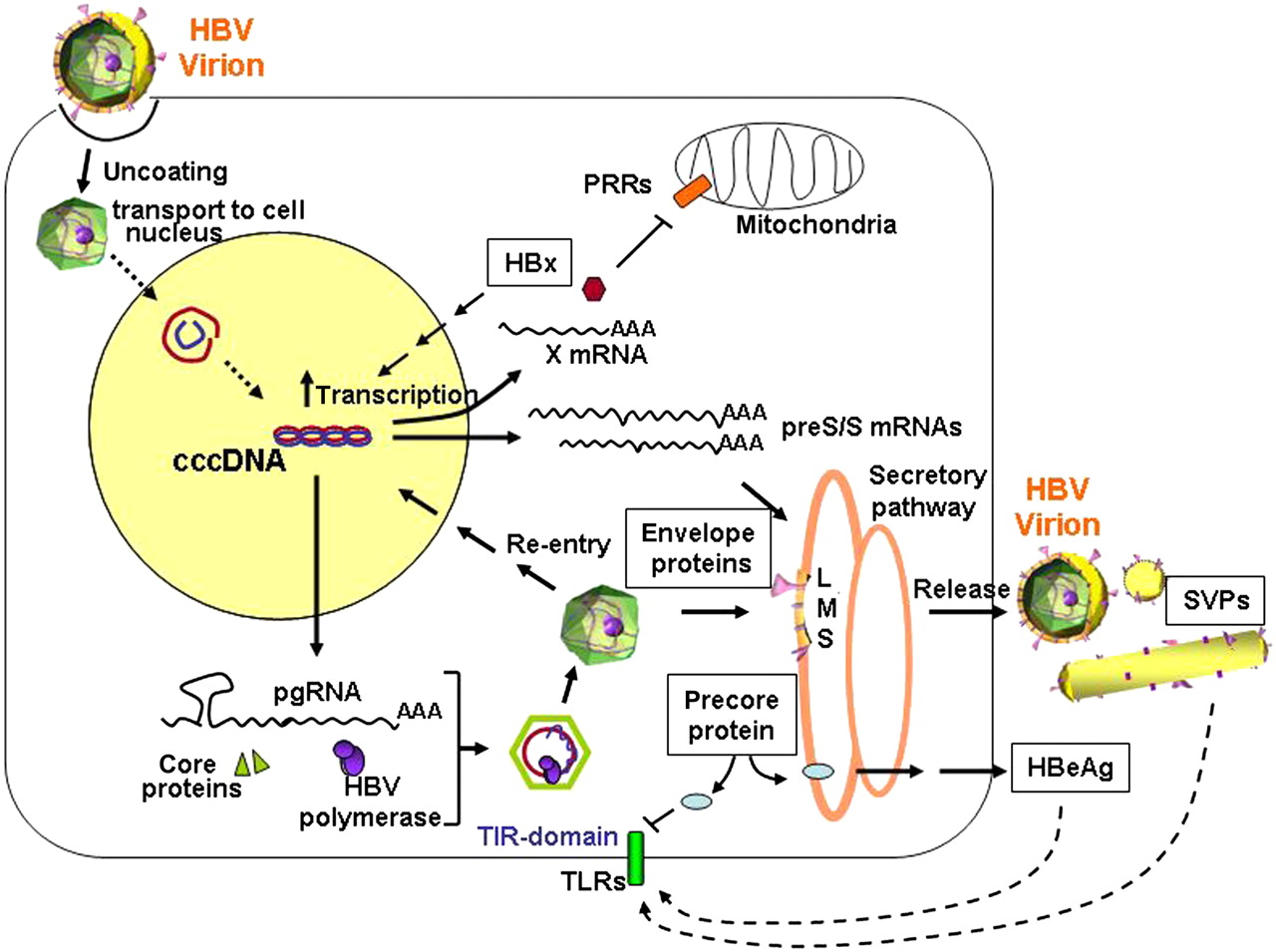
The lifecycle of hepatitis B virus (HBV). After binding to still unknown hepatocyte-specific receptor(s), the nucleocapsid is released into the cytoplasm and the HBV genome transferred to the cell nucleus, where it is converted into the covalently closed circular DNA (cccDNA) minichromosome. Viral replication takes place in the cytoplasm after encapsidation and reverse transcription of a over-length pregenomic RNA (pgRNA). Through Golgi and endoplasmic reticulum apparatus the core particles acquire the envelope and are secreted. Via viral entry and re-transporting of newly synthesised relaxed circular partially double-stranded DNA (rcDNA) into the cell nucleus, the cccDNA pool can be amplified. Transcription of the preS/S mRNAs leads to the production of L, M, S envelope proteins, which are needed for virion secretion and to produce the non-infectious spherical and filamentous subviral particles (SVPs). The regulatory HBx protein, which can affect gene expression and various cellular pathways, is translated from a shorter X mRNA and is essential for cccDNA-driven HBV replication. The secreted e-antigen (HBeAg) is a non-structural form of the nucleoprotein which, like the SVPs, was shown to display immunomodulating functions (see also figure 3). TIR, toll-interleukin-1 receptor; TLR, toll-like receptor; PPR, pattern recognition receptor.
All three envelope proteins, large (L), middle (M) and small (S) share the same C-terminal domain, which contains the HBsAg, while the L and M proteins display progressive N-terminal extensions.5 The anti-HBs neutralisation domain has been mapped to amino acids 99–170 of the S-protein and is referred to as the α or ‘a’ determinant.10 Characteristic of HBV infection is the presence of non-infectious structures, the spherical and filamentous subviral particles (SVPs), which are exclusively composed of viral envelope proteins and host-derived lipids5 and are secreted in a 103–106-fold excess into the blood of infected individuals (figure 1).
