Article Text

Abstract
BACKGROUND Intestinal barrier dysfunction concomitant with high levels of reactive oxygen metabolites (ROM) in the inflamed mucosa have been observed in inflammatory bowel disease (IBD). The cytoskeletal network has been suggested to be involved in the regulation of barrier function. Growth factors (epidermal growth factor (EGF) and transforming growth factor α (TGF-α)) protect gastrointestinal barrier integrity against a variety of noxious agents. However, the underlying mechanisms of oxidant induced disruption and growth factor mediated protection remain elusive.
AIMS To determine: (1) if oxidation and disassembly of actin (a key cytoskeletal component) plays a major role in ROM induced epithelial monolayer barrier dysfunction; and (2) if growth factor mediated protection involves prevention of theses alterations.
METHODS Caco-2 monolayers were preincubated with EGF, TGF-α, or vehicle before incubation with ROM (H2O2 or HOCl). Effects on cell integrity, barrier function, and G- and F-actin (oxidation, disassembly, and assembly) were determined.
RESULTS ROM dose dependently and significantly increased F- and G-actin oxidation (carbonylation), decreased the stable F-actin fraction (index of stability), and increased the monomeric G-actin fraction (index of disassembly). Concomitant with these changes were disruption of the actin cytoskeleton and loss of the monolayer barrier function. In contrast, growth factor pretreatment decreased actin oxidation and enhanced the stable F-actin, while in concert prevented actin disruption and restored normal barrier function of monolayers exposed to ROM. Cytochalasin-D, an inhibitor of actin assembly, not only caused actin disassembly and barrier dysfunction but also abolished the protective action of growth factors. Moreover, an actin stabilising agent, phalloidin, mimicked the protective actions of the growth factors.
CONCLUSIONS Oxidation, disassembly, and instability of the actin cytoskeleton appears to play a key role in the mechanism of oxidant induced loss of intestinal barrier integrity. In contrast, organisation and stabilisation of actin through promotion of its assembly plays a critical role in the mechanism of growth factor mediated protection.
- western immunoblotting
- F-actin
- G-actin
- Caco-2 cells
- barrier function
- growth factors
- dinitrophenylhydrazine immunoreactivity
Statistics from Altmetric.com
- western immunoblotting
- F-actin
- G-actin
- Caco-2 cells
- barrier function
- growth factors
- dinitrophenylhydrazine immunoreactivity
The intestinal mucosa is a highly selective barrier that permits absorption of nutrients but normally restricts the passage of harmful proinflammatory agents (e.g. endotoxin). Thus maintenance of normal intestinal barrier integrity is essential for health as the loss of the mucosal barrier can result in an increase in the absorption of toxic agents into the circulation, leading to intestinal inflammation and tissue disruption.1 2 Indeed, several intestinal disorders, including inflammatory bowel disease (IBD), are characterised by abnormal mucosal barrier function.1-4The pathophysiology of mucosal barrier dysfunction in IBD is not established but it may involve oxidants. Several studies have shown that chronic gut inflammation is associated with high levels of reactive oxygen metabolites (oxidants) which may be responsible for mucosal barrier dysfunction.5 6 None the less, the precise mechanisms underlying oxidant induced mucosal barrier dysfunction remain unknown.
In recent years, the importance of the complex network of actin cytoskeleton in the maintenance of normal cellular homeostasis and barrier function has been shown.7-11 Actin is the principal protein in the cell cortex and as such plays a central role in maintaining cellular integrity, morphology, and the structure of the cytoplasmic matrix. We have previously shown the importance of cytoskeletal stability in gastrointestinal (GI) healing in vivo12 13 and under in vitro conditions.7 14-16 However, the role of the actin cytoskeleton such as oxidation, disassembly, and instability in oxidant induced loss of mucosal barrier function and as an effective means of preventing such deleterious alterations, remains elusive.
Restoration of barrier function by protective factors following insults to the mucosa, such as oxidants, is essential for re-establishing normal intestinal homeostasis. Epidermal growth factor (EGF) and transforming growth factor α (TGF-α) play a major role in regulating proliferation, differentiation, barrier function, and repair processes throughout the GI mucosa.17-19 Numerous studies, including our own, have shown that EGF and TGF-α can effectively protect GI barrier integrity against injurious agents, including oxidants.14 15 17-20 The specific mechanisms underlying this protective phenomenon have remained poorly defined. We hypothesised that oxidant induced mucosal barrier dysfunction is caused by oxidation, disassembly, and instability of the actin cytoskeleton and that growth factors elicit protection by maintaining normal actin assembly and integrity. Accordingly, the objective of our study was to investigate the inter-relationships between actin stability, mucosal barrier function, oxidant insult, and EGF or TGF-α protection using a human intestinal cell line, Caco-2.
Materials and methods
CELL CULTURE
The Caco-2 (a human colonic) cell line was obtained from ATCC (Rockville, Maryland, USA) at passage 15. These cells resemble intestinal cells in that they have defined apical brush borders, form tight junctions, and exhibit a highly organised actin network on differentiation.21 22 Cells were maintained at 37°C in complete Dulbecco's minimum essential medium (DMEM).21They were split at a ratio of 1:6 on reaching confluency every six days and set up in either six or 24 well plates (at a density of 200 000 cells/cm2) for experiments or T-175 flasks for maintenance of stocks. Cells grown for barrier integrity work were also seeded at a density of 200 000 cells/cm2 into 0.4 μM Biocoat Collagen I Cell Culture Inserts (0.3 cm2 growth surface; Becton Dickinson, Massachusetts) and used at least seven days post-confluence. Cell passages were maintained between 25 and 40 for all work, and media were changed every two days. The volume of media was 400 μl per well during the experimental procedures (see below).
EXPERIMENTAL DESIGN
We initially evaluated the effect of a 30 minute incubation period with graded concentrations (0.01–5 mM) of oxidants (H2O2, HOCl) or vehicle (isotonic saline/DMEM) on barrier integrity, cell integrity, and actin stability, as described below. Preliminary studies showed that the presence of FBS in the medium did not influence the effects of oxidant on cell integrity or monolayer barrier function. For example, H2O2(or HOCl) 0.5 mM caused similar levels of monolayer barrier disruption in medium with FBS (750 (23) nl/h/cm2) and medium without FBS (762 (12) nl/h/cm2). Thus all subsequent studies were done in DMEM without FBS. To evaluate the protective effects of EGF and TGF-α against oxidant induced cell injury, barrier disruption, and actin instability, EGF or TGF-α (10 ng/ml, Sigma, St Louis, Missouri) was added to the cell culture media 10 minutes before exposing cells to damaging concentrations of oxidants for 30 minutes (0.1–5 mM). These concentrations of EGF and TGF-α have previously been shown to be protective in GI epithelial cell cultures.7 14 15 18 To ascertain the specificity of the protective actions of growth factors, monoclonal anti-EGF receptor antibody (anti-EGFR 1 μg/ml) was also administered with the growth factors.
