Article Text

Abstract
BACKGROUND There is evidence for genetic susceptibility to Crohn’s disease, and a tentative association with tumour necrosis factor (TNF) and HLA class II alleles.
AIMS To examine the potential of genetic linkage between Crohn’s disease and the MHC region on chromosome 6p.
METHODS TNF microsatellite markers and, for some families, additional HLA antigens were typed for 323 individuals from 49 Crohn’s disease multiplex families to generate informative haplotypes. Non-parametric linkage analysis methods, including sib pair and affected relative pair methods, were used.
RESULTS Increased sharing of haplotypes was observed in affected sib pairs: 92% (48/52) shared one or two haplotypes versus an expected 75% if linkage did not exist (p=0.004). After other affected relative pairs were included, the significance level reached 0.001. The mean proportion of haplotype sharing was increased for both concordant affected (π=0.60, p=0.002) and unaffected sib pairs (π=0.58, p=0.031) compared with the expected value (π=0.5). In contrast, sharing in discordant sib pairs was significantly decreased (π=0.42, p=0.007). Linear regression analysis using all three types of sib pairs yielded a slope of −0.38 at p=0.00003. It seemed that the HLA effect was stronger in non-Jewish families than in Jewish families.
CONCLUSIONS All available analytical methods support linkage of Crohn’s disease to the MHC region in these Crohn’s disease families. This region is estimated to contribute approximately 10–33% of the total genetic risk to Crohn’s disease.
- Crohn’s disease
- HLA
- linkage
- inflammatory bowel disease
- tumour necrosis factor
- genetics
Abbreviations
- MHC
- major histocompatibility complex
- MZ
- monozygotic
- TNF
- tumour necrosis factor
Statistics from Altmetric.com
Crohn’s disease is one of the two major forms of chronic inflammatory bowel disease (IBD). The important role of genetic factors in the aetiology of Crohn’s disease has been strongly supported by genetic epidemiological studies, including consistent ethnic differences, dramatic familial aggregation,1 and a significantly increased concordance rate in monozygotic twins (0.44) compared with that in dizygotic twins (0.04).2 However, the exact genetic factors and their influence on the susceptibility of an individual to Crohn’s disease are still unclear.
To date, genes within the major histocompatibility complex (MHC) region have been the most intensively studied in Crohn’s disease. Genes in this complex are critical in antigen presentation and recognition and thus fundamental to the immune response. However, HLA association studies in Crohn’s disease revealed somewhat confusing results, as some studies have shown positive associations with HLA genes and some have not. Even among studies that showed a positive association, different HLA alleles have been associated with Crohn’s disease.3-8 Variations in association results could conceivably be due to inadequate sample sizes, inappropriate controls, or genetic heterogeneity. However, the observation that Crohn’s disease is associated with different HLA DR and DQ alleles suggests that this gene itself is unlikely to be the disease susceptibility gene, but is in linkage disequilibrium with other more specific Crohn’s disease susceptibility genes within the HLA complex and that this linkage disequilibrium may vary among different populations.
Other candidate genes residing in the MHC region are the genes for tumour necrosis factor α (TNF-α) and TNF-β. Both cytokines are potent modulators of the immune response and mediators of inflammation. A central role for TNF-α in the pathogenesis of Crohn’s disease has been shown by dramatic clinical responses following infusion of an anti-TNF-α monoclonal antibody (cA2, Centocor, Inc., Malvern, Pennsylvania) in open label and placebo controlled trials.9 ,10 Furthermore, in a case control study evaluating genetic markers at five loci flanking and within the TNF locus (TNF microsatellites), a specific TNF microsatellite haplotype, a2b1c2d4e1, was found to be associated with Crohn’s disease.11 In contrast, polymorphisms of the TNF-α gene promoter region (TNF-308) were not associated with either Crohn’s disease as a whole,12 or a subset of Crohn’s disease.13
One way to evaluate the importance of the MHC region in Crohn’s disease is to perform a linkage study. A linkage study asks whether a genetic marker (for example, MHC gene) travels together with the disease (for example, Crohn’s disease) in families. If a Crohn’s disease susceptibility gene is located close enough to the marker gene (for example, TNF, HLA)—that is, within less than 10 million base pairs, then in a particular family, the disease phenotype and a particular marker allele will travel together from parent to offspring, although in different families there may be different marker alleles travelling together with the disease. Thus, a genetic linkage study will help clarify the potential role of the MHC region in the susceptibility to Crohn’s disease, even when no population associations or differing associations are identified.
