Article Text

Statistics from Altmetric.com
Colorectal cancer is the second most common cause of cancer deaths in the USA. Most colorectal cancers develop from adenomatous polyps, and morphological and genetic progression in an adenoma-adenocarcinoma sequence and in hereditary colorectal cancer is well described.1-4
The molecular genetic alterations in sporadic and hereditary colorectal neoplasms are well understood and involve two distinct pathways.1-4 The genetic alterations present in most colorectal cancers are also present in adenomas and include truncating mutation or deletion of the adenomatous polyposis coli (APC) gene on chromosome 5q,5-7 point mutation of K-ras proto-oncogene,8-16 loss of the deleted in colorectal cancer (DCC) gene and nearby genes on chromosome 18q,7 ,17-19 and mutation and/or deletion of the p53 gene on chromosome 17p.8 ,9 ,20-23 Germline mutation of APC causes familial adenomatous polyposis (FAP).5 ,6 Alteration of the APC gene and K-ras mutations are genetic events observed early in the adenoma-carcinoma sequence,5 while loss of chromosome 18q and alteration of p53 are late events. A K-ras independent pathway of colonic carcinogenesis pathway may also exist in right sided colon carcinomas, which can develop from flat adenomas.11 ,24 ,25
In the second pathway to colorectal neoplasia, DNA replication errors (RER; microsatellite instability, ubiquitous somatic mutations) are caused by mutations in nucleotide mismatch repair genes, including hMSH2, hMLH1, PMS1, PMS2, and GTBP.1-4 Germline mutation of one of the mismatch repair genes causes hereditary non-polyposis colorectal cancer syndrome (HNPCC). RER is characterised by additions and deletions of nucleotides in numerous repeated nucleotide sequences (microsatellites). RER positive colorectal carcinomas are more commonly right sided and poorly differentiated with a diploid cell population; patients with these cancers have better survival. Alteration of a 10 basepair (bp) polyadenine tract in the transforming growth factor β type II receptor (TGFβRII) gene is present in more than 90% of RER positive carcinomas.26 ,27 RER and alterations of TGFβRII are also reported in colorectal adenomas.27-33
Although these genetic events have been studied extensively in sporadic colorectal carcinomas, in adenomas with a contiguous or synchronous carcinoma, and in patients with familial colorectal tumours and precursors (FAP and HNPCC),1-3 ,5 ,6 ,31 these events have not been investigated in a prospective study of sporadic colorectal adenomas removed at colonoscopy from patients without cancer. We therefore evaluated K-ras point mutations, loss of chromosome 18q, overexpression of p53 gene product, RER phenotype, and mutations of the TGFβRII gene in 201 sporadic colorectal adenomas from such a cohort and compared the results with demographics and adenoma characteristics.
Materials and methods
PATIENT POPULATION
This study included endoscopic biopsy specimens (n=154), polypectomy specimens (n=44), and resection specimens (n=3) of 201 colorectal adenomas which were collected from 60 patients prospectively enrolled at the endoscopy unit of The Johns Hopkins Hospital. Thirty one patients (52%) had specimens from two or more colonoscopies. The Joint Committee of Clinical Investigations (institutional review board) of The Johns Hopkins University School of Medicine reviewed and approved the consent forms and procedures used in this study, and written informed consent was obtained from each patient.
ELIGIBILITY CRITERIA AND EXCLUSION CRITERIA
Eligibility criteria for recruitment of colonoscopy patients were: (1) history of colorectal adenoma, symptoms and/or signs of colorectal neoplasia, positive faecal occult blood test, or family history of colorectal neoplasia; (2) age 18 years or over; and (3) ability to understand the study. The patients filled out a detailed questionnaire which included presenting complaints, past medical history, and family history.
The exclusion criteria were: refusal to participate, absence of colorectal adenoma at colonoscopy, inability to give informed consent or complete the questionnaire due to lack of English language skills or unsatisfactory mental status, adenomatous polyposis syndrome, presence of or history of colorectal carcinoma, and unsatisfactory genetic analysis of adenoma specimens (no amplification of DNA by polymerase chain reaction assay).
FAMILIAL HISTORY OF CANCER
Each patient’s family history of colorectal cancer was assessed. The patients with a positive family history were characterised as fulfilling criteria for hereditary non-polyposis colorectal cancer (HNPCC) as defined by the International Collaborative Group34 when at least three relatives in two successive generations had histologically verified colorectal cancer; at least two were first degree relatives of the third; one member was diagnosed before 50 years of age; and FAP was excluded. The other categories of classification were hereditary colon cancer (at least two successive generations with colorectal cancer, or at least one generation with colorectal cancer and one contiguous generation with adenomatous polyps), family at risk for hereditary colon cancer (two family members with adenomatous polyps or colon cancer in the same generation, or two family members with adenomatous polyps in different successive generations), cancer family syndrome (at least two successive generations with cancer, and at least 50% of the cancers not colorectal), family at risk for cancer family syndrome (multiple non-colorectal cancers in one generation), and young colon cancer (colon cancer under age 50 years, applied alone or in combination with the above categories).