To determine the potential modulatory effects of cytochalasin-D (an actin disruptive drug, 39 μM, incubated for four hours) and phalloidin (an actin stabiliser, 1 μM, incubated for one hour) (Sigma) on barrier function and actin, we added each compound (in DMEM or vehicle) to monolayers before exposure to growth factor (10 ng/ml, 10 minutes) or vehicle, and subsequently to oxidant or vehicle, as described above. Concentrations of these agents have previously been shown to be effective under similar in vitro conditions.7 16
To determine the role of actin in maintaining intestinal epithelial barrier function, monomeric (G-) and polymerised (F-) fractions of actin cytoskeleton were initially isolated following the aforementioned protocols (above) and subsequently analysed using two sensitive immunoblots (below). In all experiments actin integrity was determined using three different procedures: (a) fluorescent labelling of actin to determine the percentage of cells with normal actin by fluorescence microscopy; (b) detailed analysis of actin integrity by high resolution laser scanning fluorescent microscopy (LSCM); and (c) state of the art quantitative western immunoblotting analysis of monomeric and polymerised fractions of actin.
FLUORESCENCE STAINING AND HIGH RESOLUTION LASER SCANNING FLUORESCENCE MICROSCOPY OF ACTIN
Cells were fixed in cytoskeletal stabilisation buffer and subsequently stained for actin with FITC-phalloidin (specific for F-actin; 1:40 dilution; Sigma).12 16 After staining, cells were observed in a blinded fashion with LSCM (argon laser, wavelength 488 nm) using a 63 X oil immersion plan-apochromat objective, NA 1.4 (Zeiss). The cytoskeletal elements were examined for overall morphology, orientation, and disruption (%), as previously described.7 16
ACTIN FRACTIONATION AND QUANTITATIVE WESTERN IMMUNOBLOTTING OF F- AND G-ACTIN
F- and G-actin were isolated using a modified method for isolating cytoskeletal pools described previously.7 16 Briefly, cells were pelleted by centrifugation at low speed (700 rpm, seven minutes, 4°C) and resuspended in actin stabilisation-extraction buffer (0.1 M Pipes, pH 6.9, 30% glycerol, 5% DMSO, 1 mM MgSO4, antiprotease cocktail (10 μg/ml), 1 mM EGTA, and 1% Triton X-100) at room temperature for 20 minutes. F- and G-actin were separated following a series of centrifugation and extraction steps. Fractionated actin samples were flash frozen in liquid N2 and stored at −70°C until immunoblotting. For immunoblotting, samples (5 μg of protein) were placed in a standard SDS buffer, boiled for five minutes, and then subjected to PAGE (7.5% gel).16 The protein content of samples was estimated using the Bradford method.23 The optical density of the autoradiographic bands were measured as previously described.16
IMMUNOBLOTTING DETERMINATION OF ACTIN CARBONYLATION (F- AND G-ACTIN OXIDATION)
This immunoblotting procedure was similar to the quantitative blotting15 16 24 except for some differences in primary antibodies and buffers (to avoid unwanted oxidation of actin samples, all buffers contained 0.5 mM dithiothreitol (DTT) and 20 mM 4,5-OH2-1,3-benzene sulphonic acid). Briefly, actin samples were blotted to a polyvinylidene difluoride (PVDF) membrane followed by successive incubations in 2 N of HCl and 2,4,dinitrophenylhydrazine (DNPH, 100 μg/ml in 2 N HCl) for five minutes each. Immunological evaluation of carbonyl formation was performed for one hour in 1% BSA/PBS-Tween 20 buffer containing anti-dinitrophenylhydrazine (DNP) antibody (1:25 000 dilution) (Molecular Probes, Oregon), conjugated to peroxidase by protein cross linking with 0.2% glutaraldehyde. Cell free controls (actin ± growth factors ± oxidants) were performed and confirmed the specificity of the primary antibody. Wash steps, film exposure, and quantification were as previously described.15 16
DETERMINATION OF CELL INJURY AND CELL DEATH
Live/injury/dead assay kits (Molecular Probes) were used according to the manufacturer's instructions. This assay measures: (1) parameters of cell injury, such as intracellular esterase inactivation and plasma membrane integrity; and (2) parameters of cell death, such as nuclear membrane integrity and nuclear chromatin condensation. Following treatments, cells were loaded with two fluorescent probes, calcein-AM (2 μM) to determine cell injury and ethidium homodimer-1 (4 μM) to assess cell death, for 20 minutes at 37°C and then examined using a diphot inverted fluorescent microscope. All experiments were performed by counting 100–200 cells in four different fields from each slide. Cell injury was determined by calcein-AM and expressed as follows: % injured cells=(injured cells/(normal cells+injured cells))×100. Cell death was assessed by the number of nuclei stained by ethidium homodimer-1 and calculated as follows: % dead cells=(dead cells/(normal cells+dead cells))×100.
DETERMINATION OF BARRIER INTEGRITY
Barrier integrity was determined by measuring apical to basolateral flux of a fluorescent marker (fluorescein sulphonic acid (FSA 200 μg/ml; 478 Da)).11 15 Fresh phenol free DMEM (800 μl) was placed into the basolateral chamber and phenol free DMEM (400 μl) containing FSA was placed in the apical chamber. Following treatments, fluorescent signals from samples were quantitated using a fluorescence multiplate reader (FSA excitation, 485 nm; FSA emission, 530 nm). Clearance (Cl) was calculated using the following formula: Cl (nl/h/cm2)=Fab/([FSA]a×S), where Fab is the apical to basolateral flux of FSA (light units/h), [FSA]a is the concentration at baseline (light units/nl), and S is the surface area (0.3 cm2).11 Simultaneous controls were performed during each run.
STATISTICAL ANALYSIS
Data are presented as mean (SEM). All experiments were performed with a sample size of at least six observations per group. Statistical analysis was carried out using analysis of variance followed by Dunnett's multiple range test.25 A p value <0.05 was considered statistically significant.