HBV also produces and secretes a non-structural form of the nucleoprotein, the PC protein or HBeAg, which is not required for viral replication, but displays immune-modulating functions and hence contributes to viral persistence. The HBeAg is translated initially as the PC protein P25, and then undergoes cleavage of the N-terminal signal sequence in the ER/Golgi complex, producing a 22 kDa protein (P22), which can either undergo further processing to form the secreted 17 kDa HBeAg (P17), or traffic to the cytosol where it remains localised.11 Except for a 10-amino-acid N-terminal extension, which is highly conserved in all orthohepadnaviruses and contains a putative toll-interleukin-1 receptor (TIR) domain,12 HBeAg shares significant homology with the core protein (P21), which is translated using the core AUG codon of the same ORF.13
The first step in HBV infection involves a non-cell-type specific primary attachment which is then followed by an irreversible binding of the virus to a still unknown hepatocyte-specific receptor.5 14 Following viral entry, the capsids accumulate at the nuclear membrane, where interactions with the nuclear pore complex may favour the release of the HBV genome (rcDNA) into the cell nucleus.15 To establish a productive HBV infection host components of the cellular replicative machinery are needed to remove the covalently attached terminal protein and to complete the positive-DNA strand to form a super-coiled cccDNA molecule, which after being associated with histone and non-histone proteins, is incorporated into the host chromatin.6 9 16 Unlike the provirus DNA of retroviruses, the cccDNA does not need to be integrated into the host genome. Albeit integrations of HBV-DNA sequences do occur in infected hepatocytes, particularly in the presence of DNA damage17 and cell turnover,18 19 these are typically truncated.
Camouflaged as a minichromosome, and hence undetectable by innate immune defense mechanisms, the cccDNA utilises the cellular transcriptional machinery to produce all transcripts necessary for protein production and viral replication, which takes place in the cytoplasm after reverse transcription (rt) of a over-length pregenomic RNA (pgRNA).9 Viral transcription is under the control of two enhancer elements and four distinct viral promoters. Similarly to cellular chromatin, the transcription of the different viral genes is regulated by the activity and dynamic interplay of numerous cellular transcription factors, coactivators, corepressors and chromatin modifying enzymes.6
Experimental studies in the duck model indicated that the cccDNA can be formed from incoming virions and from newly synthesised nucleocapsids, which instead of being enveloped and secreted into the blood, are rather transported into the nucleus to ensure accumulation, and later maintenance, of the cccDNA pool9 20 (see figure 1).
Although a high cccDNA copy number is detected in chronically infected ducks and woodchucks,21 22 lower cccDNA intrahepatic loads are generally determined in human liver biopsies (median 0.1–1 cccDNA copy/cell)23–28 and in HBV chronically infected human liver chimeric uPA/severe combined immunodeficient (SCID) mice,29–32 suggesting that different viral and host mechanisms may control cccDNA dynamics and cccDNA pool size in human infected hepatocytes.6 In this regard, a recent study elegantly showed that HBV converted the rcDNA into cccDNA less efficiently than DHBV in the same human cell background.33 Although the cccDNA pool appears to be very stable in the absence of cell division, accumulating evidence indicates that hepatocyte proliferation can induce drastic reduction of the intrahepatic cccDNA loads in vivo.19 32 Since the cccDNA lacks centromere structures ensuring correct migration during mitosis, immune-mediated cell death and compensatory hepatocyte regeneration may facilitate cccDNA loss. This could represent a weak point in HBV persistence deserving further investigation as new therapeutic concepts are explored.32
The next crucial step in HBV replication is the packaging and rt of pgRNA within the newly formed capsids. After rt and concomitant degradation of the pgRNA, first a single-stranded DNA of minus polarity, whose 5′ end remains covalently linked to the terminal protein of the polymerase, and then a complementary plus-strand DNA are synthesised to form the rcDNA.9 34 Assembly and release of the DNA-containing nucleocapsids appears to require a balanced co-expression of the small and large envelope proteins to recruit the nucleocapsid to the site of budding. Although the role of the envelope proteins in regulating viral particle release and cccDNA amplification is not well understood, recent studies indicate that the lack of envelope protein expression increases cccDNA levels, while co-expression of the envelope proteins favours viral secretion with limited completion of the plus strand.35
Phases of CHB
The natural history of CHB
The immunopathogenesis of hepatitis B depends on a complex interplay of host and viral factors. Factors such as age, gender and immune status are important. Perinatal and childhood infection results in chronic infection in 90–95% and 50% of cases respectively, whereas greater than 95% of immunocompetent adults with acute HBV spontaneously clear the infection. Furthermore, CHB infection is more common in immunocompromised hosts (eg, HIV infection). Viral factors such as HBeAg status are also important. Under normal circumstances viral replication is not cytopathic, and it is the host's immune response, which is typically ineffective and inappropriate, that causes much of the damage associated with CHB.
The natural history of CHB is generally regarded as consisting of four phases36: immune tolerant; HBeAg-positive CHB (immune clearance); immune control (low or non-replicative); and HBeAg negative CHB (immune escape). These phases have been identified on the basis of specific biochemical, serological and virological characteristics, including serum alanine aminotransferase (ALT) levels, HBeAg serostatus, HBV DNA titre, and more recently, HBsAg level (figure 2). It is important to note that these phases do not occur in all individuals with CHB and do not always occur sequentially.37 It is patients in either the immune clearance or immune escape phases that are potential candidates for the currently approved antiviral and immune modulatory treatments.

Phases of chronic hepatitis B (CHB) infection: serum and liver compartment. ALT, alanine aminotransferase; cccDNA, covalently closed circular DNA; HBeAg, hepatitis B secreted e-antigen; HbsAg, hepatitis B surface antigen; HBV, hepatitis B virus; ORF, open reading frame; PC/C, precore/core; PE, Paul Ehrlich Institute; rcDNA, relaxed circular partially double-stranded DNA.