Linkage analysis utilises families and thus is free from potential bias due to population stratification (inappropriate controls). However, the results of linkage analysis can be affected by other factors such as sample size (number of families), genetic heterogeneity (linked forms and unlinked forms within Crohn’s disease), clinical heterogeneity (ulcerative colitis and Crohn’s disease), and analytical approaches (parametric and non-parametric methods). Some cosegregation studies between Crohn’s disease and HLA antigens in families with multiple members affected with Crohn’s disease have shown evidence for linkage,14-17 while others did not.18-23The reasons for such inconsistent results are unclear, but could be due to any combination of factors listed above. Genetic heterogeneity within Crohn’s disease has been evidenced both by epidemiological observations,24 and genetic marker studies.5 ,6 ,13 ,25 In an epidemiological study,24 we observed an increased empiric risk for IBD in relatives of Jewish probands compared with the relatives of Caucasian non-Jewish probands. Genetic marker studies have observed different associations not only between different populations (for example, DQB1*04 association in a Japanese population,25 but DQB1*0501 in Caucasian populations from France6 and North America5), but also within patients with Crohn’s disease (for example, DRB1*03 was negatively associated with fistulising Crohn’s disease13).
The use of non-parametric methods allows linkage to be evaluated without imposing any specific genetic model and without assuming related parameters, such as gene frequency and degree of penetrance. While sib pair methods are most often used for non-parametric linkage analysis, linkage analysis with other affected relative pairs can increase the statistical power to identify linkage.26 ,27
Concurrence of ulcerative colitis and Crohn’s disease in the same family exceeds the expected frequency by chance alone, suggesting these two disorders have some of their aetiology in common.24However, the clinical differences between ulcerative colitis and Crohn’s disease, and the differences observed in subclinical and genetic marker associations, strongly suggest that ulcerative colitis and Crohn’s disease are also to a large extent genetically distinct.4 ,5 ,28-30 Therefore, a clinically well defined homogeneous group of families with either ulcerative colitis or Crohn’s disease will maximise the chance of identifying linkage between genetic markers and ulcerative colitis or Crohn’s disease.
We therefore investigated evidence for or against the MHC linkage with Crohn’s disease in a large number of US families with at least two sibs affected with Crohn’s disease and/or other affected relative pairs, and with no known members affected with ulcerative colitis in the family (Crohn’s disease only families). Both TNF microsatellite markers and HLA class I and class II data were used to generate haplotypes to determine identical by descent status in sib pairs and relative pairs. Non-parametric linkage methods were applied to these families and significant evidence for linkage between TNF-HLA haplotypes (the MHC region on short arm of chromosome 6) and Crohn’s disease was found.
Subjects and methods
PATIENTS AND FAMILIES
The patients were ascertained through the IBD clinical programmes at Cedars-Sinai Medical Center, or referred by the Crohn’s and Colitis Foundation of America and physicians nationwide. Through these patients, we recruited families with two or more sibs affected with Crohn’s disease and with no known family history of ulcerative colitis. More distant relatives (than sibs) who were affected with Crohn’s disease and connecting relatives were also included in this study. All family members were interviewed in person or by telephone to obtain detailed family and medical histories. Medical records were obtained to confirm the diagnosis. The study protocol was approved by the institutional review board of Cedars-Sinai Medical Center.
Blood samples were obtained from 323 individuals of 49 Crohn’s disease only families. Among these families, 40 have two affected sibs, eight have three affected sibs, and one has four affected sibs. In addition, 15 of the 49 families have other distant affected relatives who were also included in this study. The sample population is composed of Caucasians from North America, 36% being of Ashkenazi Jewish origin.