SPECIMENS AND ADENOMA CHARACTERISTICS
The haematoxylin and eosin stained slides and paraffin wax embedded, formalin fixed, or Bouin’s fixed (prior to 1991) tissue blocks of adenomas were obtained from the surgical pathology files of The Johns Hopkins Hospital. A blood specimen was collected from each patient by venepuncture, processed, and frozen. Polyp size was taken from endoscopy notes (n=156) or alternatively from the pathology specimen (n=45). The site of polyps was taken from the surgical pathology requisition sheet completed by the colonoscopist. The histopathological slides were reviewed by a gastrointestinal pathologist (AR). The villous component of adenomas was estimated from the histopathological sections. The adenomas were classified as tubular if a villous component was present on less than 25% of the surface, tubulovillous if the villous component was 25–75%, and villous if the villous component comprised more than 75% of the surface. Only three adenomas had high grade dysplasia and therefore severity of dysplasia was not assessed any further in our study.
DNA PREPARATION
Genomic DNA was extracted separately from adenomatous tissue and control non-adenomatous colorectal tissue (polyp stalk or native colorectum in other specimens) by microdissection and prepared as described previously.35-37 DNA from adenomatous epithelium represented at least 50% tumour DNA. We were able to amplify DNA by polymerase chain reaction (PCR) from the majority of formalin fixed or Bouin’s fixed tissue. DNA from peripheral blood was used when non-adenomatous tissue was not available.
POINT MUTATIONS OF K-ras PROTO-ONCOGENE
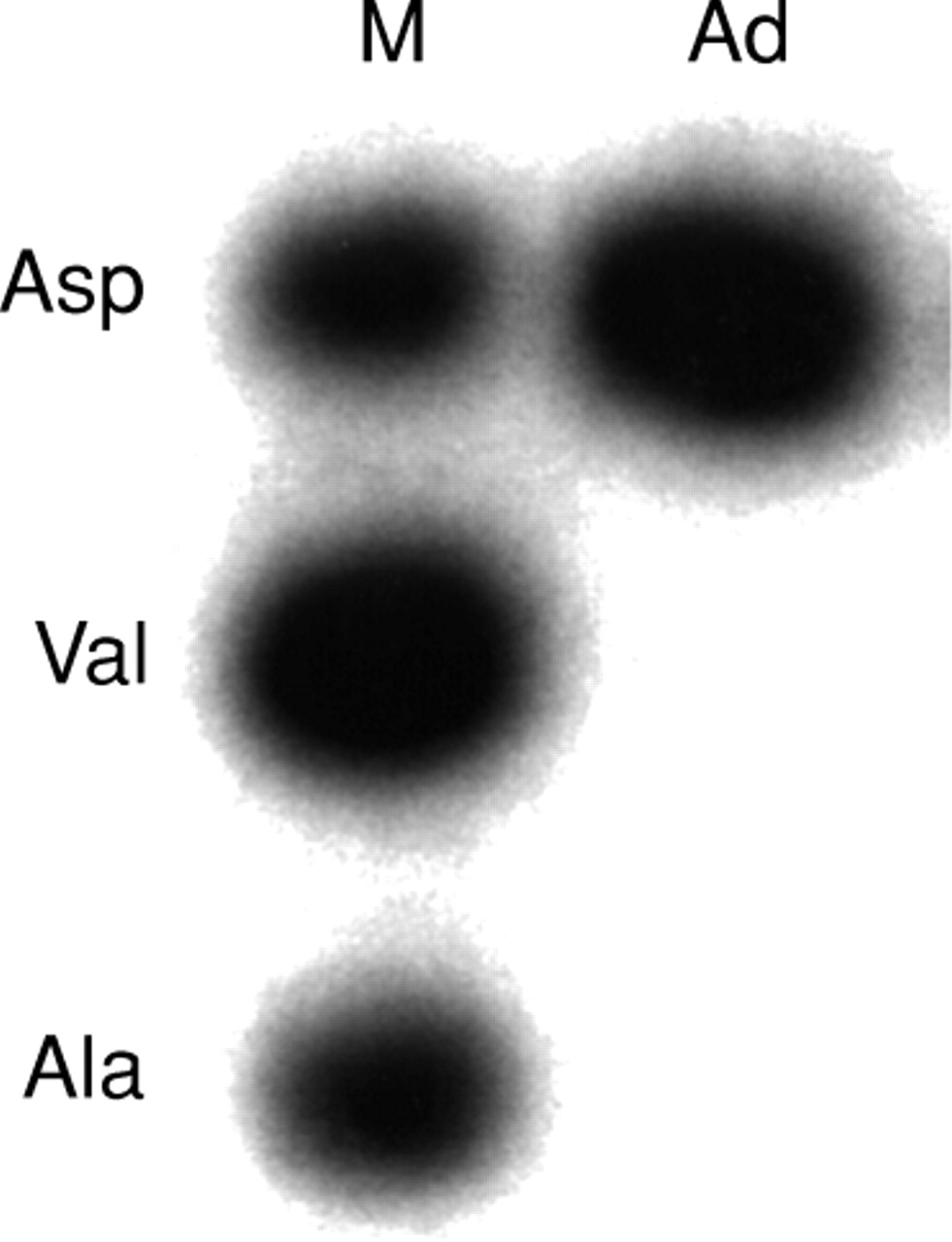
The first exon of c-K-ras was amplified and all possible point mutations in the first and second nucleotide position in codon 12 and the second nucleotide position of codon 13, which account for the majority of K-ras mutations in colorectal neoplasms, were tested by three separate ligations of mutation allele specific oligonucleotides, as described previously.36 ,37 The ligation products were analysed on a 12% denaturing polyacrylamide gel. A band in the tumour lane corresponding to a band in the positive control lane indicated a mutation (fig 1).