The immune tolerant phase is characterised by positivity for serum HBeAg, high levels of HBV DNA, normal serum ALT levels and normal/near normal histological profile in the liver. The levels of HBV DNA typically exceed 20 000 IU/ml while HBsAg levels range from 4.5 to 5.0 log IU/ml.38 39 Patients in the immune tolerant phase are usually young and were infected early in life. The immune tolerant phase can last for more than 20 years. In contrast, individuals progressing to chronic infection following adulthood infection do not typically undergo a prolonged immune tolerant phase, and may instead enter directly into the immune clearance phase.
When CHB infection is acquired from an early age, the immune clearance phase usually occurs between ages 20 and 40 years, and is characterised by HBeAg positivity, elevated serum ALT levels, fluctuating HBV DNA levels (>20 000 IU/ml), lower levels of HBsAg (approx 4.3 log IU/ml) and histological damage. During this phase, the virus is under intense immune pressure and the emergence of dominant PC and/or basal core promoter (BCP) variants may occur, with eventual HBeAg seroconversion (SC) and control of HBV replication. Alternatively, HBV escapes from immune clearance with the person then developing HBeAg-negative CHB (figure 3). Following successful HBeAg SC, the majority of patients enter an immune control or low/non-replicative phase, characterised by low/undetectable viral replication and normal ALT levels. A serum HBV DNA of <2000 IU/ml and a quantitative HBsAg level of <1000 IU/ml have been suggested to differentiate the immune control and escape phases.40 41 A subset of patients can enter into the HBeAg-negative hepatitis phase, whereby HBV DNA levels >2000 IU/ml and increased quantitative HBsAg (3.5 log IU/ml) are found with increasing serum ALT levels and consequent further histological damage (figure 2).
{kind=link}
{kind=link}
{kind=link}
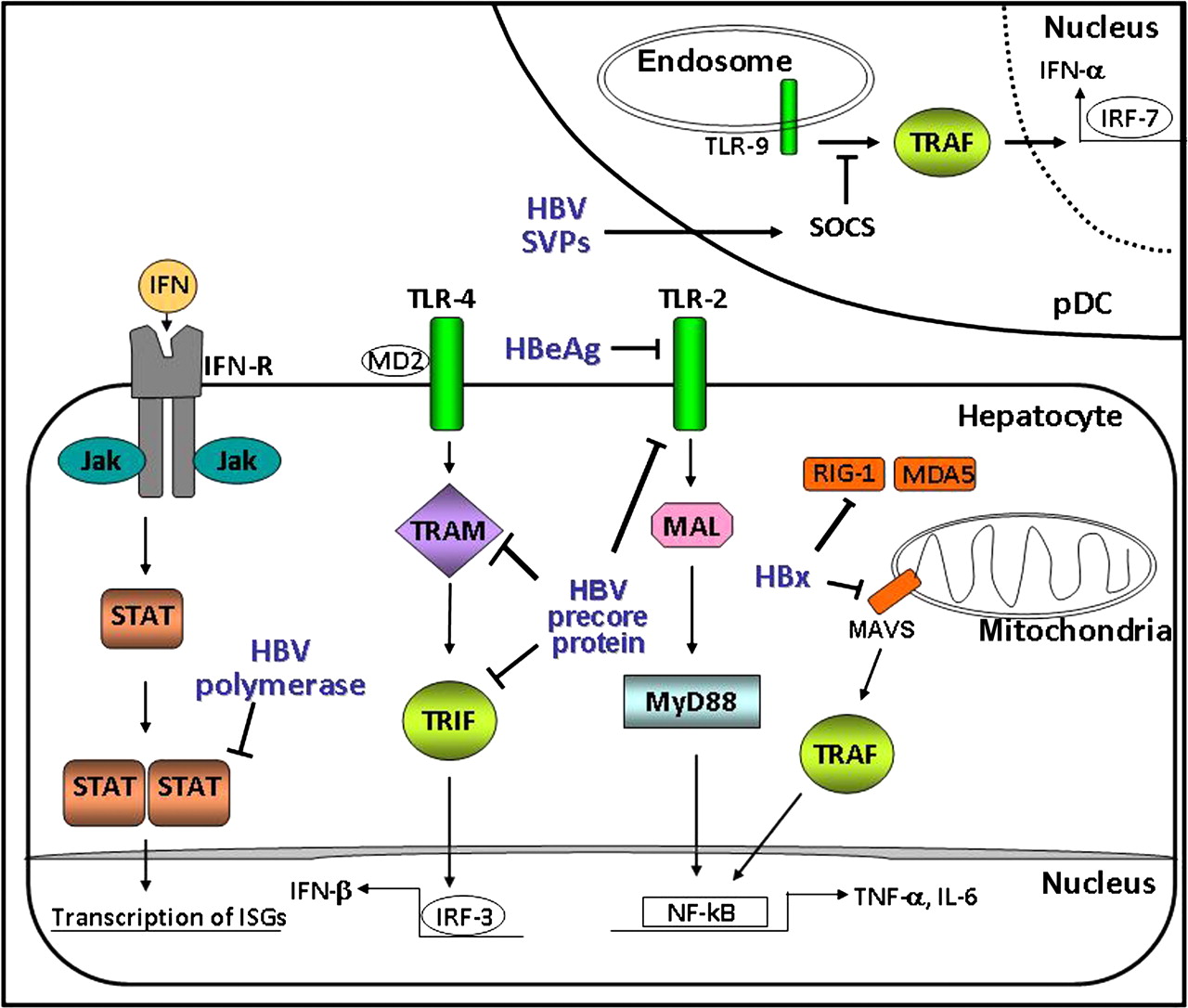
Proposed interactions between hepatitis B virus (HBV) proteins and components of the innate immune system. The diagram shows the intracellular effects of the precore/hepatitis B secreted e-antigen (HBeAg) on hepatocyte signalling via the MyD88-dependent (toll-like receptor (TLR)-2) and MyD88-independent (TLR-4) pathways. The terminal protein of the HBV polymerase was also reported to hinder the type I interferon (IFN)-mediated transcription of IFN-stimulated genes by impairing nuclear translocation of signal transducer and activator of transcription (STAT) proteins, while HBx expression was shown to inhibit the retinoic acid inducible gene I (RIG-I)–MAVS signalling. Circulating subviral particles (SVPs) were shown to repress TLR-9 signalling by enhancing SOCS expression in dendritic cells (pDCs). Furthermore, secreted HBeAg was shown to interact and downregulate TLR-2 expression on hepatocytes. In HBeAg-negative chronic hepatitis B (CHB), the absence of HBeAg results in upregulation of TLR-2 and secretion of the proinflammatory cytokines, including tumour necrosis factor α (TNFα). IRF, interferon regulatory factor; ISG, interferon-stimulated gene; MDA5, melanoma differentiation associated gene 5; NF-κB, nuclear factor κB; TRAM, TRIF-related adaptor molecule; MAVS, mitochondrial antiviral signaling; TRIF, TIR-domain containing adaptor protein inducing interferon–β; TRAF, TNF receptor-associated factor MAL; MyD88-adapter-like protein.