DIAGNOSTIC CRITERIA FOR CD
The diagnosis of Crohn’s disease was based on conventionally accepted criteria as described previously.31
TNF GENOTYPING
DNA isolation
High molecular weight DNA was isolated either from peripheral blood leucocytes (PBL) or Epstein-Barr virus transformed cell lines using the method previously described.32 ,33
TNF microsatellite typing
TNF microsatellites were determined at five loci from genomic DNA from probands and relatives as previously described34 with modifications.11 The TNF genes are tandemly arranged within a 7 kilobase (kb) region in the MHC,35 250 kb centromeric to class I and 340 kb telomeric to class III on chromosome 6.36 ,37 The TNFa and TNFb microsatellites are located 3.5 kb upstream of the TNFb gene. TNFc is located in the first intron of TNFb. The TNFd and TNFe microsatellite loci are located 8 to 10 kb downstream of the TNFa gene (fig 1). Polymerase chain reaction (PCR) products from at least one cell line of known TNF microsatellite haplotype34 per TNF microsatellite locus were analysed with the sample DNA to aid in interpretation of ambiguous alleles. Microsatellite alleles were interpreted separately by two investigators blinded to diagnosis. Successive alleles differ by one dinucleotide repeat for TNFa, TNFc, TNFd, and TNFe. For TNFb, alleles differ by one or two bases.34 The smallest allele is termed 1, with larger alleles being numbered consecutively.

Location and order of five TNF microsatellite loci within the HLA complex on chromosome 6. Arrows represent location and orientation of PCR primers used for typing. The exact orientation of TNFd and TNFe microsatellites with respect to the TNF locus has not been determined.
HLA SEROLOGICAL TYPING
Serological typing for all HLA antigens was performed using a two colour fluorochromasia, complement dependent, microcytotoxicity technique.38 Class I typing was performed directly on PBL or T cells. Class II typing was performed on B cells. T and/or B cells were obtained using Cell Prep I and/or II magnetic beads39(Dynal, Great Neck, New York, USA), respectively. All HLA-A, B, C, DR, and DQ serologically defined specificities with the exception of A43 (an antigen found only in South African Blacks) were tested for each typing. Sera were obtained by international exchange. The DR/DQ typings were supplemented by commercial trays (One Lambda, Canoga Park, California, USA).
TNF microsatellite polymorphisms and HLA class I and class II antigens were used to generate informative haplotypes so that identity by descent status could be determined, especially for distant affected relatives. Identity by descent refers to alleles that are the copies of the same ancestral allele. Both affected sibs and unaffected sibs were genotyped to increase the informativeness of the families when one or both parents were missing, and to be used for evaluation of discordant sib pairs. In four families with affected sib pairs, both parents were unavailable or non-informative in this region, and in 12 families, one parent was unavailable or non-informative. In the remaining 33 families, all four haplotypes were unequivocally determined.
LINKAGE AND STATISTICAL ANALYSIS
To increase the power of identifying linkage it is important to determine the frequency of the marker alleles or haplotypes shared by sib pairs or relative pairs identical by descent. Non-parametric linkage approaches were used to examine linkage between TNF-HLA haplotypes and Crohn’s disease. Each of these methods, though based on the same principle, has its own utility, as delineated below. Given that this study used a candidate gene approach, which tested a specific hypothesis—whether the susceptibility to Crohn’s disease is linked with the MHC haplotype—we used p<0.05 as our criterion for evidence of linkage between this region and Crohn’s disease susceptibility.
Sib pair analysis with complete informative families
The observed numbers of affected sib pairs who share zero, one, and two haplotypes identical by descent are compared with expected numbers based on null hypothesis (no linkage), which are 25%, 50%, and 25% respectively, by a χ2 goodness of fit test.
