Article Text

Abstract
Objective Sessile serrated adenomas (SSAs) are the precursors of at least 15% of colorectal carcinomas, but their biology is incompletely understood. We performed a clinicopathological and molecular analysis of a large number of the rarely observed SSAs with dysplasia/carcinoma to better define their features and the pathways by which they progress to carcinoma.
Design A cross-sectional analysis of 137 SSAs containing regions of dysplasia/carcinoma prospectively collected at a community GI pathology practice was conducted. Samples were examined for BRAF and KRAS mutations, the CpG island methylator phenotype (CIMP) and immunostained for MLH1, p53, p16, β-catenin and 0-6-methylguanine DNA methyltransferase (MGMT).
Results The median polyp size was 9 mm and 86.5% were proximal. Most were BRAF mutated (92.7%) and 94.0% showed CIMP. Mismatch repair deficiency, evidenced by loss of MLH1 (74.5%) is associated with older age (76.7 versus 71.0; p<0.0029), female gender (70% versus 36%; p<0.0008), proximal location (91% versus 72%; p<0.02), CIMP (98% versus 80%; p<0.02) and lack of aberrant p53 (7% versus 34%; p<0.001) when compared with the mismatch repair-proficient cases. Loss of p16 (43.1%) and gain of nuclear β-catenin (55.5%) were common in areas of dysplasia/cancer, irrespective of mismatch repair status.
Conclusions SSAs containing dysplasia/carcinoma are predominantly small (<10 mm) and proximal. The mismatch repair status separates these lesions into distinct clinicopathological subgroups, although WNT activation and p16 silencing are common to both. Cases with dysplasia occur at a similar age to cases with carcinoma. This, together with the rarity of these ‘caught in the act’ lesions, suggests a rapid transition to malignancy following a long dwell time as an SSA without dysplasia.
- COLON CARCINOGENESIS
- COLONIC NEOPLASMS
- COLONIC POLYPS
- COLORECTAL CANCER GENES
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Sessile serrated adenomas (SSAs) are the source of approximately 15% of colorectal carcinomas.
Cancers of the serrated neoplasia pathway disproportionately contribute to interval colorectal carcinoma.
SSAs with dysplasia or carcinoma are rarely observed.
What are the new findings?
SSAs with dysplasia or carcinoma are predominantly small (<10 mm), flat polyps of the proximal colon.
The mean age of patients with SSAs with dysplasia only and those with carcinoma is the same, but this is 17 years older than patients with SSAs without dysplasia.
SSAs progress to malignancy via a mismatch repair-deficient (75%) pathway or a mismatch repair-proficient (MMRP) (25%) pathway with WNT pathway activation and p16 silencing in both, but p53 mutation is only common in MMRP cases.
How might it impact clinical practice in the foreseeable future?
This study indicates SSAs without dysplasia have a mean dwell time of 17 years before the development of dysplasia and then rapidly converts to carcinoma. SSAs with dysplasia or carcinoma are clinically similar to SSAs without dysplasia, making endoscopic recognition difficult. Improved colonoscopic detection and removal of these lesions are likely to reduce interval cancers and understanding of the molecular drivers may allow the development of chemoprevention strategies.
Introduction
The serrated neoplasia pathway is a major contributor to colorectal carcinoma, with approximately 25% of cases arising via this route.1–4 These cancers have their origins in serrated polyps, including sessile serrated adenomas (SSAs) and traditional serrated adenomas (TSAs).4 ,5 Of these, the SSA is by far the most prevalent and accounts for most serrated neoplasia pathway carcinomas.
SSAs are subtle polyps that can be difficult to detect colonoscopically, are frequently incompletely excised and have the hypothesised potential for rapid malignant degeneration.4 ,6–8 Colonoscopically, a dysplastic component can be difficult to detect.8 ,9 Although an exophytic component has been proffered as evidence of the development of dysplasia,9 particularly in large SSAs,10 this has not been confirmed in more inclusive series. For the pathologist, misdiagnosis or underdiagnosis of SSA as a microvesicular hyperplastic polyp remains an issue.11–15 This combination of factors has clinical implications, the most significant being interval carcinoma. This can occur due to missed lesions, incompletely excised lesions, rapid progression of de novo lesions or inadequate surveillance due to misdiagnosis by the pathologist. Several studies have demonstrated that serrated pathway carcinomas are over-represented among interval cancers, confirming that some, if not all, of these factors contribute to this occurrence.16 ,17
SSAs occur predominantly in the proximal colon and in older women.11 ,18 Histologically, they are characterised by abnormal crypt architecture, but without cytological dysplasia.18 ,19 Oncogenic mutation of the BRAF gene and the development of the CpG island methylator phenotype (CIMP) are characteristic molecular features.20 ,21 Progression to the SSA with dysplasia (SSAD) heralds an aggressive phase in polyp development and is accompanied by underlying molecular events.22 ,23 The best characterised is methylation-induced silencing of the tumour suppressor gene MLH1, resulting in loss of immunohistochemical expression of the MLH1 protein and DNA mismatch repair deficiency (MMRD). Loss of mismatch repair function allows for a rapid accumulation of mutations in genes with microsatellites and thus underlies microsatellite instability (MSI).
The proportion of SSADs that are MMRD is not clear. In the literature, based on either MLH1 loss by immunohistochemistry or by significant MLH1 promoter methylation, the rates range between 15% and 72%.22 ,24–26 However, these studies are often confounded by small size and older methods of polyp classification.
There has been no large study of the clinicopathological and molecular features of SSAs with a focus of dysplasia and/or carcinoma to accurately determine what percentage of SSAs methylate MLH1 as they develop dysplasia. In those that do, the development of MSI with increased mutational load appears to drive rapid development of malignancy, but the key genes and pathways affected in this early phase are not clear. The pathways driving progression towards malignancy in SSAs which do not methylate MLH1 are even less understood. We hypothesised the pathways affected may be those involved in traditional colorectal carcinogenesis even though the order of occurrence may be different and the mechanism of perturbation may more often be silencing by DNA methylation. Thus, we examined WNT pathway activation, p16 silencing and TP53 mutation. Additionally, we compared ages of patients with SSAD with ages of patients with SSA and of patients with BRAF-mutated cancers to provide an estimate of the time course of progression.