Results
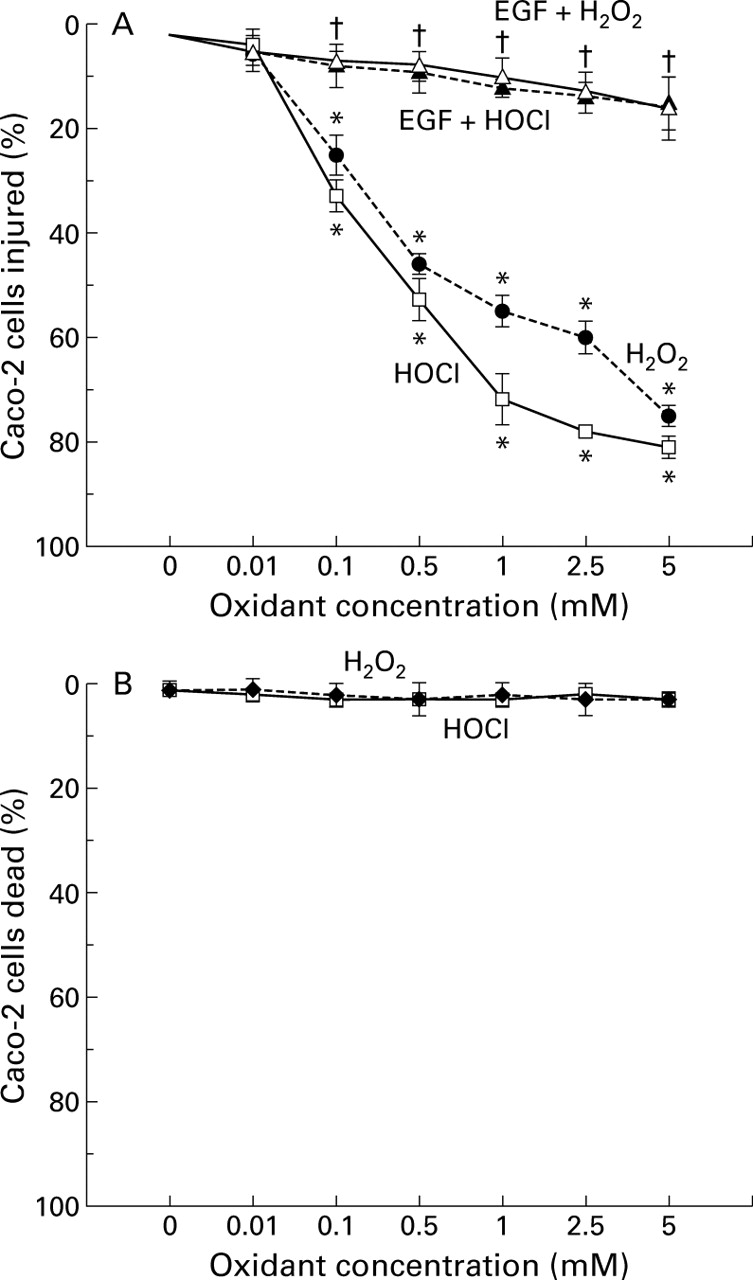
Caco-2 cells exposed to H2O2 or HOCl for 30 minutes demonstrated dose dependent disruption of monolayer barrier function (fig 1), as demonstrated by the increased clearance of fluorescein sulphonic acid (FSA). The lowest concentration of both oxidants which caused barrier dysfunction was 0.1 mM. In contrast, preincubation with EGF or TGF-α 10 ng/ml before immediate exposure to the same oxidants prevented barrier dysfunction (fig 2). To determine the specificity of protection, monolayers were preincubated with monoclonal anti-EGF receptor (anti-EGFR 1 μg/ml) antibody before exposure to growth factor and oxidant. Anti-EGFR completely prevented the protective effects of EGF or TGF-α (fig 3). To assess if changes in barrier dysfunction were caused by cytotoxicity (death) or injury, calcein-AM and ethidium homodimer-1 fluorescent probes were used. The oxidants dose dependently injured the cells (fig 4A) but did not cause significant cell death (fig 4B). The injurious effect of the oxidants on cells was completely abolished by preincubation with EGF or TGF-α 10 ng/ml (fig 4A, EGF shown).

Disruptive effects of oxidants on barrier integrity of Caco-2 monolayers, as determined by fluorescein sulphonic acid (FSA) clearance. Apical chambers were loaded with FSA before exposure to graded concentrations of H2O2 and HOCl in the range 0.01–5 mM. Barrier integrity was determined as apical to basolateral flux of probe divided by the concentration of FSA in the apical chamber, expressed as clearance. *p<0.05 compared with control (0 mM oxidant or isotonic saline) (n=9 per group). H2O2, hydrogen peroxide; HOCl, hypochlorous acid.

Protective effect of epidermal growth factor (EGF) and transforming growth factor α (TGF-α) preincubation on the barrier integrity of Caco-2 monolayers, assessed by fluorescein sulphonic acid (FSA) clearance. Monolayers were pretreated with growth factor 10 ng/ml before subsequent exposure to 0.5 or 5 mM of H2O2 or HOCl. Clearance was assessed as described in fig 1. *p<0.05 v control or growth factor pretreatment; †p<0.05 v 0.5 or 5 mM of H2O2 or HOCl alone (n=6 per group).
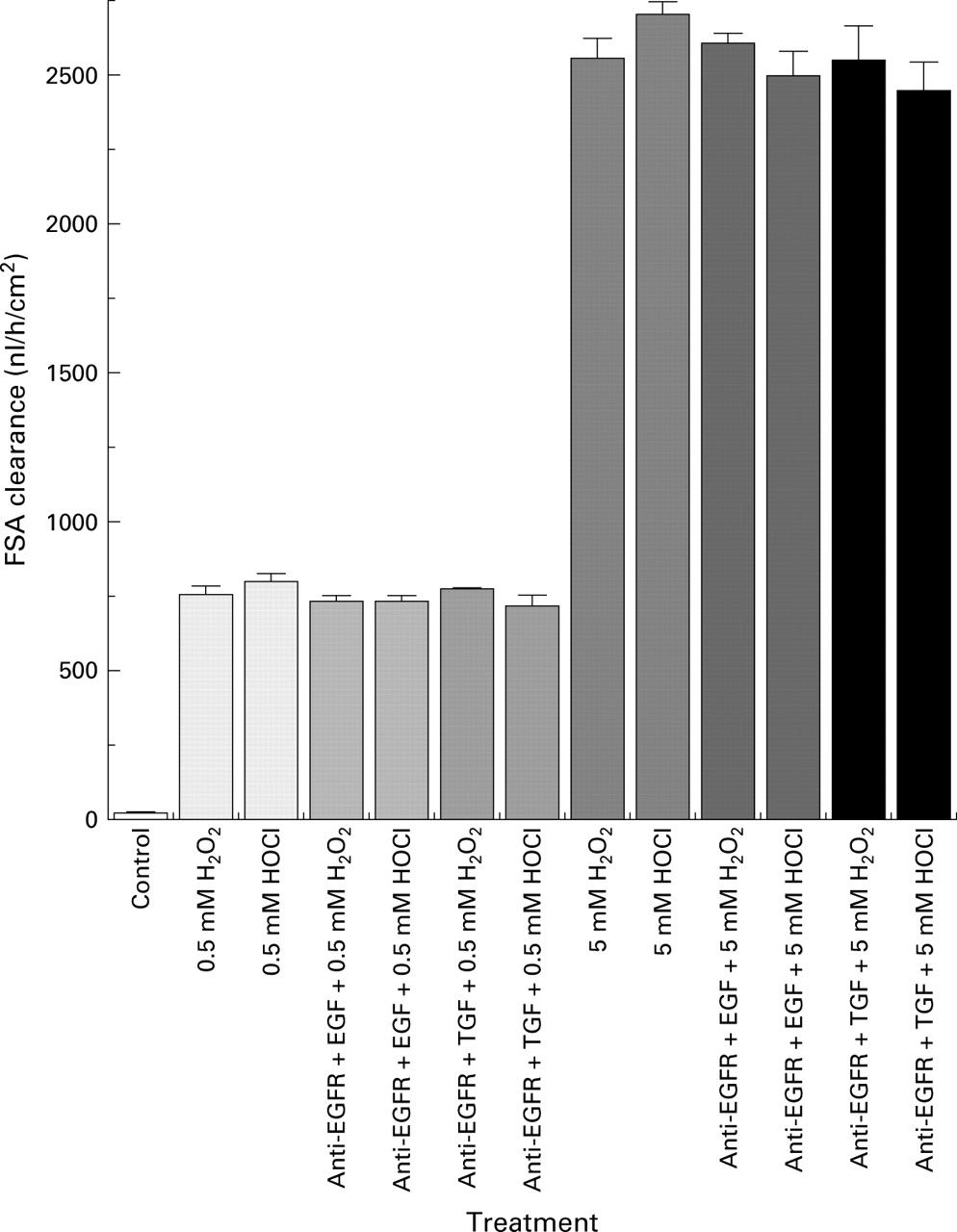
Blocking effects of epidermal growth factor receptor monoclonal antibody (anti-EGFR) on the protective action of growth factors on Caco-2 monolayer barrier function, as assessed by fluorescein sulphonic acid (FSA) clearance. Conditions as in fig 2except that anti-EGFR 1 μg/ml was pre-loaded before the growth factors. Clearance was measured as described in fig 1.