In this test, only sib pairs from complete informative families (both parents informative) were used. If a family has three affected sibs, it would contribute three sib pairs (sib 1-sib 2, sib 1-sib 3, and sib 2-sib 3); similarly, four affected sibs would provide six sib pairs. A total of 52 sib pairs were obtained from 25 families with two affected sibs, seven families with three affected sibs, and one family with four affected sibs. As the sib pairs from families with three or four affected sibs are not independent, we examined sib trios separately by comparing the observed frequencies of affected sib trios who all share zero, all share one, or all share two haplotypes identical by descent with the expected values. The expected values under the null (no linkage) hypothesis are 9/16, 6/16, and 1/16, respectively.
Allele sharing method with all informative haplotypes
In order to utilise families with only one informative parent, we calculated the mean proportion of haplotypes (π) shared identical by descent from the number of informative haplotypes (N) and the number that were identical by descent (I). Information was provided only by heterozygous parents with unambiguous parental transmission. When both parents were heterozygous and the parental origin of the haplotypes could be determined, each affected sib pair contributed two informative haplotypes (one from each parent). When one parent’s genotype was missing, she/he did not contribute any information unless the genotype could be inferred by examining at least three children, including those affected and unaffected with Crohn’s disease. This procedure does not require knowledge of marker allele frequencies in the population and uses all available information from parents. The estimated mean proportion of haplotypes shared identical by descent for a locus (π=I/N) was compared with 0.5 to test for linkage, using the statistic Z = 2(I − N/2)/N1/2 for an approximately standardised normal deviate.40 For this test, sib pairs from families with at least one informative parent (heterozygous) were used (118 informative haplotypes).
SIBPAL program in the SAGE package
Two point linkage analysis was also performed using the SIBPAL program (version 2.7) of the Statistical Analysis for Genetic Epidemiology (SAGE) package.41 ,42 This computerised sib pair analysis program estimates the proportion (π) of alleles the sib pair shares identical by descent at that locus. The observed mean proportion of alleles shared by sib pairs was compared separately for concordant affected, concordant unaffected, and discordant sib pairs with expected 0.5 by z test (one sided). If the linkage between disease and locus exists, an increase of π in both concordant affected and concordant unaffected groups, but a decrease of π in the discordant group should be observed.
Although most sib pair methods focus on affected sib pairs, discordant sib pairs add additional information. If for some reason, not related to the disease under study, particular alleles tend to be transmitted from parents to offspring with a frequency greater than 50%, there will be an increased proportion of allele sharing in all sib pairs. If only affected sib pairs are examined, this may falsely suggest that there was a linkage between disease susceptibility and the marker locus. To eliminate such false inference of linkage, we compared the mean proportion of sharing haplotypes in discordant sib pairs with that in affected concordant sib pairs. When there is linkage between the disease locus and the tested marker (the TNF-HLA region), marker allele sharing will be significantly greater when both sibs are clinically concordant than when they are discordant.
In this program, when the parents were not informative, an estimated proportion of allele sharing was used in the test. Therefore, in this analysis, all sib pairs were utilised, including affected concordant, discordant, and unaffected concordant sib pairs from fully informative and partially informative families.
Linear regression to test for possible linkage with SIBPAL in the SAGE package
Linear regression analysis with the SIBPAL program was also conducted to assess the prediction of the differences in disease status (Crohn’s disease) according to the degree of sharing the TNF-HLA haplotype. In this method, all types of sib pairs are used and the affected individuals have a trait value 1, the unaffected, 0. The y axis is the squared difference between the pairs (Y) and the x axis is the proportion of haplotypes shared identical by descent between the pairs (π). For a qualitative trait, Y varies between 0 and 1 (Y = 0 for sib pairs that have the same trait value—that is, both affected and unaffected, and Y = 1 for sibs that have different trait values). The proportion of haplotypes shared identical by descent between the pairs (π) ranges from 0 to 1 (0 for sharing none, 0.5 for sharing one, 1 for sharing two, and the values in between are derived from estimation by using non-informative parents).