Materials and methods
Case selection and study design
This study represents a prospective series collected by one author (NW) during routine reporting over 6 years at Envoi Specialist Pathologists in Brisbane, Australia. Envoi is a high-volume community GI pathology practice, staffed by expert GI pathologists. Referral cases were not included. All potential cases underwent pathological review by two authors (MB and NW). For inclusion, cases were required to show (1) a component of ordinary SSA at the edge of the lesion comprising at least three crypts, one of which must show SSA-type histology;11 (2) abrupt transition from ordinary SSA to overt cytological dysplasia or carcinoma within one tissue fragment and (3) exclusion of cases representing TSA arising in an SSA.27 These criteria were used to ensure the series represented a homogenous group. Criteria one was designed to guarantee origin in an SSA, criteria two to ensure the dysplasia or carcinoma was arising in the SSA of interest rather than being from a separate conventional adenoma collected in the same specimen jar and criteria three to exclude the phenomenon of TSA arising in an SSA.28 ,29 TSA arising in an SSA can be easily misdiagnosed as SSAD but is a separate entity with distinct clinicopathological and biological features, thus requiring exclusion from the current study.27 Cases were additionally excluded if there was insufficient material to perform the molecular and immunohistochemical analysis. The cases included lesions removed either colonoscopically or surgically and included lesions clinically considered as polyps as well as carcinomas. Patients with serrated polyposis syndrome were not excluded.
Two additional cohorts were used for comparison with the study series. These were 579 SSAs without dysplasia from a previously published series11 and 99 BRAF-mutated colorectal carcinomas also collected from Envoi as part of a study of a consecutive series of 500 colorectal carcinomas. The vast majority of BRAF-mutated colorectal carcinomas have their origins in SSAs and thus represent the end-point of malignant progression for these polyps. In particular, we aimed to compare the ages of patients from these series with the study cohort.
Clinicopathological data collection
Clinical data included patient age and gender, lesion size and lesion location. Lesion location was divided into proximal (proximal to the splenic flexure) and distal (distal to the splenic flexure). Pathological data included the nature of the advanced component (dysplasia and/or carcinoma), the size of the advanced component and the growth pattern of the advanced component (flat vs exophytic). Carcinoma was defined as invasion into submucosa and excludes intramucosal carcinoma (which is included with the dysplastic cases). The advanced component was diagnosed as flat if it was less than twice the height of the adjacent SSA.
Three pathologists (MB, NW, CR) assessed all cases with invasive carcinoma for the following features: WHO subtype, grade (high vs low), infiltrative margin, tumour-infiltrating lymphocytes (>5 per high power field)30 and Crohn-like inflammatory reaction (>4 aggregates per×4 objective field).30 This assessment was performed around a multiheaded microscope and consensus opinion was used for each feature. The pathologists were blinded to the mismatch repair status.
Immunohistochemistry
Immunohistochemistry was performed for MLH1, p53, β-catenin, p16 and MGMT as previously described on all cases.27
DNA extraction
The DNA extraction was performed on three 10 μm sections cut from formalin-fixed paraffin-embedded blocks using the Chelex method as previously described.27
Molecular analysis
All cases were assessed for the BRAF V600E mutation by allelic discrimination and for KRAS codon 12 and 13 mutations by high-resolution melt analysis as previously described.27 ,31
The CIMP status was assessed using the panel of Weisenberger et al,32 (SOCS1, NEUROG1, RUNX3, IGF2, CACNA1G) by quantitative methylation-specific PCR as previously described. CIMP-high required a percentage of methylation reference >10 in three of the five Weisenberger et al markers. Methylation in the other assessed genes (MLH1 and MGMT) also required a percentage of methylation reference >10. To ensure the validity of the results, a Ct value of <23 and an Alu representative calculated concentration of >1000 was required.27 ,33
Statistical analysis
Categorical variables were compared by Fisher's exact test and continuous variables by Student's t test. A p value of ≤0.05 was considered significant. SPSS V.19, R V.3.0.2 and GraphPad Prism V.6.02 were used for statistical analyses.
Results
One hundred and thirty-seven advanced SSAs from 132 patients met the inclusion criteria. These included 96 SSADs, 31 SSAD with carcinomas and 10 SSAs with carcinomas. One hundred and twenty-nine were clinically recognised as polyps and eight as carcinomas. Ninety-five were removed colonoscopically and 42 by surgical resection.
Clinicopathological data
The clinicopathological data are presented in table 1. The mean age of patients was 75.2 years and 61.4% were women. Of the polyps, 86.5% were proximal, the median polyp size was 9 mm (mean 10.7 mm) and 54.3% of the polyps were <10 mm. The mean age of patients with the 579 SSAs without dysplasia from our previous study was 58.6 years and the mean age of patients with the 99 BRAF-mutated colorectal carcinomas was 76.0 years. Figure 1 compares the age distribution of the different patient cohorts demonstrating that SSAs without dysplasia are found approximately 17 years earlier than SSADs, suggesting a long dwell time. The SSADs, SSAs with cancer and the BRAF-mutated carcinomas are found at the same age, suggesting rapid progression after the development of dysplasia.