It is generally accepted that the extent of chronic liver disease is directly linked to the frequency, duration and severity of hepatic flares occurring in either the HBeAg-positive (immune clearance) and/or HBeAg-negative phases. HBeAg SC is predictive of a sustained multi-log reduction in HBV DNA (>3 log10 IU/ml), a decrease in intrahepatic inflammatory activity and an improved prognosis.42 For these reasons, HBeAg SC has been traditionally used as an important therapeutic endpoint in the management of HBeAg-positive CHB. Not surprisingly, then, delayed HBeAg SC, particularly in patients >40 years old, is associated with higher fibrosis scores on liver biopsy. HBsAg SC is associated with a favourable long-term clinical prognosis,43 but unfortunately, spontaneous HBsAg loss and/or SC, now regarded as an important immune control endpoint, is rare in CHB with approximately a 1% annual rate.44
Spontaneous HBV reactivation in the low/non-replicative phase is clinically significant, since this is typically followed by increased hepatitis activity and ALT flares.45 The annual incidence of hepatic flare has been reported to be over 10% in HBeAg-negative patients.45 These hepatic flares are due to the resurgence of host immunity against HBV-infected hepatocytes and severe hepatic flares can be complicated by hepatic decompensation in 2–3% of patients. These adverse events typically drive the development of advanced fibrosis, which in time, eventually leads to cirrhosis.
Major virological events during the natural history of CHB
Viral evolution is important in the pathogenesis of chronic viral diseases such as hepatitis C,46 HIV47 and HBV.48 For example, during the immune clearance phase, Lim et al demonstrated that before HBeAg SC actually occurred, the viral sequence diversity in the PC/BCP region was significantly higher in HBeAg seroconverters compared with non-seroconverters.48 The selection pressure driving this diversity was found to be in effect at least 3 years before HBeAg SC actually occurred, and interestingly, the viral diversity increased further after viral load reduction. These results extended previous work demonstrating that viral mutation rates are increased in the immunoelimination phase compared with patients in the immunotolerant phase of CHB.49 Lim and colleagues also demonstrated that nucleotide substitution rates persisted at high levels after HBeAg SC, with viral diversity increasing further.48 Thus, it appears that this phase is far from being a ‘non-replicative state’, and that there is an ongoing dynamic post-SC process, albeit at lower levels of viral load, with viral diversity likely being driven by an immune-based selection pressure such as significant production of anti-HBe antibody.
Occult HBV infection: the final phase of CHB?
Occult HBV infection (OBI) can be defined as the presence of HBV DNA in the liver (with detectable or undetectable HBV DNA in the serum) of individuals testing HBsAg negative by currently available assays. When detectable, the amount of serum HBV DNA is typically very low (<200 IU/ml). The molecular basis of OBI is related to the long-lasting persistence of HBV cccDNA in hepatocyte nuclei.20 Although some HBV isolates from OBI cases may harbour ‘a’/α determinant mutations that prevent HBsAg detection by antibodies against the wild type virus, almost all OBI cases are infected with replication-competent HBV rather than escape variants, and so the lack of HBsAg in the peripheral compartment is regarded as a possible reflection of ‘strong immune control’ or suppression of viral gene expression of the infecting virus.50 51 The role of OBI in the development of chronic liver disease and as a risk factor for HCC development appears significant50 and further molecular studies are needed to elucidate the underlying pathogenetic mechanisms.51
Viral biomarkers in the blood compartment: research and diagnostic tools
Specific serology has proven to be the mainstay of diagnostic testing for hepatitis B. Several commercial immunoassays are available for the various hepatitis B markers of viral antigens and antibodies.52 Recently, sensitive and reliable assays have been developed to quantify serum HBsAg and HBeAg. Quantitative HBsAg levels are reported in IU/ml, with 0.1 IU/ml being equivalent to approximately 0.1 ng/ml of HBsAg,53 which is in turn equivalent to 2×107 subviral HBsAg particles or 5×106 virions. For HBeAg measurement, most quantitative assays are modified by reference to an external standard, such as the Paul Ehrlich (PE) Institute reference standard for HBeAg, and results are expressed in PE IU/ml.54
The presence of large amounts of HBsAg and HBeAg in the serum, however, may affect the ability to detect circulating antibodies and may obscure the onset of HBeAg or HBsAg SC. The available commercial assays usually detect anti-HBs and anti-HBe antibodies only after the respective antigens have been cleared from the serum. By using more sensitive immunoassays it was possible to detect antibody in the presence of excess serum antigen.55 These approaches identified serological responses in the context of active viral replication. For example, all patients with ‘active’ CHB and the majority of patients in the so-called tolerant phase demonstrated ongoing humoural immune responses, including anti-HBe and anti-HBs production. Anti-HBe and anti-HBs production may coexist with virions and subviral HBsAg particles for many years before viral clearance and loss of antigenemia eventually occur. HBeAg SC almost certainly accounts for increased quasispecies diversity in PC/core and BCP regions.48 The clinical picture of a relatively non-overlapping SC from HBeAg-positive to anti-HBe-positive status during CHB should be replaced by a more dynamic interplay of a potent host selection pressure (eg, B-cell response) targeted to key epitopes of HBV that result in emergence of escape variants that now have a replicative fitness advantage above the rest of the pool of HBV quasispecies.