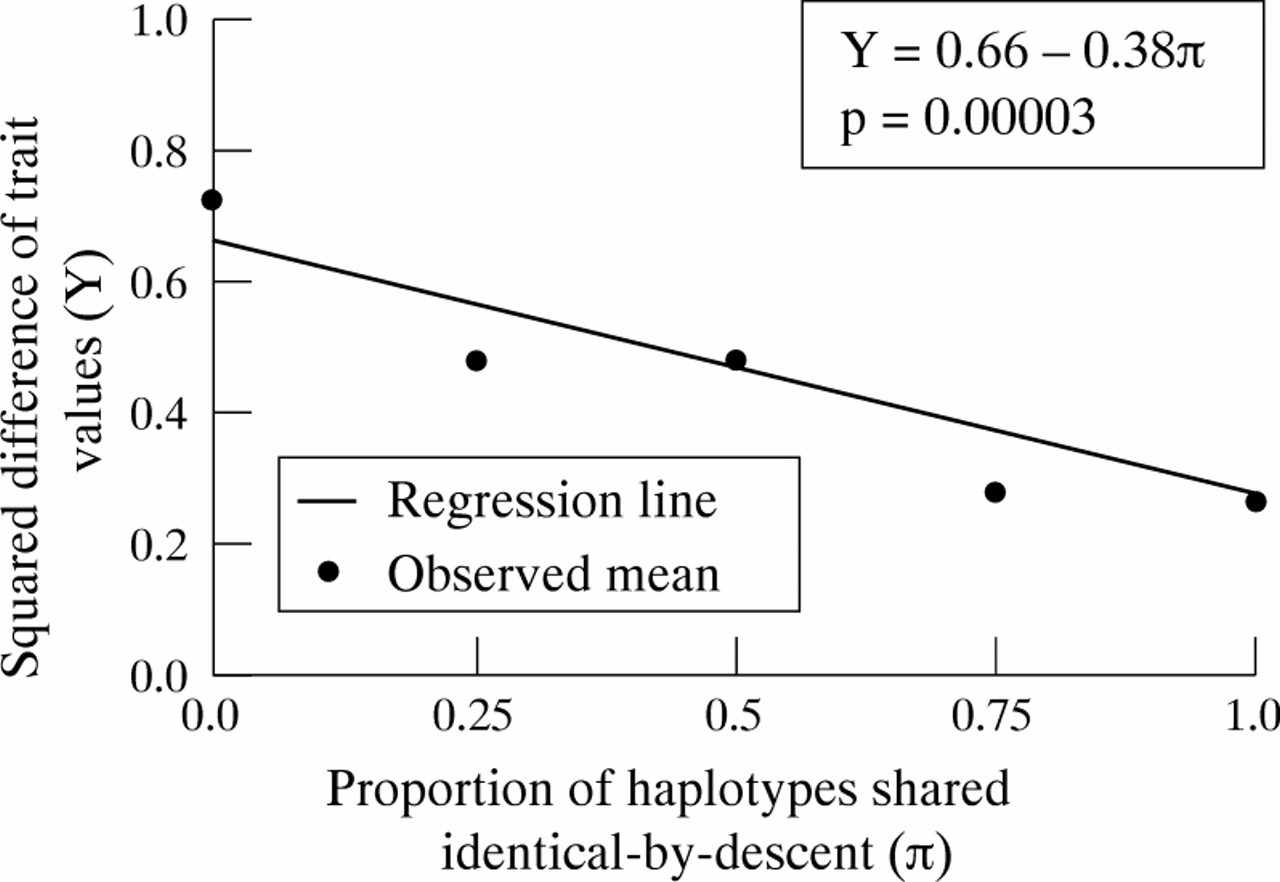
If there were no linkage, a horizontal line would be observed—that is, the degree of clinical concordance would be independent of haplotype sharing. A significantly negative regression coefficient b of the dependent variable (phenotype) Y on the independent variable (genotype) π is taken as evidence for linkage (the more alleles are shared by a sib pair, the more alike their phenotypes are). This inverse relation is expressed by a negative regression line in this analysis (fig 2).