Clinicopathological features of study lesions
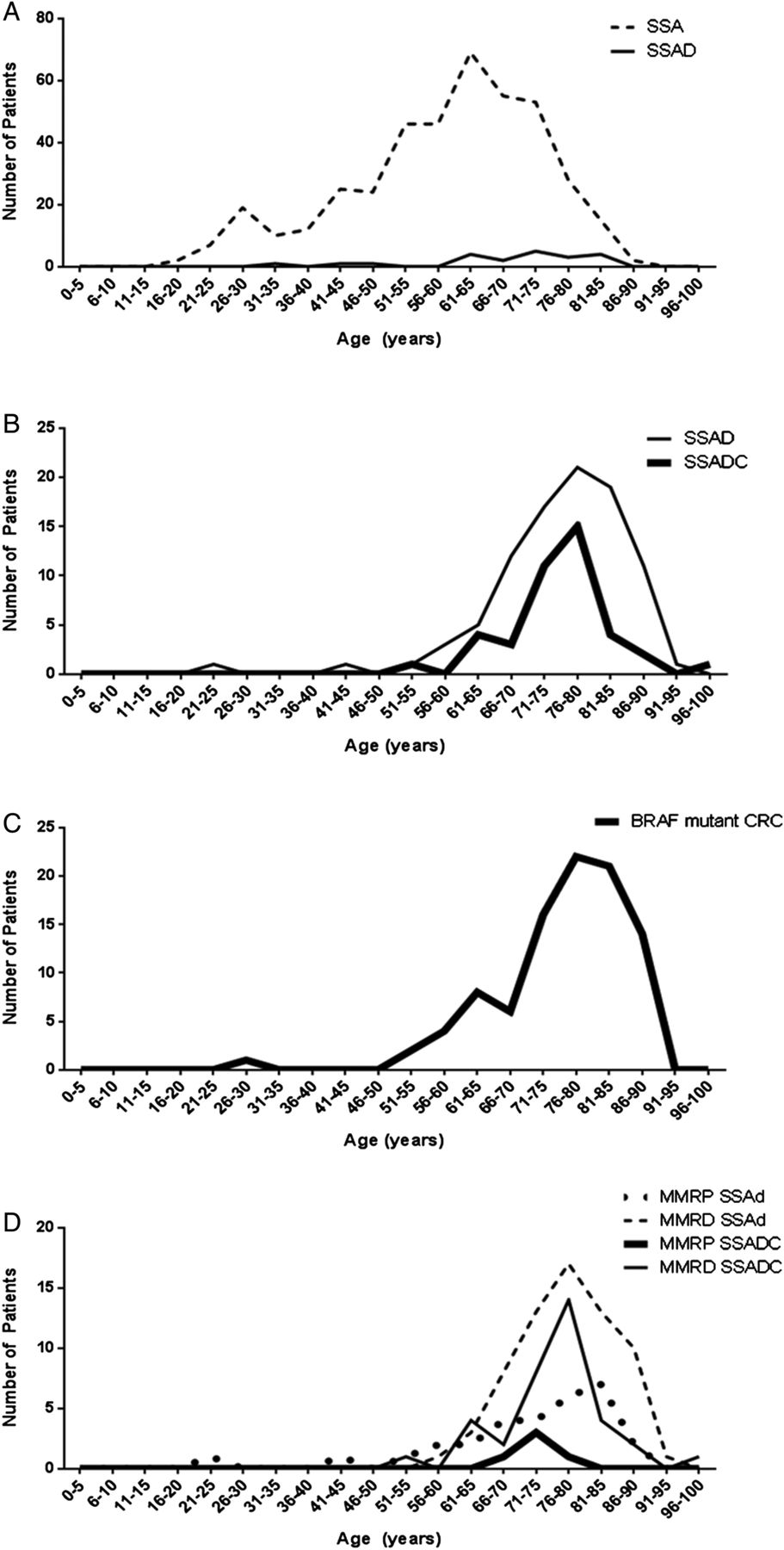
Age distribution of the patient cohorts highlighting the proposed slow transition from SSA to SSAD and then the proposed rapid transition from SSAD to carcinoma. Data from an earlier study11 of 6340 consecutive polyps (A) demonstrate that SSAs without dysplasia are much more common than SSADs and arise in significantly younger patients (p<0.001), suggesting a long dwell time prior to the development of dysplasia. Data from the current study (B) show that the mean age and distribution of SSADs and SSAs with cancer are very similar, suggesting a rapid transition from dysplasia to malignancy. The mean age and distribution of 99 BRAF-mutated colorectal carcinomas (C) are very similar to (B), providing further support for the proposal of a rapid transition to malignancy. (D) shows SSADs and SSAs with cancer in the current study stratified for mismatch repair status demonstrating that there is a trend for MMRP cancer to develop at a younger age. SSA, sessile serrated adenoma; SSAD, sessile serrated adenoma with dysplasia; MMRP, mismatch repair proficient.
By definition, all cases had a component of cytological dysplasia, invasive malignancy or both. One hundred and twenty-three/one hundred and twenty-nine cases recognised as a polyp had a dysplastic component, with a median size of 3 mm (mean 3.6 mm). Thirty-three/one hundred and twenty-nine cases recognised as a polyp had an invasive component, with a median size of 4 mm (mean 3.8 mm). A protuberant growth pattern in any part of the advanced component was present in 22 (17.0%) cases; the remainder were flat (figure 2A, B). There were no significant differences in clinicopathological features between lesions with dysplasia only and those with an invasive component.
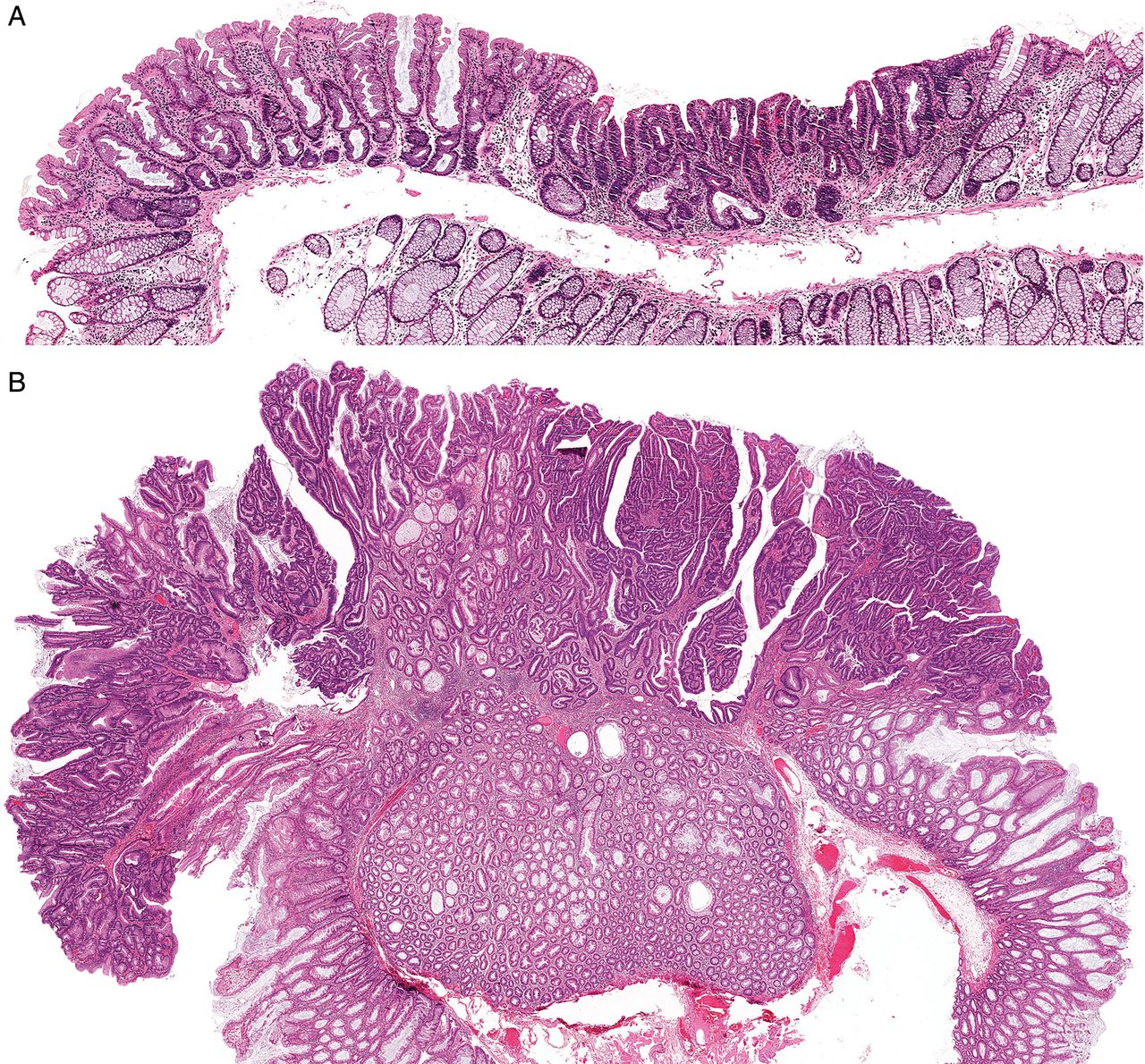