(A) Effect of graded concentrations of H2O2 or HOCl and of pretreatment with epidermal growth factor (EGF) on the integrity of Caco-2 cells, assessed by the fluorescent probe calcein-AM and expressed as percentage of cells injured. Protective EGF 10 ng/ml was added to the monolayers 10 minutes before subsequent exposure to deleterious oxidant for 30 minutes. *p<0.05 v control (0 mM oxidant or isotonic saline) or EGF pretreatment; †p<0.05 v 0.1–5 mM H2O2 or HOCl alone (n=9 per group). (B) Effect of graded doses of H2O2 or HOCl on the cell integrity of Caco-2 monolayers determined by ethidium homodimer-1, expressed as percentage of cells dead.
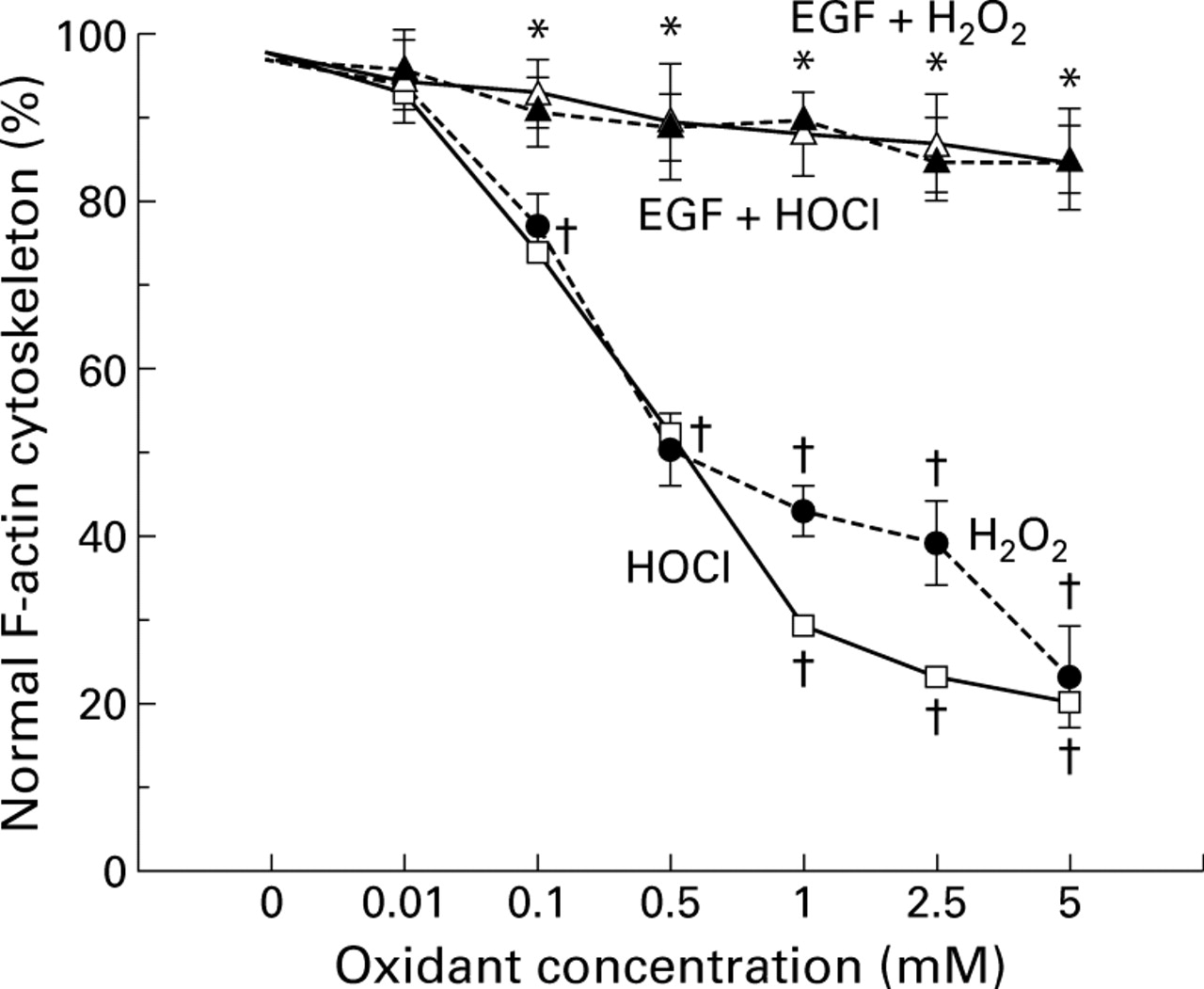
Incubation of Caco-2 monolayers with increasing concentrations of H2O2 and HOCl dose dependently elicited F-actin cytoskeletal disruption, as quantitated by reduction in the percentage of cells displaying normal actin (fig 5). The lowest concentration of either oxidant noted which disrupted the actin cytoskeleton was 0.1 mM, paralleling findings for barrier dysfunction. In contrast, pretreatment with EGF or TGF-α completely abolished the disruptive effects of oxidants on F-actin (fig 5, EGF data shown).