The molecular detection tools for HBV DNA are useful for resolving ambiguous serological patterns, in staging patients in the various phases of CHB (see figure 3), and for monitoring patients undergoing antiviral therapy. Sustained suppression of HBV replication as assessed by HBV DNA measurement currently represents the cornerstone of evaluation of antiviral efficacy which must aim to reduce HBV DNA to as low a level as possible, ideally below the lower limit of detection by real-time PCR assays (<10–15 IU/ml). Thus, the goal of antiviral therapy is to suppress viral replication to a level that will lead to biochemical remission, histological improvement and prevention of disease progression.36 However, recent studies have indicated that even potent nucleos(t)ide analogue (NA) therapy minimally impacts on the HBsAg levels in blood. This is not surprising since NAs block the HBV DNA replication pathway (via inhibiting rt) and have no direct effect on transcription or translation of the Pre-S1/Pre-S2/S, Pre-core and X genes.56 In contrast, immune-based therapies do reduce transcription57 and viral protein levels58 59 through non-cytolytic and cytolitic60 mechanisms, as part of their broader antiviral activity.
HBsAg SC is now being regarded as the new treatment endpoint because HBsAg loss has been associated with successful immunological control of HBV and durable suppression of viral replication, thereby representing an endpoint for patients to be able to cease oral NA or immune-based therapies. Assessment of on-treatment changes in HBsAg titres may facilitate new management algorithms and future trials aimed at achieving this important endpoint. For example, the role of quantitative HBsAg in predicting response to pegylated interferon (IFN) therapy has been the focus of several recent studies.61–63 In HBeAg-positive patients, reduction of HBsAg levels to <1500 IU/ml at week 12 has been shown to be an early favourable sign of future HBsAg clearance. Over 50% of patients on pegylated IFN who achieved this level at week 12 were found to have HBeAg SC 6 months post treatment and nearly 20% of these patients achieved subsequent HBsAg clearance at 6 months post treatment. In contrast, a HBsAg level of >20 000 IU/ml at week 12 was associated with a very low rate of HBeAg SC and so may become a potential stopping rule of therapy.63 64 In HBeAg-negative patients treated with pegylated IFN, the decline in HBsAg titre at week 12 has also been shown to be a useful predictor of achieving an undetectable viral load at 24 weeks post therapy.61 Among patients who achieved HBsAg decline ≥10% from baseline at week 12 of treatment almost 50% achieved HBV DNA ≤2000 IU/ml at 1 year post treatment and 40% of these individuals achieved HBsAg clearance at 5 years post treatment. Rijckborst and colleagues65 proposed a clinically useful algorithm in HBeAg-negative CHB that any HBsAg decline at week 12 with a 2 log drop or more in HBV DNA could predict almost 40% of sustained responders in their cohort. If patients did not achieve a HBsAg decline or only had a <2 log drop in HBV DNA, then the chance of responding was 0%.
Quantitative HBeAg testing has proved to be more problematic mainly because of the lack of commercially available assays and studies have used mainly in-house techniques with reference to external standards such as the one available from the PE Institute. Fried and colleagues54 demonstrated a moderately significant relationship between baseline HBeAg level and subsequent HBeAg SC in patients treated with pegylated IFN. A level of ≤31 PE IU/ml at baseline was associated with a greater than 50% chance of HBeAg SC. The on-treatment week 12 and 24 HBeAg levels were more likely to be associated with a negative predictive response; for example, only 14% of patients at week 12 with a qHBeAg of >100 PE IU/ml went on to a HBeAg SC at week 72 whereas just 4% of patients with a qHBeAg of >100 PE IU/ml at week 24 achieved HBeAg SC at week 72, possibly forming the basis of another stopping rule.
Viral biomarkers in the liver
The effects of antiviral therapy on serum HBV DNA levels are well documented; while less information is available about their effects on intrahepatic virological parameters. The development of highly selective quantitative real-time PCR assays has provided the necessary tools to determine intrahepatic HBV DNA levels, including the quantification of cccDNA loads23 27 28 66 67 and transcriptional activity25 26 in liver biopsy specimens. For example, by measuring ‘virion productivity’ (defined as the ratio of intrahepatic rcDNA/cccDNA) and ‘transcriptional activity’ (defined as the level of pgRNA) it was observed that patients with HBeAg-negative CHB had significantly lower levels of viraemia, intrahepatic cccDNA (>1 log lower) and rcDNA (>2 log lower).26 Furthermore, virion productivity per cccDNA molecule was found to be fivefold lower than in HBeAg-positive disease. Levels of pgRNA were correspondingly reduced, suggesting that the low virion productivity resulted from lower amounts of pgRNA. In contrast to patients with HBeAg-positive CHB, there was no correlation between serum viral load and the amount of cccDNA in the HBeAg-negative patients, consistent with the observed impairment of virion productivity (figure 2). Although HBsAg titres were significantly lower in the HBeAg-negative cohort, pre-S/S mRNA levels and HBsAg titres per cccDNA molecule appeared similar between HBeAg-positive and HBeAg-negative patients, indicating that the production of SVPs was not impaired in the HBeAg-negative phase, but rather that the replication pathway might be selectively blocked. The study of Volz and colleagues26 also indicated that patients' transition through SC into HBeAg-negative disease is clearly not a ‘black and white’ immunological event, as suggested by existing commercial serological assays. Rather, a progressive increase in the viral diversity with positive selection of viral quasi-species defective for HBeAg production was observed. Identifying these selection pressures will be essential for understanding the host–virus interplay determining the course of CHB.