(A) A low-power image of a sessile serrated adenoma with dysplasia (SSAD) from the proximal colon, demonstrating the frequent small size and flat nature of these polyps. Non-dysplastic sessile serrated adenoma at left, abruptly transitioning to dysplasia at right. (B) A low-power image of a larger and protuberant SSAD. This appearance is much less common.
Although both MMRD and mismatch repair-proficient (MMRP) lesions were mostly proximal, a significant minority, especially of MMRP lesions, were found in the sigmoid colon and rectum. The specific locations of the polyps are as follows (MMRD followed by MMRP): caecum, 17% and 3%; ascending colon/hepatic flexure, 34% and 38%; transverse colon, 38% and 32%; splenic flexure/descending colon, 7% and 6%; sigmoid, 2% and 6% and rectum, 0% and 13%.
When comparing the histological features of the MMRD carcinomas versus the MMRP carcinomas, the MMRD cancers were more likely to have a pushing margin (95.5% vs 20%; p=0.0014), tumour-infiltrating lymphocytes (94.4% vs 0%; p≤0.0001) and a Crohn-like inflammatory infiltrate (58.3% vs 0%; p=0.0421). The MMRP cancers were more likely to be of serrated type (100% vs 11.1%; p=0.0002). Of the 36 MMRD carcinomas, 7 were mucinous, 4 were serrated, 2 were medullary, 2 were signet ring cell and a further 12 had a mucinous component (<50%).
Immunohistochemical data
The immunohistochemical data are presented in table 2. All lesions retained a component of SSA without dysplasia and this served as a baseline against which other components could be compared. There were significant changes in staining patterns between the non-dysplastic and dysplastic/malignant components of the lesions for all immunohistochemical markers. This represented increased nuclear staining for β-catenin (figure 3C) indicating WNT pathway activation and p53 indicating an inactivating mutation stabilising the protein. Loss of staining for MLH1, if present, occurred abruptly at the transition to dysplasia. Loss of p16 (figure 3A, B) and MGMT staining in the dysplastic/malignant components indicates silencing of these tumour suppressor genes.