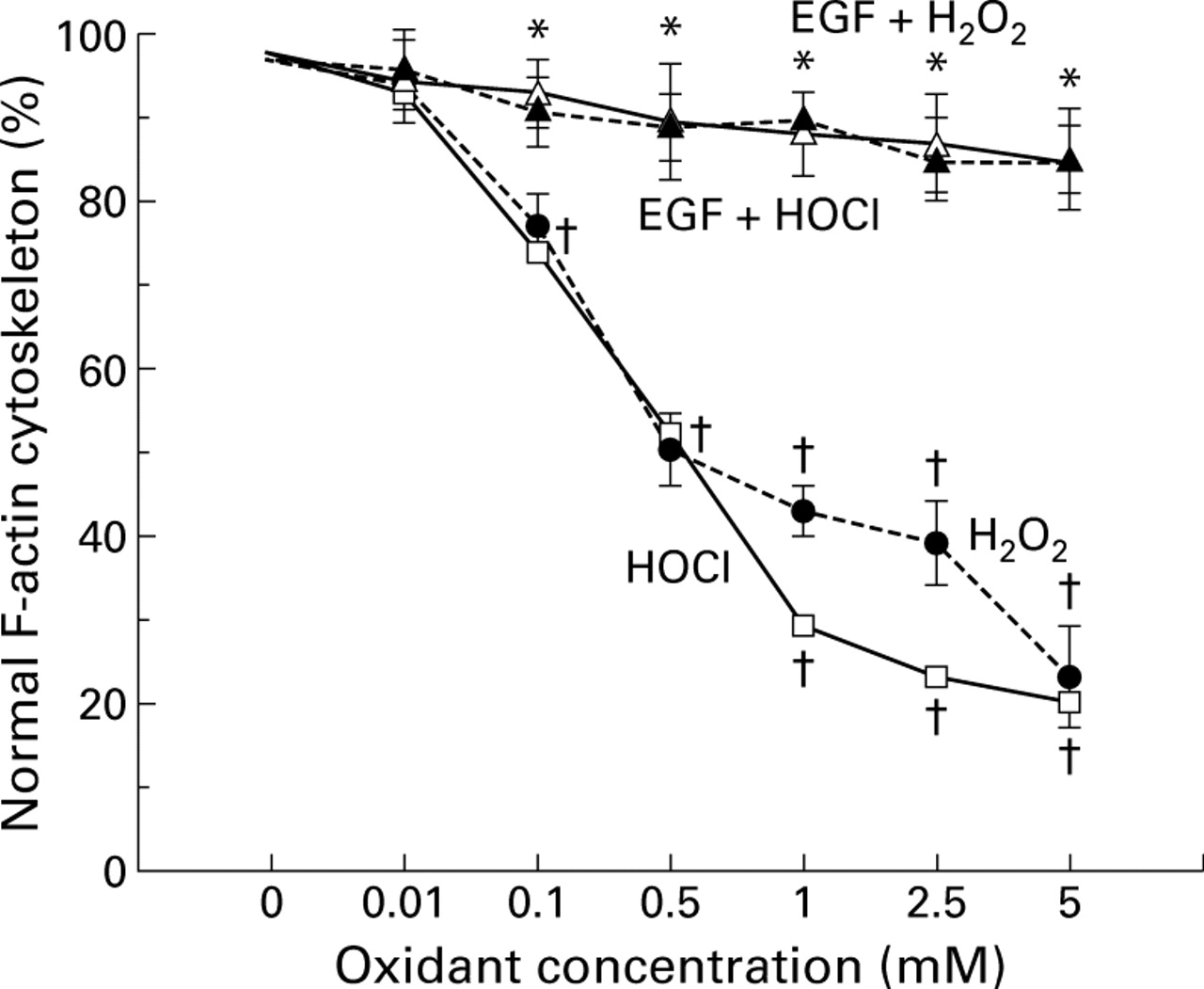
Percentage of Caco-2 cells displaying normal actin cytoskeleton, determined by high resolution laser scanning confocal microscopy. Monolayers were exposed to damaging H2O2 or HOCl concentrations or pretreated with protective epidermal growth factor (EGF) before immediate exposure to injurious oxidant for 30 minutes. †p <0.05 v control (0 mM oxidant or isotonic saline) or EGF pretreatment; *p<0.05 v 0.1–5 mM oxidant alone (n=6 per group).
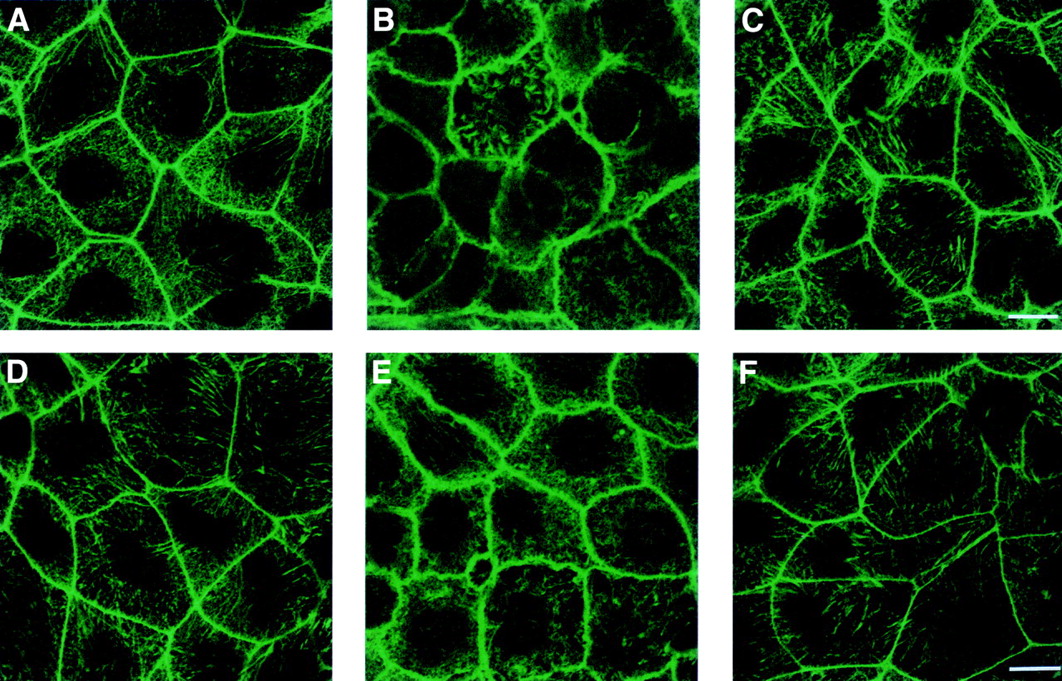
Figure 6 shows that H2O2 or HOCl exposure (0.5 mM shown) produced the disruption, condensation, and beading of the actin “ring” associated with the plasma membrane, as shown by the fluorescent staining (fig 6B and E, respectively). Pretreatment with EGF or TGF-α before exposure to injurious oxidants stabilised F-actin, as demonstrated by a normal, smooth, continuous pattern of actin ring at the areas of cell-cell contact (fig 6C, F; EGF shown). Growth factor pretreated monolayers were indistinguishable from controls (fig 6A, D).

Fluorescent staining of actin cytoskeleton by fluorescein conjugated phalloidin in intestinal monolayers following incubation with isotonic saline (control) (A), 0.5 mM H2O2 (B), pretreated with epidermal growth factor (EGF) before exposure to 0.5 mM H2O2 (C), control (D), 0.5 mM HOCl (E), and those preincubated with EGF before exposure to 0.5 mM HOCl (F). Control monolayers demonstrate a normal, continuous, smooth distribution of actin (ring or cortex) at areas of cell-cell contact whereas oxidant exposed monolayers show areas of actin disruption, condensation, beading, and fragmentation. Actin in EGF pretreated cells resembles the morphology detected in the controls and appears normal. Bar, 25 μM.
Quantitative immunoblotting of the polymerised (F- or S2) and monomeric (G- or S1) fractions of actin confirmed the fluorescent findings (fig7). Both oxidants resulted in a significant decrease in the stable F-actin and an increase in the monomeric G-actin, indicating disruption of the actin cytoskeleton. In contrast, EGF or TGF-α augmented the stable F-actin and decreased the G-actin in monolayers exposed to oxidants (fig 7). A representative western blot gel (fig 8) demonstrated that exposure to oxidants decreased the autoradiographic F-actin band density while preincubation with growth factors enhanced the F-actin band density to control levels, indicating enhanced assembly (and stabilisation) of actin.

Quantitative western immunoblotting analysis of polymerised F-actin (S2 fraction, index of stabilisation) and monomeric G-actin (S1 fraction, index of disruption) after treatment with epidermal growth factor (EGF), transforming growth factor α (TGF-α) 10 ng/ml, or vehicle before exposure to oxidants in Caco-2 cells. Percentage polymerisation of actin pool = ((S2)/total actin pool (S2+S1)). †p<0.05 v control or pretreatment with EGF or TGF-α.; *p<0.05 v H2O2 or HOCl alone (n=6 per group).