Aspartic acid mutation of codon 12 of K-ras gene by ligation of allelic specific oligonucleotide. M, size marker; Ad, adenoma sample.
MICROSATELLITE MARKERS AND PCR AMPLIFICATION
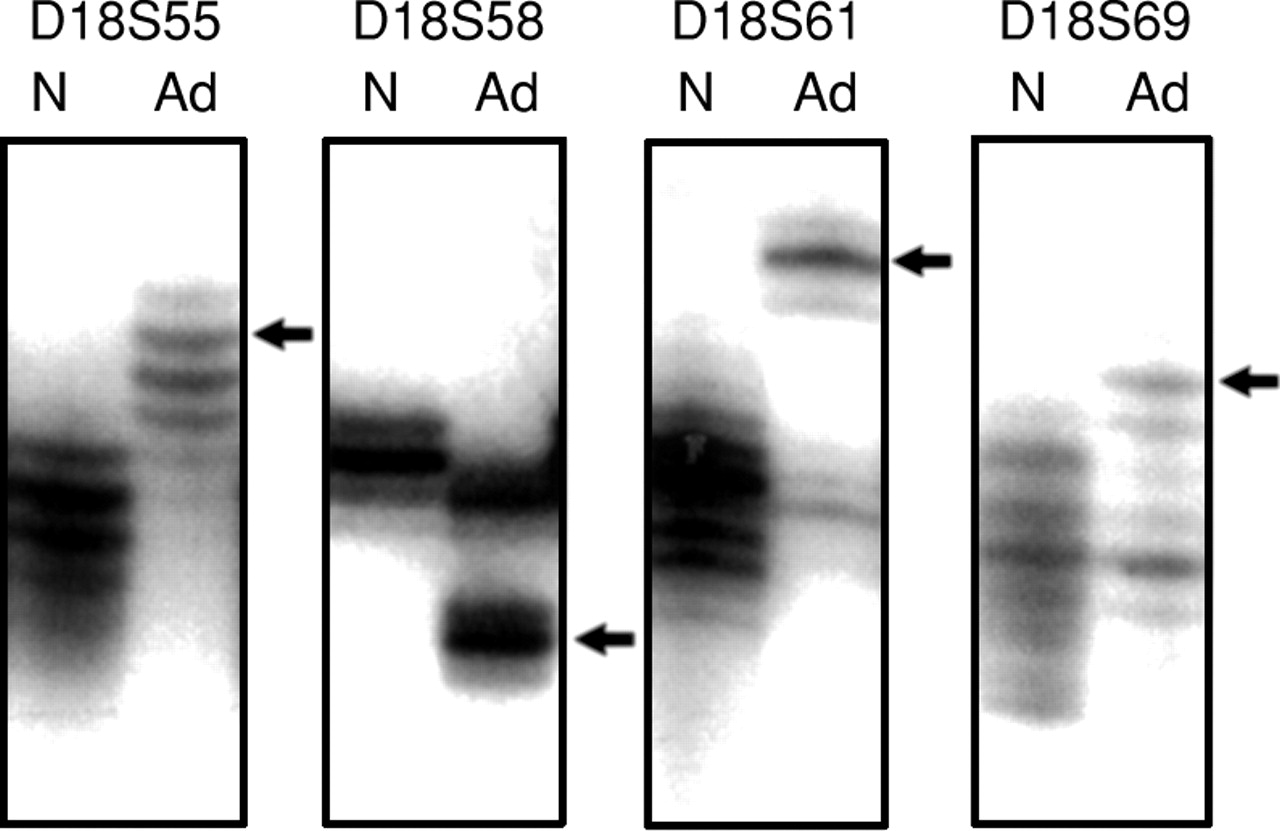
Five dinucleotide repeat microsatellite markers on the long arm of chromosome 18 (D18S58, D18S61, D18S55, D18S64, and D18S69 in order from centromere to telomere) were used to determine loss of heterozygosity (LOH) of chromosome 18q and RER status.38 PCR based dinucleotide repeat assays were carried out in 96 well plates for 35 cycles; each cycle was carried out at 95°C for 30 seconds, 50°C for 30 seconds, and 70°C for one minute, in a buffer containing 83.75 mM TRIS, pH 8.8, 20 mM (NH)2SO4, 1.8 mM MgCl2, 12.5 mM β mercaptoethanol, 125 μg bovine serum albumin (BSA), 250 μM dATP, dGTP, and dTTP, 6.25 μM dCTP, 0.05 μl [a32P] dCTP (NEN DuPont; 3000 Ci/mmol), 0.6 μM PCR primers, and 0.5 units Taq DNA polymerase in 12 μl volume. An equal volume of stop buffer (95% formamide, 20 mm EDTA, and 0.05% bromophenol blue and xylene cynanol) was added at end of amplification, and the samples were loaded onto 8% polyacrylamide gels containing 5.6 M urea. RER status or loss of heterozygosity of chromosome 18q could not be determined in 25 adenomas due to lack of non-adenomatous tissue or frozen blood sample (n=8), or lack of PCR amplification in three or more markers (n=17).
LOSS OF HETEROZYGOSITY OF CHROMOSOME 18q
Loss of chromosome 18q is associated with loss of the DCC gene and its mRNA expression,17 ,38 the DPC4 gene,18and the JV18 gene.19 Loss of a marker was considered to be present when the PCR assay showed absence or decrease in intensity by 50% of a band from a tumour sample compared with the control non-neoplastic sample. Complete or partial loss of each chromosome 18q was evaluated, based on the pattern of marker losses.