Immunohistochemical features of the lesions divided into ordinary and advanced (dysplasia and/or carcinoma) components
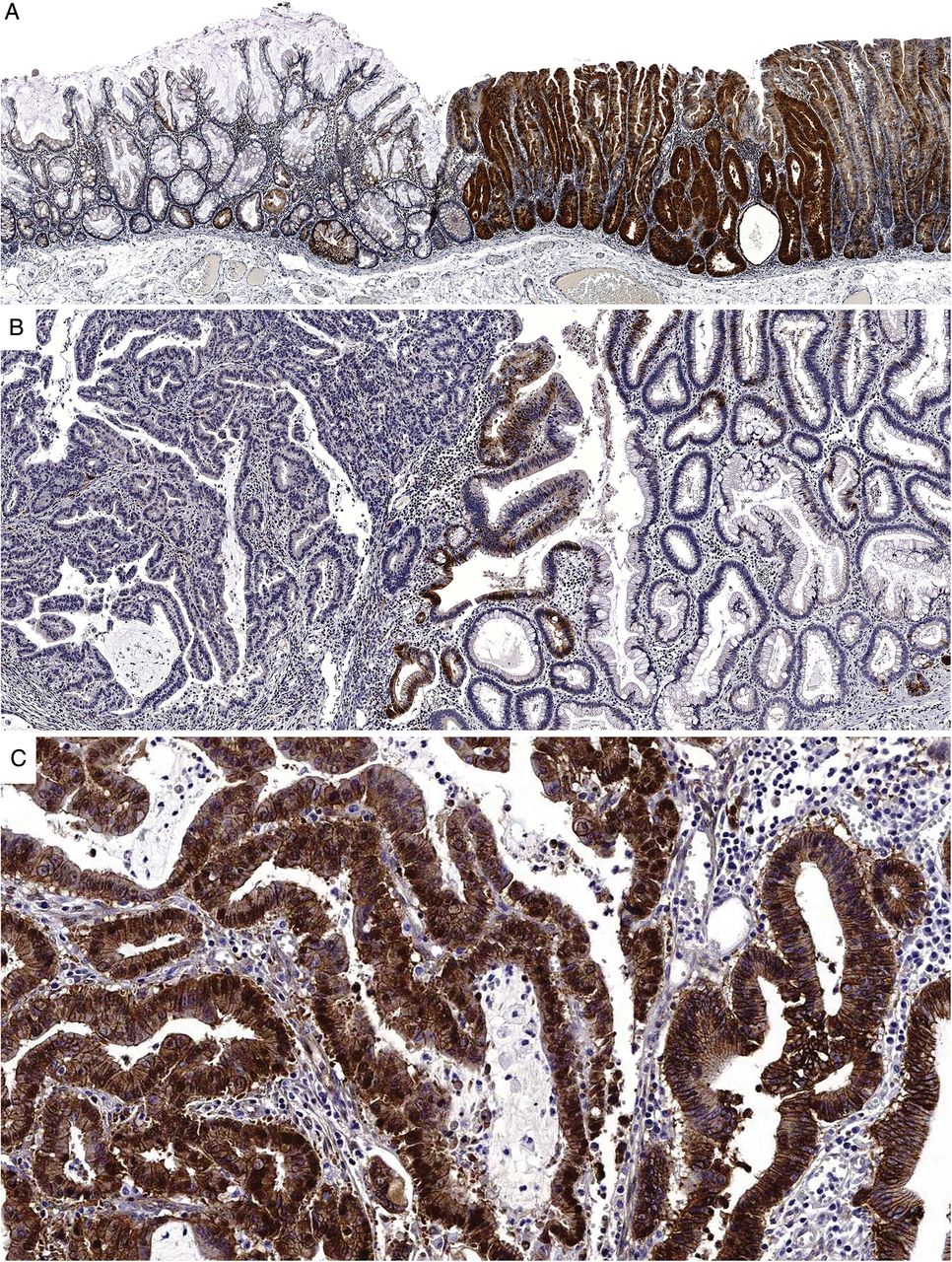
(A) A medium-power image of a p16 immunohistochemical stain of a sessile serrated adenoma with dysplasia. There is markedly increased staining for p16 in the dysplastic area at right, presumably representing cellular efforts to prevent uncontrolled proliferation. (B) A medium-power image of a different polyp containing dysplasia (right) and carcinoma (left). In this case, there is abrupt loss of staining for p16 at the transition to carcinoma. (C) A medium-power magnification of the same area as image (B) this time showing β-catenin staining. Scattered nuclear staining is present in the dysplasia but becomes uniform in the carcinoma, indicative of WNT pathway activation.
Molecular data
The molecular data are presented in table 3. Of the cases, 92.7% harboured a BRAF V600E mutation and one had a KRAS codon 12 or 13 mutation. CIMP-high (CIMP-H) was present in 92.7%.
Molecular features of the study lesions
Comparison of MMRD lesions with MMRP lesions
One hundred and two (74.5%) cases lost staining for the mismatch repair enzyme MLH1 indicating a MMRD phenotype. When comparing MMRD with MMRP cases, MMRD cases occurred in older patients (mean age 76.7 vs 71.0; p=0.0029), more often in women (70.4% vs 36.4%; p=0.0008) and more often in the proximal colon (91.5% vs 71.9%; p=0.0130). In addition, the MMRD cases were more likely to show invasive carcinoma than the MMRP cases (35.3% vs 14.3%; p=0.0196).
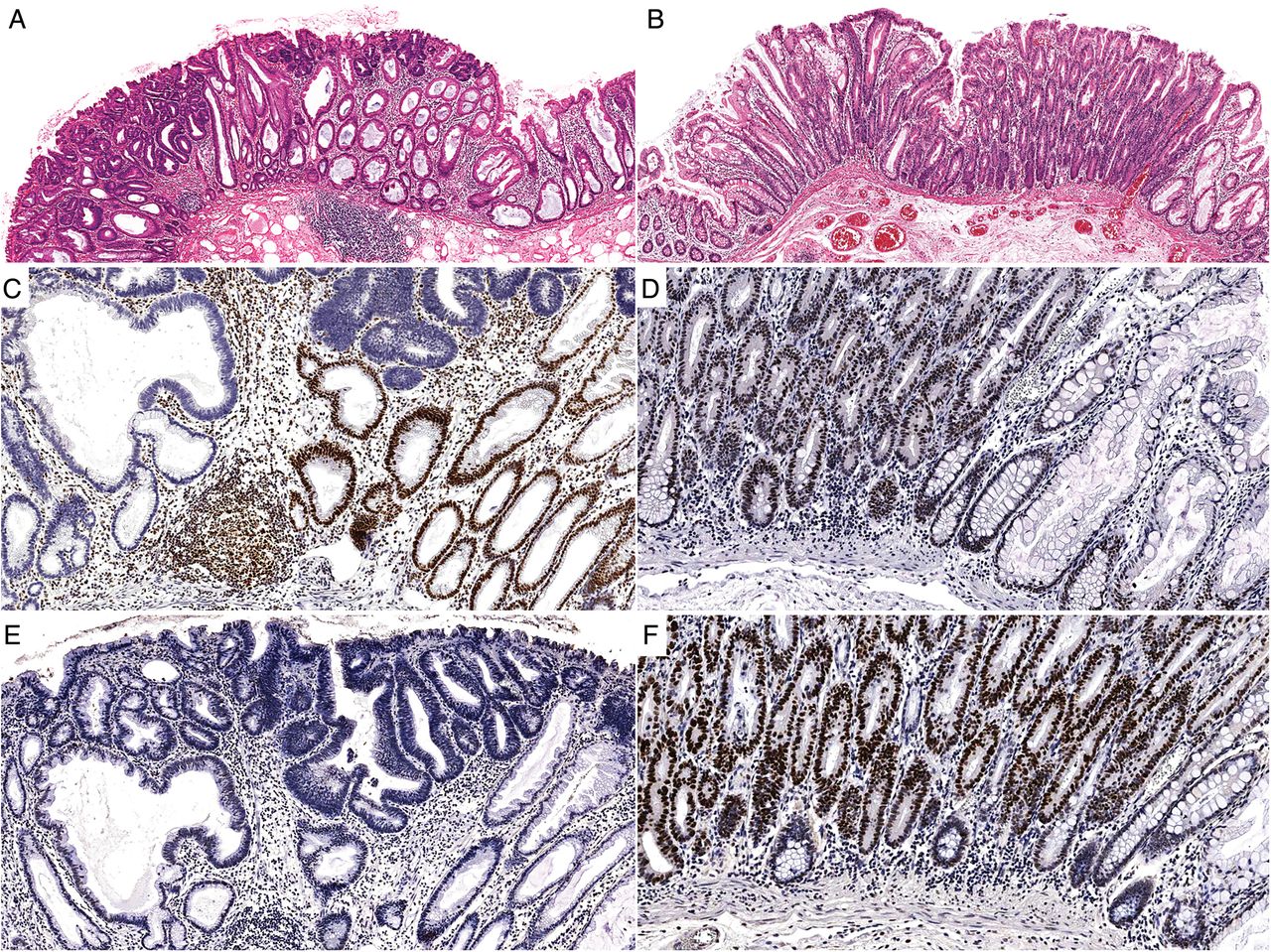
Table 4 and figure 4 compare the patterns of immunohistochemical staining in the advanced components of the lesions divided by mismatch repair status. MMRD cases were less likely to show positive p53 staining (6.9% vs 34.3%; p=0.0002). There was no significant difference in the staining patterns of β-catenin, p16 or MGMT between the MMRD and MMRP cases.