Western immunoblot micrograph of the F-actin (S2, triton insoluble) extracts following treatments. F-actin fractions were analysed by SDS-PAGE and western immunoblots using monoclonal antiactin as primary antibody followed by horseradish peroxidase conjugated-secondary antibody and subsequently processed for x ray film exposure. The lanes (corresponding optical densities) from left to right are: actin standard (10 951); 0.5 mM H2O2 (7452); 5 mM H2O2 (5611); isotonic saline (control, 10 734); EGF+5 mM H2O2 (9924); EGF alone (9865); TGF-α+5 mM H2O2 (9752); TGF-α alone (10 568).
To determine whether the disruptive effects of oxidants are caused by oxidation of actin and whether protection by growth factor involves prevention of such a change, we measured carbonyl formation (an index of protein oxidation) by immunoblotting. H2O2and HOCl (0.1 and 0.5 mM data shown) increased the oxidation (anti-DNP immunoreactivity) of F-actin while pretreatment with EGF or TGF-α prevented such oxidation (fig 9). These data parallel findings on the disassembly and disruption of actin by oxidants and promotion of its assembly and stabilisation by growth factors.

Graphical representation of western immunoblotting of anti-dinitrophenylhydrazone (DNP) immunoreactivity showing oxidation (carbonylation) of the polymerised F-actin fraction in Caco-2 cells after various treatment regimens. Monolayers were pretreated with epidermal growth factor (EGF), transforming growth factor α (TGF-α), phalloidin, or vehicle before exposure to H2O2 or HOCl. Carbonyl formation (anti-DNP immunoreactivity) was expressed as the ratio of carbonyl formation (oxidation) in the treatment group divided by oxidised actin standard. †p<0.05 v control, EGF, TGF-α, or phalloidin pretreatment;*p<0.05 v H2O2 or HOCl alone (n=6 per group).
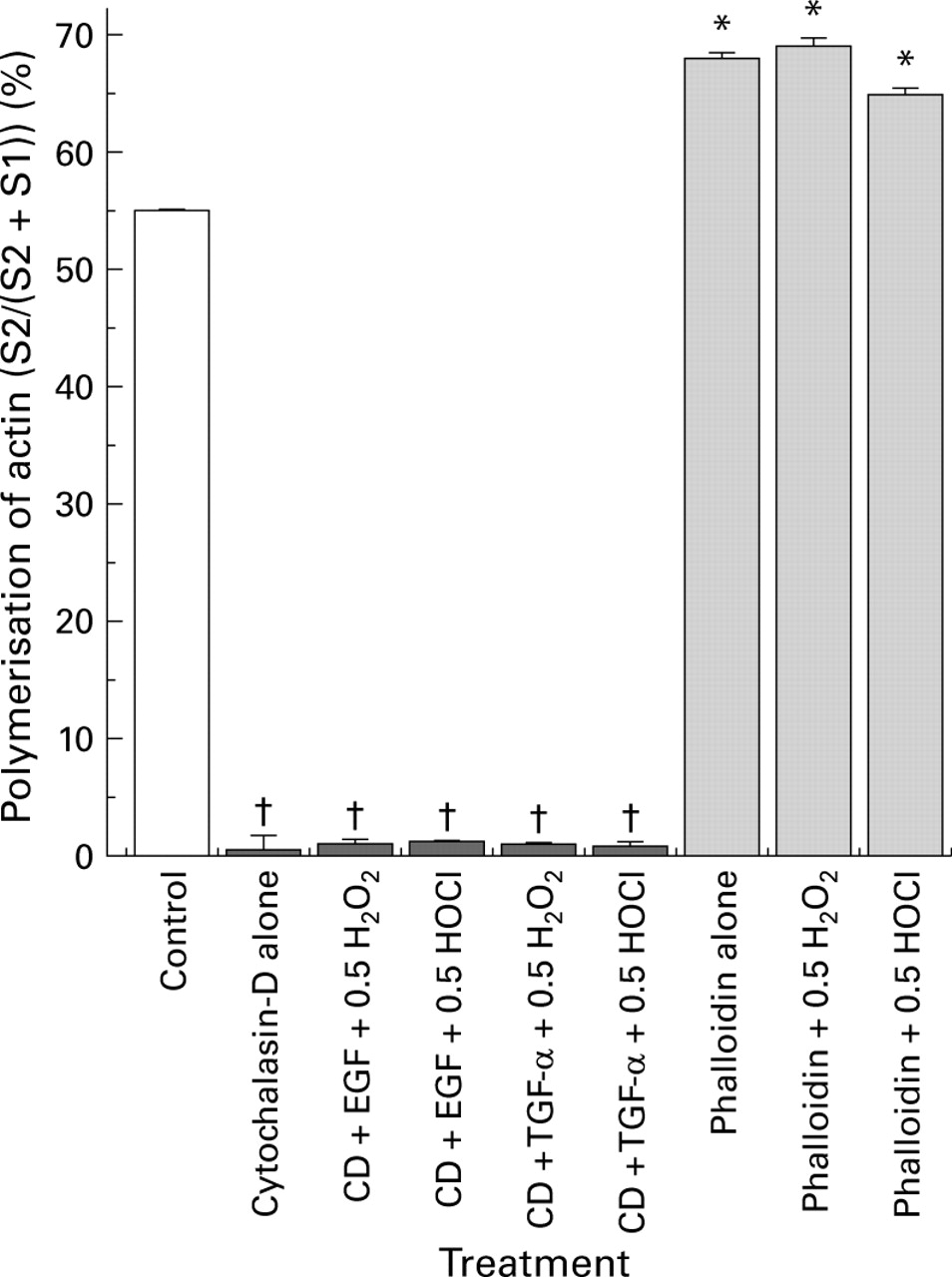
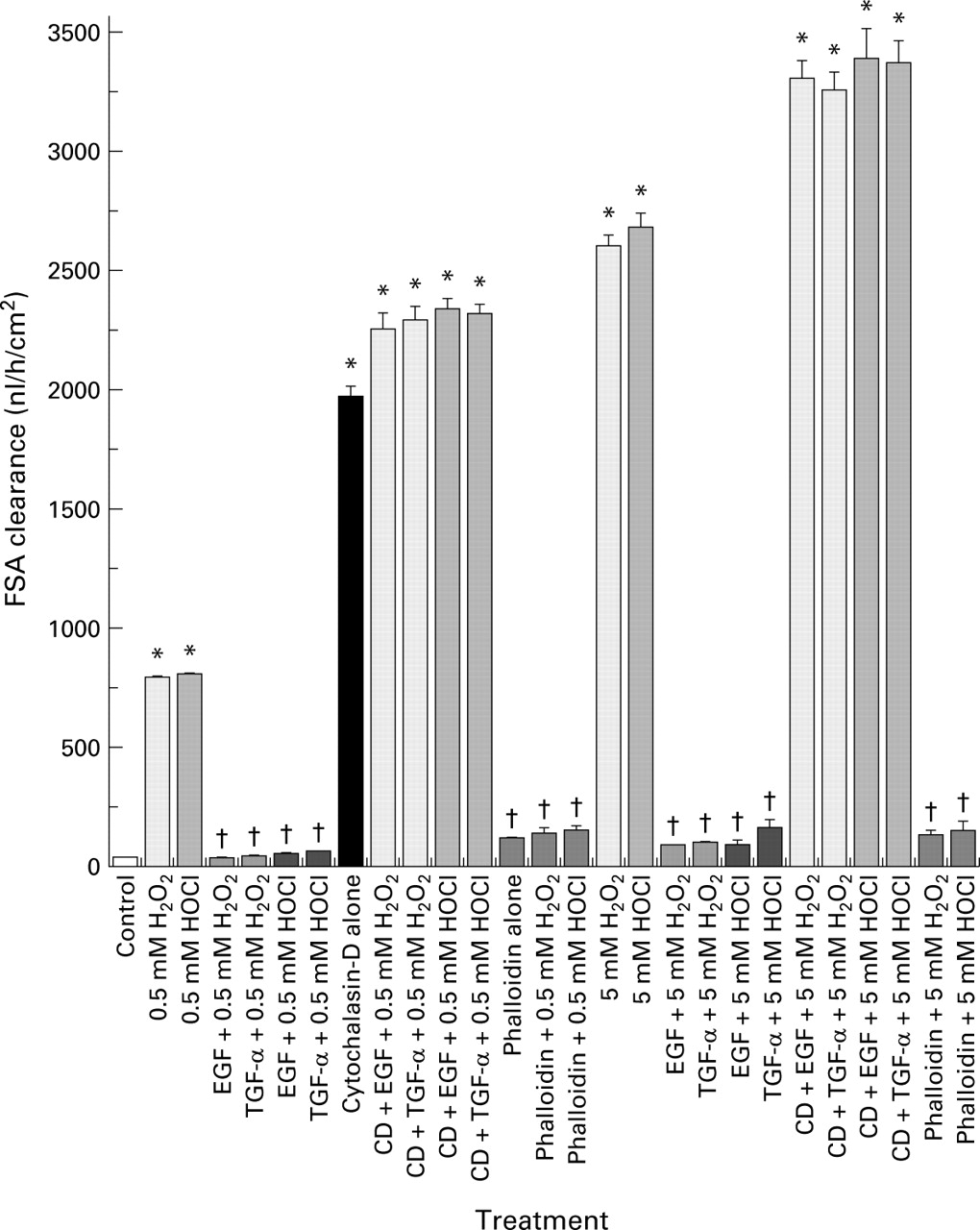