IMMUNOHISTOCHEMISTRY FOR p53 GENE PRODUCT
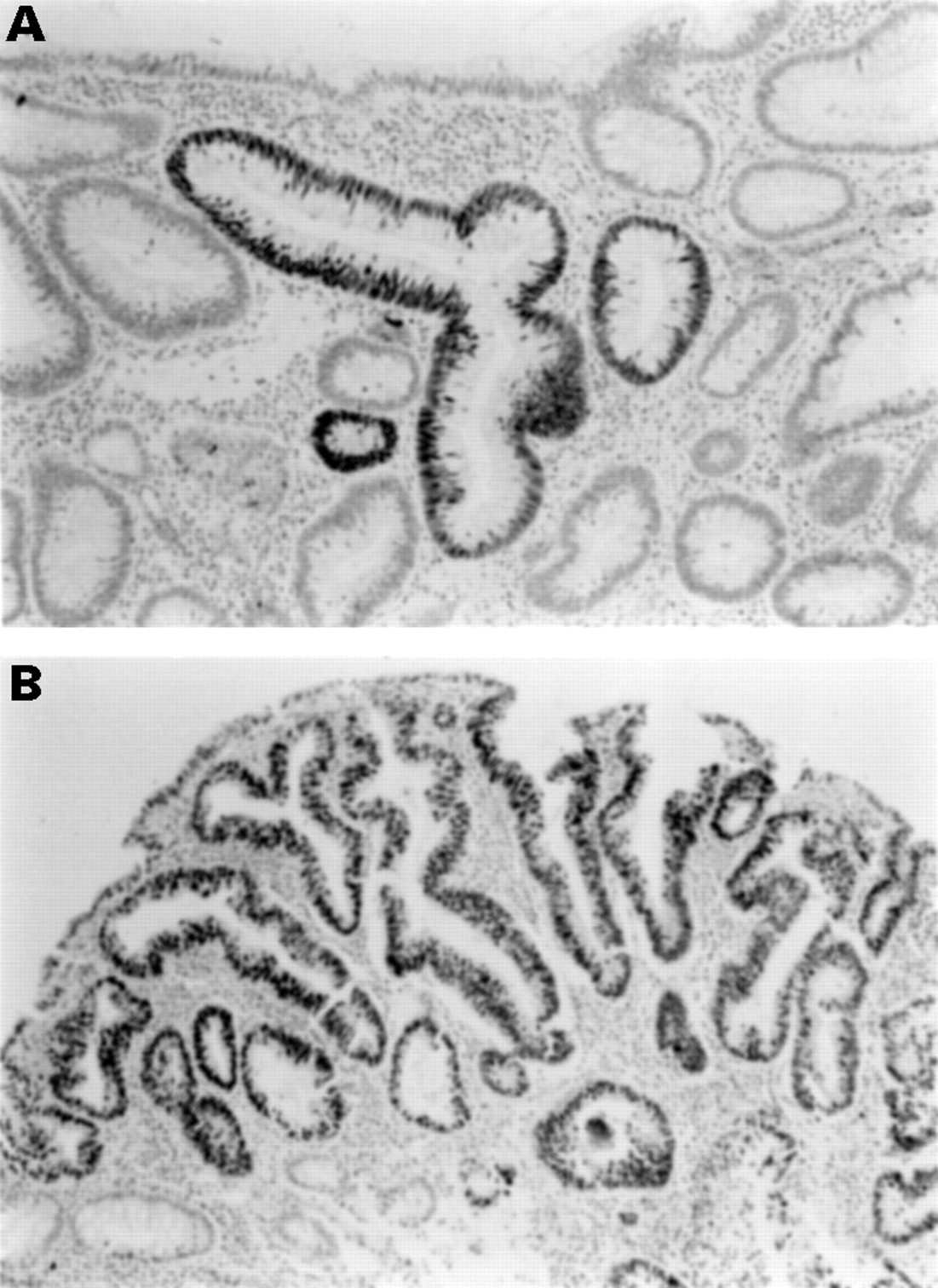
Immunohistochemistry with mouse monoclonal antibody D07 and standard techniques was used to determine p53 gene product overexpression, defined as staining of more than 50% of nuclei in adenomatous epithelium. This criterion was based on our previous studies showing this finding was strongly associated with p53 mutation in colorectal neoplasms.39 Topography of staining was defined as focal if five or fewer glands of adenomatous epithelium were intensely stained (fig 2A), regional if more than five crypts but less than 50% of adenomatous epithelium were stained, and diffuse if more than 50% of adenomatous epithelium was stained (fig 2B).

Immunohistochemistry for p53 gene products. (A) Focal staining of adenomatous epithelium. (B) Diffuse staining of an adenoma.
RER STATUS
The presence of DNA replication errors (RER) was determined from the PCR amplifications of the five dinucleotide microsatellite markers on the long arm of chromosome 18 (18q). RER positive adenomas were defined by shifts of bands as compared with control DNA in at least two microsatellite markers (fig 3). RER status could not be determined in 25 adenomas from 19 patients due to failure of DNA amplifications in three or more markers or lack of non-neoplastic tissue.

RER in colorectal adenomas. Instability in microsatellite markers is shown by additional bands (arrows). N, non-neoplastic; Ad, adenoma.
ALTERATIONS OF BAT-26 AND BAT-25 ALLELES
Alterations of BAT-26, a polyadenine tract present in the intron 5 of hMSH2 gene, and BAT-25, a polyadenine tract present in the intron of c-kit gene, were studied in RER positive adenomas as described previously.26 An unstable allele was defined by an additional band present below the wild type allelic band (fig4).