Comparison of the immunohistochemical staining patterns of the dysplastic and/or invasive components of the MMRD versus MMRP cases
{kind=link}
{kind=link}
{kind=link}
{kind=link}
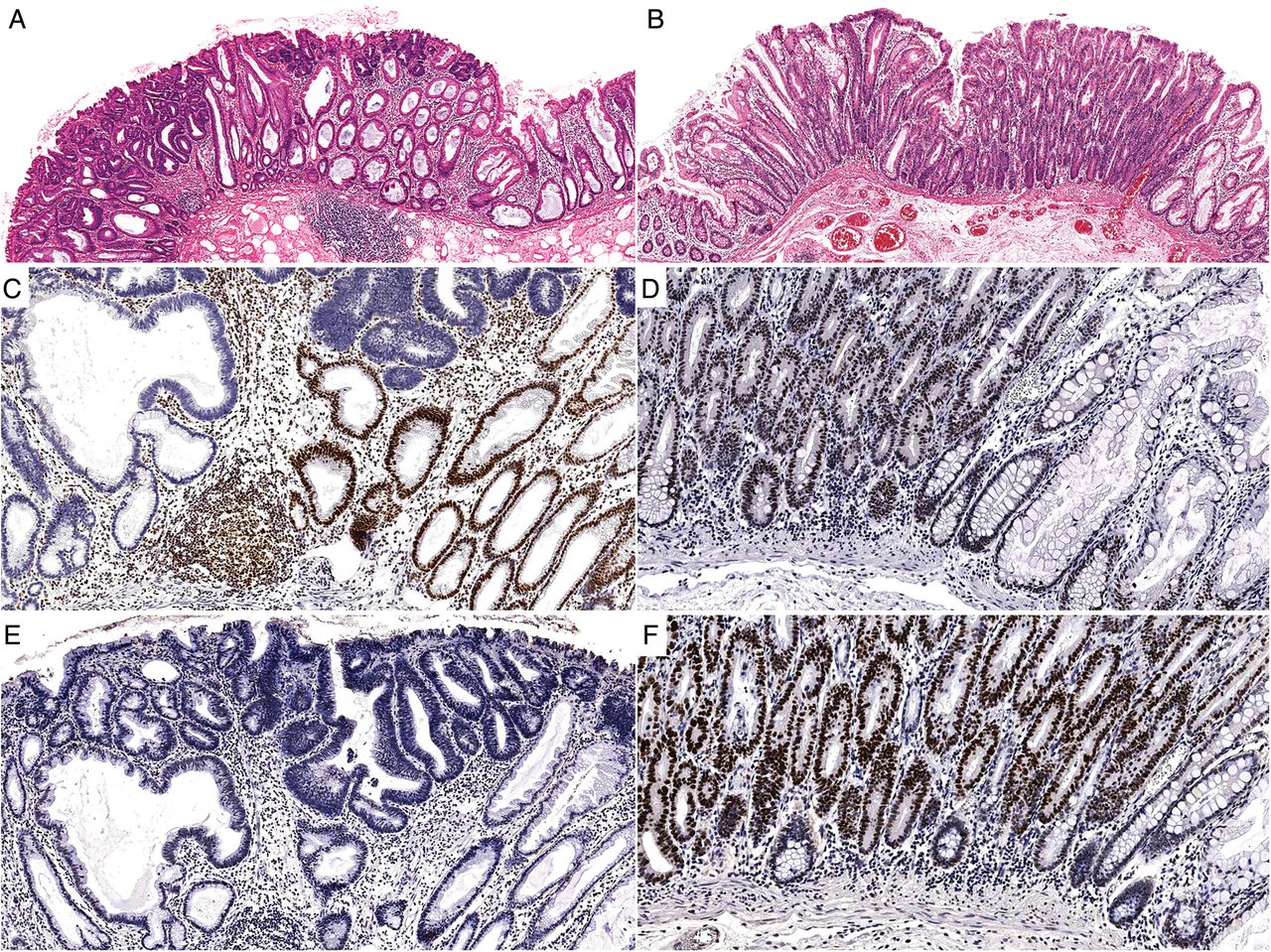
A mismatch repair-deficient (MMRD) (A) sessile serrated adenoma with dysplasia (SSAD) and a mismatch repair-proficient (MMRP) (B) SSAD. Both polyps are flat and show an abrupt transition from ordinary sessile serrated adenoma to overt cytological dysplasia. (C) and (D) are the MLH1 immunohistochemical stains for each case. Note the loss of staining in (C) compared with the retained nuclear expression in (D). (E) and (F) demonstrate the p53 immunohistochemical staining patterns for the same lesions. The overexpression in (F) is indicative of TP53 mutation and highlights a major difference between MMRD cases and MMRP cases.
As shown in table 3, MMRD cases were more likely to be CIMP-H than MMRP cases (98.0% vs 80.0%; p=0.0010), although both are heavily methylated. Unsurprisingly, they were more likely to show MLH1 methylation (94.1% vs 11.4%; p≤0.0001). The lack of complete correlation with immunohistochemistry is likely due to the contribution of the remnant non-dysplastic SSA to the total DNA analysed in MMRD cases and to partial methylation not sufficient to silence MLH1 in the MMRP cases. MGMT methylation was not significantly different between the MMRD and MMRP groups (42.2% vs 28.6%; p=0.1663) but tightly correlated with immunohistochemical expression (p<0.0001).