Although independent reports have shown that cccDNA loads vary significantly in the different phases of CHB infection, the correlation between antigenemia and intrahepatic cccDNA levels has not been well established and the results conflicting.26 68–70 The reason for such discrepancies may be multifactorial and due to virological factors (HBV genotype differences,71 emergence of mutations72 73) and host-mediated factors, which may affect cccDNA activity causing fluctuations in serum HBsAg concentrations among patients presenting similar cccDNA levels. Differences in cccDNA amounts determined by needle liver biopsy may also reflect uneven distributions of the cccDNA in different parts of the liver. The acquisition of viral DNA integrations containing an intact preS/S ORF may also contribute to the production of HBsAg and alter such correlations.
Polymerase inhibitors do not directly block formation and activity of the cccDNA, a significant decrease in cccDNA levels (approximately 1 log reduction) has often been observed after 1 year of therapy.23 28 66 67 Such reduction is supposed to derive from the lack of incoming viruses from the blood and insufficient recycling of viral nucleocapsids to the nucleus, due to suppression of viral DNA synthesis in the cytoplasm. However, mathematical modelling indicated that many years of nucleos(t)ide drug administration are needed to achieve cccDNA depletion. Thus, persistence of the cccDNA minichromosome remains the key factor responsible for failure of viral clearance and relapse of viral activity after cessation of antiviral therapy with polymerase inhibitors in chronically infected individuals. Furthermore, if viral suppression is not complete, the selection of resistant variants escaping antiviral therapy is likely to occur.20 74 Under antiviral pressure, such variants will contribute to the production and further selection of replication-competent resistant mutants, which will spread to other hepatocytes and, eventually, may replace the wild-type cccDNA molecules in the liver.
More potent HBV suppression using combination therapy has been achieved with 99% inhibition of intrahepatic virion productivity in patients who had received 1 year of pegylated IFNα and adefovir dipivoxil,28 while viral suppression appeared reduced to 76% during the following 2 years, after patients were shifted to ADV monotherapy. Recent studies also showed that reduction of circulating HBsAg is more marked in patients treated with pegylated IFNα alone or in combination with nucleoside analogues, in comparison to patients receiving monotherapy with polymerase inhibitors.62 Such differences are important and not entirely surprising because IFNα displays immune-modulating capacities and can induce direct antiviral effects, preventing the formation of viral RNA-containing core particles, accelerating pgRNA degradation and the decay of core particles.59 75 76 Furthermore, the most recent studies performed in vitro and in HBV-infected human-liver chimeric uPA/SCID mice showed that IFNα administration inhibits the transcription of all HBV-RNAs by inducing epigenetic repression of the cccDNA.57 Thus, these results identify a molecular mechanism whereby IFNα can reduce virion DNA productivity and the levels of HBsAg.
Although understanding of the molecular mechanisms influencing intrahepatic virion productivity is just starting to emerge, these studies indicate that quantitative measurements of serological viral parameters may become useful to monitor and predict treatment response within individual patients, while further studies employing better standardised intrahepatic assays and well characterised patient cohorts are needed to resolve previous differences in particular studies.
Factors influencing HBV pathogenesis
Persistence of chronic HBV infection is due to the ability of the cccDNA to survive within the hepatocytes and to the successful establishment of a complex network of virus–host interactions permitting the virus to manipulate the cellular machinery to meet its replication and propagation requirements, and to evade the antiviral response of the host. Over the years, mutual attempts to control each other may set the stage for the development of severe liver pathologies, such as liver cirrhosis and HCC.