Adenoma (Ad 2) with shortening of BAT-26 allele (arrow). The normal length PCR product (Ad 1 and Ad 2) is indicated by an arrowhead.
MUTATION IN TGFβRII GENE
Alteration in the 10 bp polyadenine tract present in the third exon of the TGFβRII gene was studied by PCR amplification of a 73 bp region, as described previously.26 Mutation was defined by an additional band present above or below the wild type allelic band (fig 5).

Adenoma (Ad 2) with one bp deletion (arrow) in TGFβ type II receptor gene shown by PCR amplification of a 73 bp region. The normal length PCR product (Ad 1 and Ad 2) is indicated by an arrowhead.
STATISTICAL ANALYSIS
The major statistical endpoint of this study was the determination of factors associated with genetic alterations in adenomas. Patients with more than one adenoma were represented multiple times in this data set. To determine whether these repeated observations could be considered independent, χ2 goodness of fit tests were performed for each type of alteration. When the frequency of observed alterations was adequate, marginal logistic regression models for correlated binary data were used to assess associations with demographic features and adenoma characteristics. Estimates were obtained using the generalised estimating equation (GEE) approach of Liang and Zeger.40 When the within subject correlation is small, the independence working model for the GEE regression analysis has been shown to be adequate. For alterations where the assumption of independence was rejected due to one or two patients and the sample of observed alterations was too small for valid GEE regression, Fisher’s exact tests were performed both with and without these patients. Factors tested for an association with K-rasincluded age, sex, site, adenoma size, villous component, 18q loss, p53 overexpression, and RER status. A formal statistical method for multiple comparisons was not performed because this was considered an exploratory study to generate hypotheses that can be tested in future studies.