Discussion
The SSA is the prototype polyp of the serrated neoplasia pathway and a major contributor to the burden of colorectal carcinoma. This series has addressed important issues relating to the clinicopathological and molecular features of the very uncommonly observed SSAs ‘caught in the act’ of developing dysplasia/carcinoma. Importantly, this study includes only cases diagnosed using strict histological criteria after central pathological review by expert GI pathologists. This ensures a pure cohort of SSAs without contamination by other polyp types. In particular, cases of TSA arising in an SSA and admixed tubular adenoma and SSA have been excluded. Furthermore, this series has sufficient numbers to identify significant subgroups.
A major problem for colonoscopists is the occurrence of interval colorectal carcinoma. As discussed, factors that may contribute to this event include missed lesions, incompletely excised lesions, rapid progression of de novo lesions and inadequate surveillance intervals due to pathological misdiagnosis.7 ,8 ,11 For some, if not all of these reasons, serrated pathway carcinomas are over-represented in a series of interval carcinomas. In this series, we identified an additional worrying feature; SSAs with dysplasia/carcinoma are predominantly small polyps (54.3% <10 mm). Small SSAs are more difficult to detect than large SSAs, and thus are more likely to be missed at colonoscopy. Furthermore, some colonoscopists may assume that the dysplastic component of an SSAD is necessarily protuberant making these lesions more obvious endoscopically.10 Unfortunately, this does not appear to be the case, with only 17.0% of the dysplastic/malignant components showing a protuberant growth pattern. This misconception may have developed because of the misdiagnosis of TSA arising in an SSA as an SSAD, in which the TSA component will frequently display protuberant growth.27
Apart from the small size and flat nature of most of the polyps in this series, numerous other clinicopathological features have been demonstrated. Many are expected based on previous studies of either SSAs without dysplasia or of BRAF-mutated carcinomas.34–37 In particular, the older age and female predominance of the patients were confirmed and the predilection for the proximal colon was again striking.
SSAs without dysplasia or malignancy were not included in this study; however, previous studies have examined the clinicopathological features of these polyps.11 ,36 ,37 In a recent study from our group that reviewed an unselected consecutive series of 6340 polyps, 579 patients with SSAs without dysplasia were diagnosed using the criteria of the WHO Classification. The mean age of patients with these SSAs was 58.6 years, 80% of polyps were proximal, 56% were women and the mean polyp size was 8.5 mm.11 Thus, SSAs without dysplasia have a similar site and gender distribution to SSAs with dysplasia and/or carcinoma but occur at a younger age and are slightly smaller. Based on our work, the lag between SSA without dysplasia and SSAD is approximately 17 years. It is somewhat shorter in MMRP lesions with a lag of 12 years. In a previous study by Lash et al,37 interrogating a pathology database, the lag period between SSA and SSAD was only 5 years. The reason for the difference between the current study and the study of Lash et al is not certain but could relate to the era from which the samples were obtained. The prevalence of SSAs was lower in the Lash et al study as colonoscopists were not detecting and removing them so frequently at that time and the mean ages may have been influenced by missed lesions. Importantly, in our study, there is no significant difference in age of patients with dysplasia versus those with carcinoma or between the study lesions and the separate 99 consecutive BRAF-mutated carcinomas. This, together with the rarity of detection of SSADs (27/6340 (0.4%) in our consecutive series), provides further support for the theory that once dysplasia develops there is a rapid progression to malignancy.
A BRAF mutation effectively confirms the serrated origin of a colorectal carcinoma. The BRAF mutation status is also becoming increasingly relevant as an adverse prognostic factor.38 This finding is particularly powerful when combined with the mismatch repair function of the carcinoma. MMRD cancers have an MSI phenotype, which confers a good prognosis. When combined with a BRAF mutation, these cancers still tend to behave well with a reduced propensity to nodal and systemic metastases.39 In contrast, BRAF-mutated and MMRP (microsatellite stable) tumours are the most aggressive molecular subtypes of colorectal carcinoma.40 ,41
Because of this critical dichotomy between MMRD and MMRP serrated pathway carcinomas, these two groups were separated for the clinicopathological and molecular analysis. At a clinicopathological level, the most striking feature was the difference in gender distribution. Seventy per cent of MMRD SSAs occurred in women compared with only 34.3% of MMRP cases. In addition, only 8.5% of MMRD cases arose in the distal colorectum compared with 28.1% of the MMRP cases. These findings should be borne in mind during screening/surveillance colonoscopy. In particular, SSAs in men tend to develop into an aggressive subtype of carcinoma. Furthermore, small and distal SSAs should not be disregarded, particularly in men. Troublingly, 12.9% of the MMRP cases arose in the rectum. By contrast, none of the MMRD lesions did so. The association of SSADs which progress down the MMRD pathway with older age, female gender and the proximal colon may be due to a greater propensity for CIMP in that environment, which leads to a higher chance of methylating MLH1. Indeed, we observed a higher level of CIMP in SSADs with MMRD. Additionally, there were more invasive cancers in the MMRD group than the MMRP group. The reasons for this are not clear, but we doubt that this reflects a greater propensity for the MMRD group to progress to carcinoma. One possible explanation is that the MMRP group gives rise to an aggressive subtype of cancer that rapidly overruns the SSA component meaning cases with both components are rarely encountered. Finally, the MMRD carcinomas, as anticipated, showed more tumour-infiltrating lymphocytes and a more prominent Crohn-like reaction than the MMRP carcinomas. Interestingly and perhaps reflecting their more aggressive biology, the MMRP carcinomas were more likely to be infiltrative and to have a serrated morphology.
The molecular steps involved in progression from SSA to cancer are incompletely understood and may be of relevance to future screening tests and chemoprevention. BRAF mutation is an early and likely initiating event.20 ,42 Recent evidence suggests that BRAF mutation may direct the development of CIMP.43 Our study shows the occurrence of cytological dysplasia is accompanied by demonstrable molecular events. In MMRD cases, methylation-induced silencing of the MLH1 gene occurs at this transition. In this study, MLH1 promoter methylation tightly correlated with immunohistochemical staining.