Immuno-pathogenesis of HBV infection: general considerations
The innate immune system represents an immediate, first-line defence against foreign pathogens and plays a fundamental role in regulating the adaptive immune response. While the role of the adaptive immune system in the resolution of HBV infection has been studied extensively,77–79 knowledge of the innate immune processes occurring in HBV infection has been limited by the restricted availability of in vitro and in vivo infection systems and technical difficulties in recruiting patients at the earlier stages of infection. Despite these limitations, studies in acute infected patients80 and chimpanzees81 documented a lack of type I IFN induction and proinflammatory cytokine production in acute HBV infection.81 In contrast to these observations, recent in vitro studies indicated that the innate immune response of the hepatocytes may sense the infection and limit the spread of HBV.82–84 A modest activation of IFN-stimulated genes was also shown in human hepatocytes after HBV infection in chimeric mice.30 Induction of the antiviral state and production of type 1 IFN (α/β) can be mediated by pathogen recognition receptors, such as toll-like receptors (TLRs).84 In turn, type 1 IFN production stimulates antigen-presenting cells such as dendritic cells and Kupffer cells (resident intrahepatic macrophages) to produce interleukins (IL-8, IL-12, IL-18) and other cytokines.85 IL-12 and IL-18 subsequently induce natural killer (NK) and natural killer T (NKT) cells.86 NK cells can also be activated by the major histocompatibility complex I molecules on the surface of infected hepatocytes or by recognition of stress-induced molecules.87 Although NK and NKT cells can promote liver damage through direct cytotoxic effects,88 their role in HBV persistence and mechanisms regulating NK cell function in HBV infection are still controversial.89
The hepatocellular injury caused by HBV infection is predominately immune mediated.90 91 The immune attack by the host against HBV is mainly mediated by a cellular response to small epitopes of HBV proteins, especially HBcAg, presented on the surface of liver cells. HLA class I restricted CD8 cells recognise HBV peptide fragments derived from intracellular processing and presentation on the liver cell surface by class I molecules. This process leads to direct cell killing by the CD8 cytotoxic T lymphocytes. It is important to note that B and T lymphocytes are only activated following recognition of HBV by the innate immune system. Ultimately, it is the breadth and strength of this specific adaptive immune response which determines the outcome of acute and chronic HBV infection. Animal and clinical studies demonstrated that in acute self-limited HBV infection, the CD8 T-cell and CD4 T-cell responses to HBV proteins92 are strong, polyclonal and multispecific, while in chronic HBV infection, the responses are weak and narrowly focused.89 Besides having a direct cytolytic effect, as discussed above, CD8 T cells also have a non-cytolytic effect through the production of IFNγ and tumour necrosis factor α (TNFα) (also produced by NK and NKT cells), which are known to elicit antiviral effects via multiple mechanisms.93 Indeed, studies have shown that IFNα and IFNβ significantly suppress HBV gene expression in vivo, indicating that non-cytolitic direct antiviral effects elicited by the immune response may play a determinant role in controlling HBV infection in immunological normal adults.30 57 75 Thus the current concept is that non-cytolytic and cytolytic mechanisms are requisite for successful HBV control and eventual clearance.
HBV interactions with innate immune responses of the hepatocytes
The limited induction of innate immune responses determined upon HBV infection may also be due to recently recognised abilities of HBV to evade innate immune recognition. TLRs are evolutionary conserved germline-encoded pattern recognition receptors that play a crucial role in early host defences by recognising pathogen-associated molecular patterns and also serve as an important bridge between the innate and the adaptive immune response. HBeAg was shown to act as an immune decoy for the core antigen by depleting HBeAg-specific and HBcAg-specific T-helper cells and exerting a tolerogenic effect in utero.94 Furthermore, HBeAg has been shown to potentially downregulate genes involved in cell signalling, RNA transport and processing, cytosol-nuclear trafficking and innate immune responses.95 Lang et al12 confirmed the presence of a Box-2-like TIR motif within the unique N-terminal 10-amino-acid sequence of PC/HBeAg protein which could antagonise TLR signalling pathways via blocking the adapter proteins Mal, TRAM and TRIF but not MyD-88, thereby suppressing TIR-induced nuclear factor κB and IFNβ activation (table 1). These findings provide a possible mechanism of the ‘HBeAg immunomanipulation’ of the innate immune response, including the induction of ‘tolerance’ in the first phase of CHB, especially following perinatal HBV transmission. In patients with HBeAg-positive CHB, the expression of TLR-2 on hepatocytes, Kupffer cells and peripheral blood monocytes was shown to be significantly downregulated, while upregulation of the TLR-2 pathway and increased TNFα production was found during HBeAg-negative chronic hepatitis96 (Table 1). Although these clinical studies have indicated a role for HBeAg in downregulating immune surveillance of HBV, loss of HBeAg synthesis commonly occurs during chronic HBV infection and the emergence of HBeAg-negative variants may present selective advantages, possibly by limiting the cytotoxic T-lymphocyte (CTL)-mediated clearance of infected hepatocytes.97 However, the selection of HBeAg variants is associated with increased viral activity,26 more severe liver disease and worse prognosis.44
In support of the concept that HBV has evolved mechanisms to sabotage pathways of the IFN response, a recent study in human-liver chimeric mice showed that administration of regular IFNα failed to promote detectable nuclear translocation of signal transducer and activator of transcription 1 (STAT-1) in HBV-infected human hepatocytes, although STAT-1 nuclear accumulation and enhancement of antiviral defence mechanisms were promptly induced in uninfected human hepatocytes.30 Furthermore, in vitro studies indicated that HBV may affect STAT methylation98 and activity of cellular DNA methyltransferases (DNMT).99 100 The non-structural HBx protein has been shown to also interfere with the innate immunity by downregulating mitochondrial antiviral signalling protein101 by suppressing the retinoic acid inducible gene I (RIG-I)–melanoma differentiation associated gene 5 (MDA5) pathway in vitro. Interactions between HBx, mitochondrial antiviral signalling and members of the cellular epigenetic machinery have been also reported.101 Furthermore, plasma-derived HBsAg SVPs were reported to inhibit TLR-9 mediated IFNα production by plasmacytoid dendritic cells, probably via stimulation of suppressor of cytokine signalling 1 expression.102 Although most of these analyses were performed in systems that overexpressed HBx and further analyses are needed in more relevant infection systems, taken together, these studies suggest that distinct virus-mediated mechanisms may contribute to the limited effectiveness of the innate immune responses and subsequent adaptive immune responses in HBV infection and thereby may significantly contribute to viral persistence.
Main factors that influence HBV persistence and pathogenesis
Mutual interplays influencing HBV activity and cellular pathways
The incidence of HCC development has been shown to correlate with the levels of HBV replicative activity.103 This highlights the importance of elucidating the mechanisms modulating viral activity because these factors will affect both cccDNA loads and cellular pathways, thus influencing the clinical course of infection.