Results
Table 1 summarises the demographics of the patient population and adenoma characteristics. Table 2 and fig 6 summarise the genetic alterations in colorectal adenomas. Table 3 compares the size, histopathology, and anatomical site of the adenomas.
Patient demographics and adenoma characteristics
Prevalence of genetic alterations in patients and adenomas

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Genetic alterations in adenomas from female (A) and male (B) patients.
Prevalence of genetic alterations compared with size, histopathology, and site of adenomas
CONCORDANCE OF GENETIC ALTERATIONS IN PATIENTS WITH MULTIPLE ADENOMAS
Forty one patients (68%) had multiple adenomas which provided an opportunity for assessing within subject correlation of genetic alterations. In patients with multiple adenomas, K-ras mutation was present in 36% of adenomas (65/181), loss of heterozygosity of chromosome 18q in 2% (4/163), p53 overexpression in 6% (11/182), and RER in 7% (12/163). p53 overexpression was not independent in multiple adenomas (p=0.001, χ2 goodness of fit)—that is, the occurrence of p53 overexpression in one adenoma significantly increased the probability of p53 overexpression in a second adenoma. This was primarily due to one patient with p53 overexpression in three of five adenomas. Within patient correlation for RER was also significant (p=0.001, χ2 goodness of fit) due to two patients with RER alterations in both of two adenomas. By contrast, there was no statistically significant within subject correlation for K-ras mutation (p=0.55, χ2goodness of fit). Of the patients with two or more adenomas, 15% (6/41) had K-ras mutations in all adenomas, 17% (7/41) had no K-ras mutation, and 68% (28/41) had K-ras mutations in one or more but not all of their adenomas.
C-K-ras MUTATIONS
Mutation in K-ras was the most frequent genetic alteration, occurring in 35% of colorectal adenomas (70/200) representing 65% of the patients (table 2). The most frequently altered nucleotide position was the second nucleotide of codon 12, and the most common mutation was substitution of asparatic acid for glycine due to G to A transition (49% of K-rasmutations; table 4). Two ras mutations were found in 7% of adenomas (15/201), all involving codon 12. In 53% of adenomas with two mutations, the same nucleotide position showed different mutations, indicating two subclones in the adenoma.
K-ras point mutations in codons 12 and 13 in colorectal adenomas
Patients 65 years of age and older had a significantly decreased probability of K-ras mutation in their adenomas (26% versus 45%, p=0.01, GEE regression model; table 5), but there was no association of K-ras mutations with sex or site of adenoma. There was a slightly increased prevalence of K-ras mutations in villous or tubulovillous adenomas compared with tubular adenomas (43% versus 33%, p=0.33, GEE regression model; table 3), and in larger adenomas (31% in adenomas 0.5 cm or less in size versus 41% in those more than 0.5 cm, p=0.16, GEE regression model; table 3), but the differences were not statistically significant.
Odds ratios and 95% confidence intervals (CI) for K-ras, from GEE marginal regression models
LOSS OF HETEROZYGOSITY OF CHROMOSOME 18q
Loss of heterozygosity of chromosome 18q was rare, present in only 2% of colorectal adenomas (4/176), and was significantly less frequent than p53 overexpression (6%, 13/201, p=0.05).
p53 OVEREXPRESSION
Overexpression of p53 gene product was present in 6% of colorectal adenomas (13/201). The staining was focal in seven adenomas and regional in four, suggesting the presence of subclones with p53 alteration, and diffuse in two. There was no difference in p53 overexpression in males and females (10/128 adenomas in males versus 3/73 in females, p=0.4). By contrast, in patients 50–75 years of age, p53 overexpression was present exclusively in adenomas from male patients (10/128 adenomas in males versus 0/39 adenomas in females, p=0.06, Fisher’s exact test; fig 5). p53 overexpression was seen in three adenomas from the left colorectum in two females over 75 years of age. There was increased frequency of p53 overexpression in tubulovillous adenomas compared with tubular adenomas (19% versus 3%, p=0.001, Fisher’s exact test; table 3).
RER STATUS
RER was present in 7% of adenomas (13/176 tested) from eight patients. Of the 13 adenomas with RER phenotype evaluated by dinucleotide markers, one had mobility shifts in all five markers, one in four, two in three, and the remaining nine in two markers (at least 40% of dinucleotide markers). An unstable shortened allele of BAT-26 was present in 36% of RER positive adenomas (4/11 adenomas with PCR amplification) from three patients. No unstable allele of BAT-25 was detected in 11 RER positive adenomas.