We then directly studied the role of actin stability in the maintenance of barrier function using two actin modulators, cytochalasin-D (a potent actin disruptive drug) and phalloidin (an actin stabiliser). We surmised that cytochalasin-D might not only disrupt barrier function itself but also abolish the protection afforded by growth factors. Indeed, treatment with cytochalasin-D caused loss of monolayer barrier integrity and prevented the protective actions of EGF or TGF-α on barrier function in monolayers exposed to oxidants (fig 10). Furthermore, preincubation with phalloidin prevented the loss of barrier function induced by oxidants, exhibiting effects not dissimilar to the growth factors (fig 10). Western blotting demonstrated (fig 11) that cytochalasin-D reduced the polymerised F-actin fraction and augmented the monomeric G-actin, indicating complete depolymerisation of the actin cytoskeleton. In contrast, phalloidin stabilised actin and blunted the destabilising effects of oxidants, as shown by the presence of a large F-actin pool (fig 11).
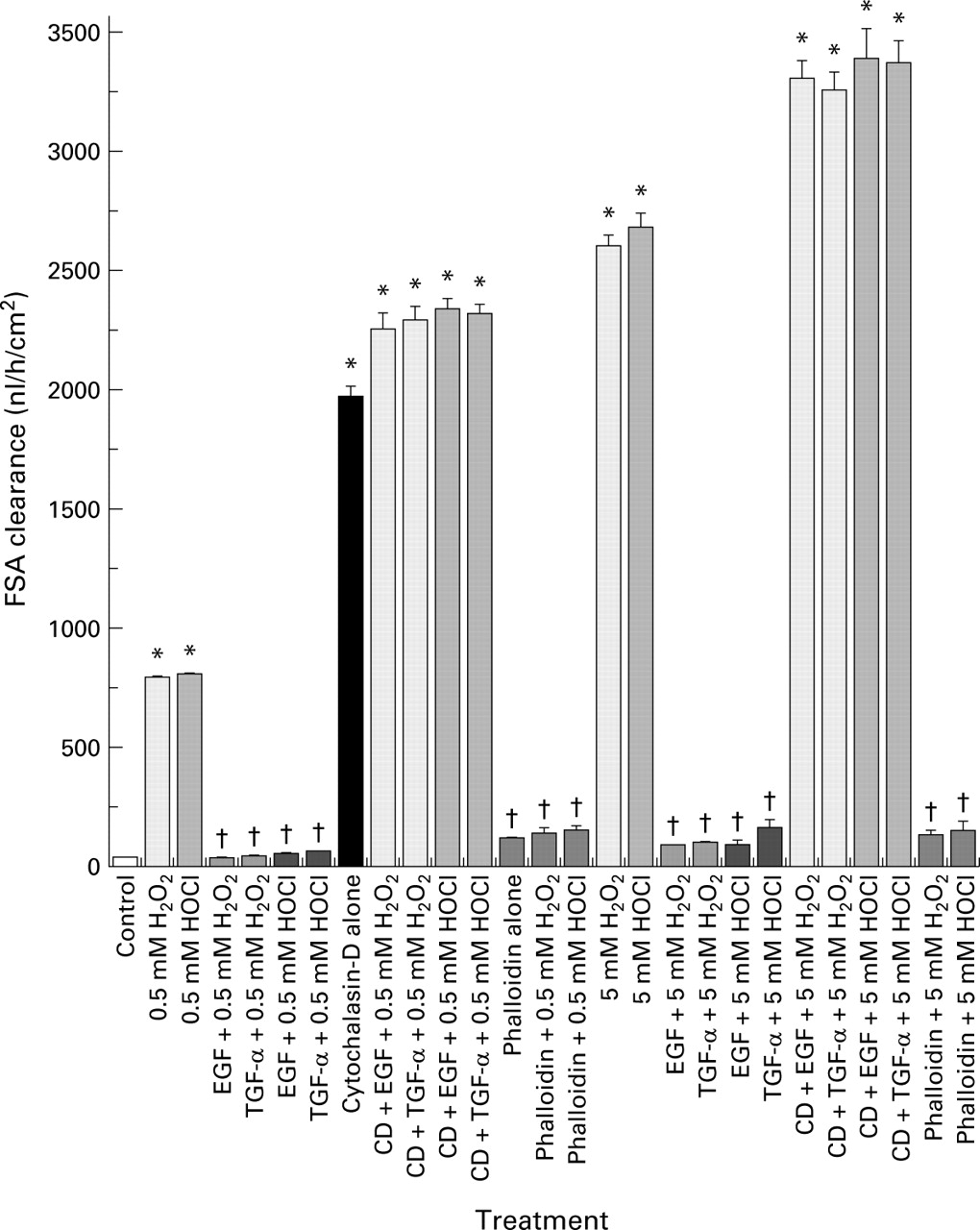
Disruptive effect of cytochalasin-D (CD) and protective action of phalloidin on the barrier integrity of Caco-2 monolayers, as determined by fluorescein sulphonic acid (FSA) clearance. Monolayers were preincubated with actin modulators, cytochalasin-D, or phalloidin, and then exposed to H2O2 or HOCl in the presence or absence of epidermal growth factor (EGF) or transforming growth factor α (TGF-α) pretreatment. Clearance was determined as described in fig 1. †p<0.05 v corresponding cytochalasin-D or oxidant alone; *p<0.05 v control (isotonic saline) or corresponding EGF (or TGF-α) + H2O2 (or HOCl), or corresponding phalloidin treatment (n=6 per group).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytochalasin-D (CD) induced actin depolymerisation and phalloidin induced actin assembly, as determined by western immunoblotting analysis of the distribution of polymerised F-actin (S2, index of stabilisation) and monomeric G-actin (S1, index of disruption) in Caco-2 cultures. Monolayers were pretreated with cytochalasin-D or phalloidin before incubation with or without epidermal growth factor (EGF) (or transforming growth factor α (TGF-α)) followed by exposure to H2O2 or HOCl. Percentage polymerisation of actin pool=((S2)/total actin pool (S2+S1)). †p<0.05 v control or phalloidin alone or pretreated groups; *p<0.05 v cytochalasin-D alone or pretreated groups (n=6 per group).