{kind=link}
{kind=link}
Significant linear relation between phenotypic concordance and proportion of shared TNF-HLA haplotypes identical by descent among sibs. Those who share no haplotypes have the greatest degree of phenotypic discordance and those who share both are the most concordant. Thus, among sibs, similar genotypes (haplotype sharing) are correlated with similar phenotypes (both affected with Crohn’s disease or both unaffected). These data are consistent with linkage between the MHC region and Crohn’s disease.
Affected relative pair method
The concept of the affected relative pair method is the same as that of the sib pair method; the only difference is the degree of relation. That is, in the affected relative pair method, one compares the observed frequency of affected relative pairs sharing alleles/haplotypes identical by descent with the expected value if there was no linkage and the degree of the specific relations.26 ,43 This method assesses whether a disease susceptibility gene has travelled through the pedigree to both affected individuals from their common ancestor together with the same marker allele. The advantage of using affected relative pairs is that the statistical power is greater with more distant relations between an affected pair given the same number of pairs analysed, and thus additional family members can be utilised, increasing the efficiency of individual pedigrees. The z statistic was used to compare the observed number of sharing, Ti, with the expected number of sharing haplotypes identical by descent between relative pairs, Pi, z = (ΣTi − ΣPi − 0.5)/[ΣPi (1 − Pi)]1/2.43
Estimation of TNF-HLA contributions
The increased risk over population prevalence to sibs due to a disease susceptibility gene in the MHC region was estimated using the equation from Risch44: λMHC = φ0/p (share 0 haplotype identical by descent given both are affected), where φ0 is the probability that two relatives share zero marker alleles or haplotypes identical by descent under the assumption of no linkage between the marker and the disease (φ0 = 0.25 for sib pairs).
Under the multiplicative model,44 the proportion (PMHC) of total relative risk in siblings (λs) (or other relatives) contributed by the gene linked to the MHC region (λMHC) was determined by the following equation: PMHC = log λMHC/log λs × 100%.
The lower bound of genetic contribution was estimated by a method derived by Rotter and Landaw.45 This method requires monozygotic (MZ) twin concordance data for Crohn’s disease, the empiric sibling recurrence risks, and the sharing of identical by descent genes at the MHC by affected sib pairs as shown below:
Results
AFFECTED SIB PAIR ANALYSIS WITH COMPLETELY INFORMATIVE FAMILIES
Of 52 sib pairs affected with Crohn’s disease, four shared no TNF-HLA haplotypes, 30 shared one, and 18 shared two. As shown in table1, the observed frequency of sharing one or two haplotypes was significantly increased compared with the expected number under the no linkage assumption (p=0.013). After pooling the latter two groups (sharing one or two haplotypes), we observed 48 sib pairs sharing at least one haplotype which was significantly greater than the expected 39 (p=0.004).
Haplotype sharing in concordant affected sibs with Crohn’s disease
Examining affected sib trios separately, we observed that in two out of seven informative families, both haplotypes were shared by all three sibs, in three families one haplotype was shared by all three sibs, and in two families, no haplotypes were shared by all three sibs (table 1). Although the number of sib trios was too small (n=7) to perform statistical testing, the observed number of sharing among all three affected sibs seemed to be increased compared with the expected value. In the family with four affected sibs, one haplotype was shared by all four.
ALLELE SHARING METHOD WITH INFORMATIVE HAPLOTYPES
Of 118 informative haplotypes from 45 families, 72 haplotypes were shared by affected sib pairs. The observed haplotype sharing (61%) was significantly greater than that expected (50%) (p=0.008 for the one sided test).
SIB PAIR ANALYSIS WITH THE SIBPAL PROGRAM
Both concordant affected sib pairs (n=70) and concordant unaffected sib pairs (n=64) had an increased mean proportion of sharing TNF-HLA haplotypes compared with the expected 0.5 (0.60, p=0.002; and 0.58, p=0.031, respectively; table 2). In contrast, sharing in discordant sib pairs (n=117) was significantly decreased (0.42) compared with expected (0.5) (p=0.007) (table 2).
Mean proportion of haplotypes shared in each type of sib pair (π)
The difference between the two means in discordant sib pairs and in affected concordant sib pairs was highly significant (p=0.0001 for one sided test).