Forty four per cent of adenomas from three HNPCC patients (2/2, 2/5, and 0/2, respectively) were RER positive versus 5% in non-HNPCC patients (table 2; p=0.002, Fisher’s exact test). All four adenomas with RER phenotype from HNPCC patients had mobility shifts in two microsatellite markers and one had a shift in BAT-26. Another patient who had one of five adenomas with RER had a family history of hereditary colon cancer (see Materials and methods for definition). By contrast, four patients with RER in 3/12, 2/2, 1/11, 1/5, and 1/1 adenomas had a negative family history. Seven of eight patients with RER positive adenomas had more than one adenoma (range 2–12). There was a correlation between RER in an adenoma and RER in a metachronous or synchronous adenoma (p=0.001, χ2 goodness of fit). Although RER positive cancers are commonly right sided,41the RER positive adenomas were not significantly more common in right colon than left colorectum (9% versus 6%, p=0.56, Fisher’s exact test). Similarly, percentage of markers with shift was not higher in right sided adenomas (45% versus 50%, p=0.81). Among seven RER positive adenomas from HNPCC patients or adenomas with alteration of BAT-26 allele, four were right sided.
ALTERATION IN TGFβRII
Mutation in the TGFβRII gene was very rare, present in only one of 13 RER positive adenomas among 201 adenomas studied. This alteration was present in an RER positive tubulovillous adenoma from a patient without HNPCC.
MULTIPLE GENETIC ALTERATIONS IN ADENOMAS
Adenomas with RER were more likely to have a K-ras mutation than adenomas without RER (62% versus 33%, p=0.04, GEE regression model; tables 5 and 6). K-ras mutation was present in 57% of RER positive adenomas from HNPCC patients or with alteration of BAT-26. Adenomas with K-ras mutation had more genetic alterations than adenomas without a K-rasmutation (21% versus 11%, p=0.09, GEE regression model) including RER in eight adenomas, p53 overexpression in five, and loss of chromosome 18q in two. However, this marginally increased probability of K-ras mutation when any one of these alterations was present was due to the association of K-ras with RER positivity, and was not associated with p53 overexpression or chromosome 18q loss.
Prevalence of multiple genetic alterations in colorectal adenomas
Discussion
The molecular genetics of colorectal neoplasms, including adenomas, have been studied extensively, but few studies have addressed clinical and pathological associations in prospectively defined patient populations. We therefore studied adenomas from patients without cancer in a cohort undergoing colonoscopy. The limitations of design in our study include multiple statistical comparisons and low frequency of genetic alterations in adenomas of patients without colorectal cancer. The significant associations generated in this study should be considered exploratory due to the low number of genetic events and should be addressed in future studies.
K-ras mutations were present in 35% of adenomas in our study. The reported prevalences of K-ras mutations in adenomas vary widely in the literature, from 20% to 70%.8-16 These variations in K-ras mutation rates in colorectal adenomas may reflect geographical and dietary variations in exposure to putative carcinogenic agents in the study populations in different countries or use of different assay techniques in these studies.42Clinical associations with K-ras mutations in adenomas are also variable in the literature. K-ras mutations in adenomas are reported to be more common in patients with a prior colorectal cancer.10 ,14 Lack of K-rasmutations in right sided colon carcinomas from patients younger than 70 years has been reported,43 but we found a lower prevalence of K-ras mutations in adenomas from older rather than younger patients. Our results corroborate a previous study which showed a low prevalence (18%) of K-rasmutations in adenomas without a synchronous carcinoma in patients over 70 years of age.14 Correlations of K-ras mutation with size or histopathology show conflicting results in different studies,9 ,10 and we found no significant relation. A lower prevalence of K-ras mutations occurs in flat adenomas.11 ,24
Heterogeneity of K-ras mutations in microdissected areas within an adenoma and multiple K-ras mutations in individual colorectal adenomas have been reported,16 ,44 and we found two mutations in 7% of our adenomas. Two mutations in an adenoma may represent either two individual subclones with two different mutations or mutations in both copies of K-ras. The type of mutations may be important as G-C or G-T transversions are present in colorectal adenomas with DNA aneuploidy,13 and transversions are more common in Dukes’s C colorectal carcinomas in comparison to Dukes’s B carcinomas45 and in recurrent colorectal carcinomas.46 By contrast, most of the mutations in our study were G-A transitions. Patients with a large adenoma (more than 1 cm) harbouring a K-rasmutation are expected to have a significantly higher risk of developing a large metachronous adenoma,14 but our study showed no significance of K-ras mutation as a predictor of a K-ras mutation in synchronous or metachronous adenomas in patients with multiple adenomas. Non-concordance of K-ras mutation in multiple adenomas in the same patient raises questions about the pathogenesis of K-ras alterations in colorectal neoplasms. In addition, our findings suggest that K-rasmutation in an adenoma is unlikely to be a reliable predictor of recurrence of adenomas in an individual patient.