Numerous transcription factors implicated in the activation of hepatic metabolic processes, such as hepatocyte nuclear factor, cAMP responsive element binding protein, retinoid X receptor and peroxisome proliferator-activated receptors, are known to bind the HBV genome and the recruitment of liver-enriched transcription factors on the cccDNA minichromosome appears essential for controlling viral gene expression.6 104 However, interactions between viral components and cellular factors may also impact the liver metabolism. Glucocorticoids are steroid hormones playing major roles in glucose homeostasis and the presence of a glucocorticoid response element on HBV genome can explain the so-called HBV reactivation observed in patients receiving steroid-based immunosuppressive agents. Although a clinical association between HBV and glucose homeostasis is lacking,104 HBV was shown to promote lipogenesis and fatty acid accumulation in HBV-replicating transgenic mice105 and in the liver of HBV-infected patients.106 Such metabolic alterations are likely to contribute to disease progression. Furthermore, a multitude of in vitro transfection experiments showed that HBx can modify cellular pathways by altering the activity of numerous transcription factors, kinases of the Src family, of the Ras-Raf-mitogen-activated protein kinase cascade and members of the protein kinase C pathway, and by affecting mitochondria function and protein degradation.107 108 Whether HBx, expressed at levels comparable to those present in infected human livers, also similarly affects these cellular functions awaits further investigation.
CccDNA-based chromatin immunoprecipitation (ChIP) assays indicated a correlation between viraemia levels and acetylation status of cccDNA-bound histones,109 suggesting that epigenetic modifications may affect the cccDNA transcriptional status in the different phases of chronic infection. Using ChIP assays HBx was shown to be recruited to the cccDNA minichromosome,110 where by favouring cccDNA-bound histone acetylation, HBx appears to play an essential role in initiating the cccDNA-driven transcription of the HBV RNAs (figure 1).8 However, epigenetic modifications, such as DNA hypermethylation, may induce suppression of cccDNA transcription, since increased expression of DNMTs has been correlated with increased cccDNA methylation and decreased HBV transcription in advanced HBV infection.111 Moreover, HBx was shown to act as a potent epigenetic modifying factor in human liver by modulating the transcription of DNMT required for maintenance of hypomethylation of tumour suppressor genes.99 The HBx-promoted hypermethylation of tumour suppressor genes suggests a novel mechanism by which this promiscuous transactivating protein may accelerate hepatocarcinogenesis.
Numerous cellular factors, such as microRNAs, may affect HBV productivity by binding directly to HBV transcripts or by targeting cellular transcription factors relevant for cccDNA transcription.112 Further studies based on a combination of bioinformatic and miRNA expression screenings are needed to determine whether specific patterns of miRNA expression could serve as diagnostic biomarkers for liver disease prediction.
A number of studies identified host variations that may contribute to the outcome of HBV infection. Genome-wide association studies determined, for instance, polymorphisms in the human leucocyte antigen (HLA) allele DRB1,113 HLA-DP114 and the oestrogen receptor115 that were associated with the susceptibility to HBV clearance or persistence. Similarly, naturally occurring sequence variations in the promoter region of CXCL10 gene were shown to result in the altered expression of the CXCL10 protein, which is important for the intrahepatic recruitment of inflammatory cells, thereby impacting disease progression of patients with chronic HBV infection.116
Emergence of virus quasi-species and infection with other viruses
The selective pressure induced by host immune responses and/or during antiviral therapy with polymerase inhibitors promotes the emergence of mutations. For instance, single amino acid substitutions have been shown to impair HBsAg recognition and lead to the development of acute or chronic HBV infection despite immunoprophylaxis. The region of the genome encoding for the rt domain of the HBV polymerase overlaps completely with the envelope; therefore, mutations emerging during nucleos(t)ide analogue treatment may lead to changes in the viral envelope. In vitro studies have shown that the rtA181T mutation, which introduces a nonsense mutation in the overlapping envelope gene, leads to intracellular retention of the truncated surface proteins and viral particles.72 As discussed above, during the immune competent HBeAg-positive phase, the host immune system may set the stage for the selection of PC/BCP mutations with reduced or absent HBeAg secretion.25 117 Sequencing of cccDNA amplicons indicated that the replacement of wild type cccDNA with populations of BCP mutations was associated with the re-establishment of high virion productivity26 (figure 3). In agreement with these observations, studies in chimpanzees indicated that HBeAg-negative viruses induce more severe hepatitis118 and the accumulation of BCP mutations have been shown to be associated with worsening of clinical manifestations and development of HCC.119
HBV/HCV and HBV/hepatitis D virus (HDV) co-infection are known to lead to a rapid and more severe progression of chronic liver disease. Although the virological profiles in the setting of co-infection appears to be widely divergent and fluctuating levels of viraemia120 are observed, information about the mechanisms responsible for such variations are scant. Although immune-mediated mechanisms may play a major role in suppressing HBV replication in the setting of co-infection,69 121 122 recent studies in chimeric mice31 showed that HDV lowered HBV productivity even in the absence of adaptive immune responses, indicating that complex virus–virus and virus–host interactions may affect replication levels and disease progression in the course of HBV/HDV co-infection.
Concluding remarks and future direction
In the past decade, there has been an explosion of knowledge and research in the field of hepatitis B molecular diagnostics. The availability of innovative human hepatocyte-based HBV infection systems and the development of molecular techniques permitting molecular analysis, such as cccDNA quantification, ChIP and transcriptome assays, in patient liver biopsies have started to provide new insight about host and viral factors regulating the activity of the cccDNA minichromosome. Furthermore, results showing the existence of interferences between HBV and pathways of the innate immune response suggest that manipulation of TLRs via TLR ligands may have therapeutic potential for the treatment of CHB by reducing viral replication and by promoting improved immune control. Because of the limitations encountered by using different experimental systems, cellular and molecular analyses based on human blood and liver biopsy samples remain indispensable to improve our knowledge of the pathobiology of HBV infection. It can be envisaged that the future understanding of the natural history of CHB will continue to evolve and that management algorithms will become more individualised, incorporating quantitative serology and possibly genotyping and markers of the innate and adaptive immune responses.
References
Footnotes
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.