Jewish and non-Jewish families were also analysed separately with the SIBPAL program and the results are summarised in table 3. It seems that the linkage effect of the HLA region is stronger in non-Jewish than it is in Jewish families, as indicated by the greater distortion from expected 0.5 in all sibling groups. Nevertheless, the allele sharing distortion in Jewish families is still apparent and consistent with linkage. It should be noted that the sample size in Jewish families is relatively small and that it is not surprising that the distortions in the Jewish group alone do not attain statistical significance. The observed proportion of allele sharing in affected sib pairs in Jewish families (π=0.55) is smaller, but is not significantly different from that in non-Jewish families at this sample size (π=0.63, p=0.3).
Mean proportion of haplotypes shared in each type of sib pair (π) for Jewish and non-Jewish families separately
LINEAR REGRESSION ANALYSIS USING THE SIBPAL PROGRAM
Using all types of sib pairs we performed regression analysis with the trait difference between a sib pair as the dependent variable and the proportion of haplotype sharing between a sib pair as the independent variable. A highly significant negative regression line slope was observed (−0.38, p=0.00003; fig 2). In other words, those who shared zero haplotypes were the most often clinically discordant, and those who shared both haplotypes were the most often clinically concordant.
AFFECTED RELATIVE PAIRS
When expanding sib pairs to other relative pairs as shown in table4, we observed that distant affected relative pairs also have increased frequencies of sharing TNF-HLA haplotypes than would be expected if no linkage existed (Z=3.07, p=0.001).
Observed and expected haplotype sharing between various affected relative pairs
ESTIMATION OF TNF-HLA CONTRIBUTION
As only four of 52 affected sib pairs share zero TNF-HLA haplotypes (0.08), the estimated relative risk of Crohn’s disease due to the MHC region (not due to any specific alleles) in siblings of patients with Crohn’s disease was estimated to be 3.13 (0.25/0.08).
If we assume that the risk to siblings of patients with Crohn’s disease is approximately 3–5%,24 and the risk to the general population is 100/100 000 (summarised by Calkins and Mendeloff for North America regions46), the total relative risk in sibs over the population risk is as high as λs=30–50. Thus, the 3.13 relative risk due to the gene linked to the MHC region will contribute an estimated 29–33% of the total increased risk over the population risk of Crohn’s disease under a multiplicative model.
To estimate the coefficient of the genetic contribution provided by the MHC region, we used the monozygotic twin concordance rate of Crohn’s disease (44%) from Sweden,2 the sibling recurrence risk of 4% in our families,24 and observed percentage of MHC haplotypes shared identical by descent by affected sib pairs in this study. By this method, we obtained a lower bound estimate of 10%, that is, the MHC region provided at least 10% of the genetic contribution to Crohn’s disease. This is a lower bound estimation because: (1) the MZ twin concordance rate includes both genetic and shared environmental effect; and (2) the twin data used for calculation are not from North America where the families for the linkage study were ascertained, but from a Scandinavian population who have an increased prevalence of inflammatory bowel diseases.
Discussion
In this study, we critically examined the hypothesis of linkage between Crohn’s disease and the MHC region on the short arm of chromosome 6. We did so by limiting our analysis to Crohn’s disease only families, using a series of analytical methods, and maximising informativeness by combining both TNF microsatellite markers and HLA class I and class II serological markers. Using these approaches, we consistently observed linkage between Crohn’s disease and the TNF-HLA region of MHC with all methods of analysis. It seems that the HLA linkage effect is stronger in non-Jewish families than it is in Jewish families, but is observed in both groups.