Allelic loss of chromosome 18q is an event late in colorectal carcinogenesis as it is present in 70% of colorectal carcinomas,38 but is present in less than 20% of stage I carcinomas17 and 10% of adenomas.7 The loss of chromosome 18q is associated with loss of DCC, DPC4, and JV18 genes.17-19 ,38 Our results show that loss of chromosome 18 is present in only 2% of sporadic adenomas in a colonoscopic series. This low rate could reflect the small size of polyps in this study (97% of polyps were 2 cm or less in size).
p53 overexpression of the type associated with p53 gene mutation was present in 6% of adenomas, representing a higher frequency than 18q loss in our study. The distribution of overexpression is of interest because the abnormality was usually present regionally in neoplastic epithelium, suggesting that a subclone with p53 overexpression emerged late in tumourigenesis. Similar results have been reported in the literature, with alterations in p53 present in 5–26% of colorectal adenomas,8 ,9 ,20-23 in 53% of carcinomatous foci in adenomas,22 ,23 and in 70% of carcinomas.20 ,21
We observed RER in 7% of adenomas representing eight patients, based on the criterion of at least two of five dinucleotide markers with allelic shift. Five of the patients had two or more adenomas with RER by our criterion, and two of these patients were members of HNPCC kindreds. RER has been observed in up to 3% of sporadic adenomas29-32 and 60–95% of adenomas from HNPCC families.27 ,29-31 Alteration in BAT-26 alleles was present in 36% of RER positive adenomas in our study, which is similar to the frequency of 50% in RER positive adenomas reported in the literature.47 Frameshift mutation in TGFβRII has been observed in 57% of adenomas from HNPCC families,27 but infrequently in sporadic adenomas,33 and is present late in the adenoma-carcinoma sequence.47 We found mutation in the TGFβRII gene in only one RER positive adenoma (8%) and in none of the RER negative adenomas.27 We therefore confirmed previous studies indicating that alteration of mononucleotide markers (BAT-26, TGFβRII, etc.), which is a sensitive predictor of high microsatellite instability in colorectal adenocarcinomas, is not frequent in adenomas.33 ,47 Thus, the distinction between “low” microsatellite instability and “high” microsatellite instability48 ,49 is problematical in adenomas.
The occurrence of K-ras mutations in 50% of RER positive adenomas from HNPCC patients has been reported,27 and we found that RER positive adenomas had increased probability of K-ras mutation. The frequency of K-ras mutations in RER positive colorectal tumours is not settled because the mutations are reported to occur at frequencies similar to microsatellite stable cancers in some series,50-54 but at low frequency in others.31 It is speculated that K-ras may not have a major role in progression of RER positive colorectal tumours.55
We found a high proportion of RER positive adenomas in the left colorectal tumours as well as a high frequency of K-ras mutation in RER positive adenomas. By contrast, RER positive colorectal cancers are strongly right sided.41 Thus, a right sided predilection or a lower frequency of K-ras mutation was not observed even in RER positive adenomas from HNPCC kindreds or adenomas with alteration of BAT-26. The differences between the features of RER positive adenomas and cancers remain to be explained.
In conclusion, our study shows the complexity of the genetic alterations involved in colorectal tumourigenesis. Factors related to patient age and RER are involved in the occurrence of K-ras mutations, as evidenced by the lower frequency of ras mutations in older patients and the higher frequency in RER positive adenomas which are associated with HNPCC in some patients. By contrast, p53 alterations have less intraindividual variation, favouring different aetiological factors than those for K-ras mutations.
Acknowledgments
We acknowledge Ms Rahj Robinson for recruitment of patients for this study, Mrs Nancy Folker for typing the manuscript, and Ms Ellen Winslow for photographs. This study was supported in part by the Clayton Fund and grants CA63718, CA62924, and CA41108 from the National Cancer Institute, National Institute of Health. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
Abbreviations
- APC
- adenomatous polyposis coli
- DCC
- deleted in colorectal cancer
- FAP
- familial adenomatous polyposis
- HNPCC
- hereditary non-polyposis colorectal cancer
- LOH
- loss of heterozygosity
- PCR
- polymerase chain reaction
- RER
- replication error
- TGFβRII
- transforming growth factor β type II receptor