Besides methylation of MLH1, other oncogenic pathways are involved in serrated neoplasia. WNT pathway activation is present in 93% of colorectal carcinomas,44 although the precise role of WNT signalling in the serrated pathway is not clear and it is not an early driver event as it is in conventional adenomas. Several studies have used immunohistochemistry for β-catenin to assess this issue. Nuclear translocation of β-catenin is a key event in the canonical WNT signalling pathway and is a useful surrogate marker for WNT pathway activation. A shift from the normal (membranous) pattern of staining to nuclear staining indicates activation of the WNT pathway. Most studies addressing the topic have demonstrated nuclear β-catenin staining in a proportion of SSADs, although the range is quite variable (50–100%).45–47 In a thorough recent paper, strong or intermediate staining was demonstrated in 60.9% of advanced SSAs (similar to our results) and correlated with methylation of the upstream WNT antagonists SFRP, MCC and AXIN2.47 This upstream methylation is postulated to be the mode of WNT pathway activation in the serrated neoplasia pathway.47–49 In the current series, nuclear β-catenin staining was uncommon in the non-dysplastic SSA component, consistent with previous results, and supporting the notion that WNT signalling is not a major factor in the development of SSAs. However, it became frequent in the dysplastic and invasive components of both the MMRD and MMRP lesions, supporting the concept that WNT signalling plays a role in polyp progression.
Methylation-induced silencing of CDKN2A (encoding p16) is also postulated to play a role in progression to malignancy in serrated pathway carcinomas.1 P16 is an important tumour suppressor that can induce cell cycle arrest at the G1/S checkpoint in response to uncontrolled proliferation and induces the phenomenon of oncogene-induced senescence.50 Loss of function of p16 has been postulated as a requirement to overcome the senescence barrier, which is critical for malignant progression.51 Immunohistochemistry for the p16 protein has been demonstrated to be an effective method to interrogate the function of this critical tumour suppressor gene. Kriegl et al,51 previously demonstrated aberrant p16 staining in SSAs with dysplasia/carcinoma. In that study, they showed increasing p16 expression until the development of either high-grade dysplasia or invasive carcinoma, when it was suddenly lost in a definite subset of cases, presumably secondary to methylation-induced silencing. We have previously demonstrated loss of p16 staining late in the malignant progression of BRAF-mutated (but not KRAS-mutated) TSAs.27 Similarly in the current study, loss of p16 staining tended to occur late in the progression of SSAs, often at the step between high-grade dysplasia and carcinoma. Interestingly, despite the difference in CIMP-H status between MMRD (98.0%) and MMRP (80.0%) cases, the rates of p16 loss were almost identical between the two groups.
TP53 is a critical tumour suppressor gene. Mutation was assessed using the surrogate of p53 immunohistochemistry. Most substitution mutations of TP53 result in markedly increased nuclear expression by immunohistochemistry.52 Overall, p53 immunohistochemistry has a specificity of 90% and a sensitivity of 67% for detecting TP53 mutation.52 Bond et al,53 have demonstrated significant differences in TP53 mutation rates between BRAF-mutated microsatellite stable and unstable carcinomas (40.6% vs 16.9%), with the microsatellite stable group having more frequent mutations. Our study is in agreement with this finding and the presence of a TP53 mutation may be part of the explanation for the poor prognosis associated with the MMRP cancers.
The role of MGMT in SSA progression is less clear and has not been extensively examined. In this study, 27.7% of cases had areas with loss of staining for MGMT by immunohistochemistry. MGMT is a DNA repair enzyme separate from the mismatch repair system. It has been correlated with KRAS mutations previously, but obviously this is not a factor in SSAs.54 ,55 Loss of function of MGMT may allow additional accumulation of mutations and may contribute to malignant progression.
The final point of discussion relates to the comparison between MMRP SSAs and advanced BRAF-mutated TSAs. This is relevant because both of these polyps give rise to the aggressive BRAF-mutated, microsatellite stable subtype of colorectal carcinoma. In a previous study, we demonstrated that BRAF-mutated TSAs have frequent origin in SSAs.27 Thus, we would expect that the advanced BRAF-mutated TSAs are quite similar to the MMRP polyps in this study. In fact, this seems to be the case. When comparing age, gender, location, size, staining for p53, β-catenin and p16 and CIMP status, only location, size and p16 staining showed a statistically significant difference between these groups. Furthermore, the discrepancy in location and size may reflect ascertainment bias in the previous study, as it was not a consecutive series of cases and was intrinsically biased towards larger polyps. The advanced BRAF-mutated TSAs showed more frequent p16 loss by immunohistochemistry (73% vs 43%; p=0.0329) but the reason for this is not clear. We hypothesise that the MMRP SSAs and the advanced BRAF-mutated TSAs are similar polyps, but the SSAs have ‘skipped’ the TSA stage in their evolution. However, the discrepancy in p16 staining suggests there are some genuine differences in their biology.
This study has limitations. First, some cases were excluded because of insufficient material for complete analysis. However, this is unlikely to have adversely impacted on the key findings. If anything, the requirement for adequate material for molecular analyses will have resulted in a bias towards larger polyps. Second, the cases were not microdissected for separate molecular analysis between the non-dysplastic and dysplastic/malignant components of the polyps. This is unlikely to have had a material impact on the results as BRAF mutation status and CIMP are established early, although it is possible that CIMP continues to evolve as the polyps progress. Because immunohistochemistry stains at a single-cell level, microdissection is not required when using this technique.
In summary, we addressed the clinicopathological and molecular features of a large series of SSAs with dysplasia and/or carcinoma. We found that these are mostly small polyps, only slightly larger than SSAs without dysplasia. They are uncommonly observed and occur in patients 17 years older than those with SSAs without dysplasia, suggesting a long dwell time with little change in size before rapid progression to malignancy. Approximately three-quarters progress through a MMRD pathway. Although a minority, MMRP cases are the precursors of an aggressive subtype of colorectal carcinoma. They occur more often in men at a younger age than MMRD cases and a significant subset occurs distally. Malignant progression of SSAs occurs through a combination of MLH1 and CDKN2A silencing, WNT pathway activation and TP53 mutation.
References
Footnotes
Contributors MB, NW, CR, IB, AC, BL and VW collaborated to develop the study design. MB, S-AP and DM performed the experimental work. MB and NW performed the histopathological analyses. MB, S-AP, DM and VW interpreted the data. MB wrote the manuscript. NW, CR, IB, AC, BL and VW amended the manuscript.
Funding This project was supported by a grant from the National Health and Medical Research Council (NHMRC) of Australia (ID: 1063105). Mark Bettington received PhD scholarship funding from Cancer Council Queensland.
Competing interests None declared.
Ethics approval The ethics committee of the QIMR Berghofer Medical Research Institute (P1298).
Provenance and peer review Not commissioned; externally peer reviewed.