In our population, we have identified both association and linkage between the HLA region genes and Crohn’s disease. Our previous case control studies (unrelated patients with Crohn’s disease and controls) showed an association of HLA class II genes (DR1/DQ5) and a TNF haplotype (a2b1c2d4e1) with Crohn’s disease.5 ,11Fourteen of 49 families in the current study have the Crohn’s disease associated TNF haplotype and only in two of these families was this haplotype shared by the affected sibs. Although the number is too small to provide any conclusive results about the association, these data do suggest that the observed TNF or HLA class II gene associations are due to linkage disequilibrium between these genes and putative Crohn’s disease susceptibility genes located close to the MHC region. In other words, the tested TNF haplotypes and DR1/DQ5 themselves are probably not the disease causing gene(s). There could be more specific variations in the TNF or class II loci; or a combination of alleles at different loci which provide susceptibility. This concept of a disease predisposing haplotype has been observed in other HLA linked diseases such as insulin dependent diabetes mellitus.47
For this study, we included Crohn’s disease only families in order to reduce disease heterogeneity and increase the power for identifying linkage. In these Crohn’s disease only families, we observed excessive sharing of TNF-HLA haplotypes in concordant affected and concordant unaffected pairs, as well as reduced sharing in discordant pairs. This pattern provides strong evidence for linkage. As Crohn’s disease has variable age of onset, some clinically normal sibs may have the disease susceptibility gene and develop the disease later in time. Therefore, unaffected sib pairs may contain some future discordant sib pairs and discordant sib pairs may contain some future concordant affected sib pairs. Distant affected relatives, who are unlikely to share household environmental factors, also provided further supportive evidence for linkage.
In our study population, it seems that both ulcerative colitis (data not shown) and Crohn’s disease show the same trends of cosegregation with HLA haplotypes. Although we do not have enough ulcerative colitis families to firmly substantiate the linkage between the HLA and ulcerative colitis at this time, a recent genome scan study from England48 observed a tentative linkage between a marker, D6S276, lying close to the MHC complex, and ulcerative colitis.
Although the evidence for linkage is clear in our Crohn’s disease multiplex families, it is quite possible that some populations may not exhibit the same effect of the MHC region. This can be due to genetic aetiological heterogeneity. That is, it is increasingly apparent that several different genes and mechanisms can cause an individual to develop Crohn’s disease. It has been shown in animal models that knock outs of the T cell receptor gene, the interleukin 2 (IL-2) gene, or the IL-10 gene, can all lead to clinical colitis in mice.49-51 Thus, the effects of certain genes may be more predominant in one population than in another population. The genome scan results in Crohn’s disease seem to support this hypothesis as a chromosome 16 gene (IBD1) is important in French29and US populations30 ,52 but not as important in the English population.48 In contrast, chromosome 3, 7, and 12 markers show significant linkage in the English population but were not identified in the French population. Furthermore, IBD1 is linked with Crohn’s disease only in non-Jewish Caucasian families, but not in Jewish families30 drawn from the same geographic areas. Though such data are not available at this time, detailed clinical characterisation of affected patients from the linked families versus unlinked families might provide some insight on aspects regarding potential genetic heterogeneity.
Given certain assumptions, the gene(s) linked to the MHC region in these data was estimated to contribute at most 29–33% of the total increased risk in sibs over the population risk of Crohn’s disease. Applying the method of Rotter and Landaw45 with monozygotic twin concordance data from Sweden,2 we obtained a lower bound for the estimate of the MHC contribution (10%). This potential range of genetic contribution could account, at least in part, for the difficulty of observing linkage in every study. Furthermore, even in our population, other genes, some of which may contribute an even greater risk to Crohn’s disease, remain to be identified. The power of identifying such genes may be augmented by detailed studies of the phenotype, both at the clinical and subclinical level.
Acknowledgments
The authors wish to thank patients, their families, and referring physicians for their support of our ongoing genetic studies of inflammatory bowel disease. This work was supported by the National Institutes of Health grant DK46763, a grant from the Crohn’s and Colitis Foundation of America (SEP, SRT), and the Cedars-Sinai Board of Governors’ Chair in Medical Genetics (JIR). Some of the results in this paper were obtained by using the program package SAGE, which is supported by a US Public Health Service Resource Grant (1P41RR03655) from the National Center for Research Resources.
Abbreviations
- MHC
- major histocompatibility complex
- MZ
- monozygotic
- TNF
- tumour necrosis factor