Discussion
Abnormal intestinal mucosal barrier integrity in concert with a high level of mucosal oxidants has been documented in patients with IBD.1-6 Despite substantial accumulation of evidence implicating oxidants in the pathogenesis of tissue damage in IBD5 6 the precise mechanism of oxidant induced loss of mucosal barrier function is poorly defined and merits further investigation.
The actin component of the cytoskeleton is one of the most abundant proteins in the eukaryotic cytosol with the ability to polymerise into filaments of highly dynamic alpha double helices.8 9 In epithelial cells such filaments make up a dense cross linked actin cortex located on the inner side of the plasma membrane. Actin also comprises filamentous stress fibres which traverse the cytosol and constitute short fibres extending into the lamellipodia in motile cells.10 This structural element is essential in maintaining normal cellular physiology, structure, locomotion, and support functions.7-11 The actin based skeleton also plays a critical role in regulating the permeability of epithelial monolayers.11 These functions are based on the ability of monomeric G-actin to polymerise and on the stability of F-actin polymers to resist disassembly. Despite the crucial role of actin in cell integrity, little is known of the role of F- and G-actin in oxidant induced loss of mucosal barrier function.
In the present investigation, both H2O2 and HOCl at physiologically relevant concentrations dose dependently and significantly caused cellular injury and monolayer barrier dysfunction. We previously demonstrated that oxidants caused oxidation and disassembly of the microtubule component of the cytoskeleton in Caco-2 cells and that these effects were prevented by EGF and TGF-α.14 15 Presently, we have further extended these findings to include the role of the actin element of the cytoskeleton in the processes of disruption and protection. Specifically, oxidants decreased the level of stable F-actin, increased the unstable G-actin, and caused actin oxidation. Parallel to these alterations was a dose related reduction in the percentage of cells displaying normal actin. Moreover, our data showed that there was a significant difference between the high concentration (1 or 2.5 mM) of HOCl and H2O2 on FSA clearance. HOCl had significantly more disruptive effects than H2O2. A possible explanation for this is catabolism of H2O2 by cellular peroxidases such as catalase and glutathione peroxidase. To our knowledge, our findings are the first demonstration that noxious oxidants cause oxidation and disassembly of actin while concomitantly leading to its disruption in a cellular model of intestinal epithelial barrier. Our data suggest that the underlying mechanism of intestinal barrier dysfunction induced by oxidants seen in disorders such as IBD may be due to the oxidation, disassembly, and disruption of the cytoskeletal network. Indeed, our previous in vivo animal study supports this possibility. Specifically, we demonstrated that mucosal injury induced by a hypertonic solution was associated with extensive disorganisation and remodelling of actin.12 However, further studies on inflamed and non-inflamed mucosal tissues from patients with IBD are required to demonstrate the pivotal role of the cytoskeletal network in mucosal barrier dysfunction in these patients.
It is well documented that EGF and TGF-α prevent the development of GI mucosal injury in animal models, as well as under in vitro conditions.17-20 26-28 EGF also abolishes the transepithelial resistance drop and barrier dysfunction in Caco-2 monolayers exposed to toxins, ethanol, or oxidants.14 15 20 28 These in vitro studies indicate that the mechanisms responsible for growth factor protection are independent of intact blood flow, neural innervation, and circulating humoral agents, and suggest that a basic cellular mechanism is orchestrated by these growth factors. None the less, the mechanisms by which EGF and TGF-α elicit protection have yet to be identified.
In the current study, we have demonstrated that EGF or TGF-α protected Caco-2 monolayers from oxidant induced injury. Moreover, such protection was specific and mediated by EGF receptor (EGF-R). It was our belief that EGF or TGF-α should prevent the oxidation, disassembly, and disruption of actin if this cytoskeletal component in fact plays a key role in the mechanism of protection. Indeed, we showed that EGF or TGF-α prevented the oxidation of actin and, in concert, abolished actin depolymerisation and enhanced the stable F-actin in monolayers exposed to damaging oxidants. These beneficial effects of growth factors were associated with maintenance of a significantly high percentage of colonocytes displaying intact actin cytoskeleton. Our findings show for the first time that EGF and TGF-α promote the remodeling of actin, protect actin against oxidative injury, stabilise the cytoskeletal network, and prevent intestinal monolayer barrier dysfunction in vitro.
Our studies using actin stabiliser (phalloidin) provided evidence for the importance of intact actin in maintaining normal barrier integrity. Moreover, using cytochalasin-D, an inhibitor of actin assembly, we provided further evidence on the key role of actin in barrier function and in EGF and TGF-α protection. These conclusions are based on the findings that phalloidin prevented disassembly and disruption of actin, and protected monolayers despite exposure to damaging oxidants. Additionally, cytochalasin-D potentiated oxidant induced disruption of the monolayer barrier and abolished the protective effects of growth factors. Our immunoblotting studies provide direct evidence that phalloidin stabilised actin while cytochalasin-D caused its disassembly. Hence the modulatory actions of these agents on barrier function was mediated by their effects on the actin network.
Our investigation indicates that actin stability is vital for intact monolayer barrier function in the face of an injurious challenge. These findings also suggest that actin can be a potential target for the development of new therapies for disorders associated with oxidative injury and barrier dysfunction, such as IBD. Furthermore, our findings suggest that EGF and TGF-α apparently evoke their protective effects, at least in part, by their unique ability to enhance actin polymerisation and integrity. The mechanism of this protective effect of growth factor remains unknown. One proposed mechanism is through possible alterations in the ratio of ATP/ADP actin bound to the end of fragmented or disassembled G-actin.9 10 Additionally, growth factors may help anchor F-actin via formation of nucleation sites at the site of membrane and by regulating the formation of anchoring sites at areas of cell-cell contact.9 10 Another possibility is activation of a monomeric G protein, Ras, which is believed to be involved in the rapid assembly/disassembly of actin through EGF-R signal transduction processes.29 30 These actions by growth factors may allow the actin cytoskeleton to maintain the shape of the plasma membrane and to reinforce membrane support, and in turn maintain a normal barrier in the face of an oxidative challenge. Further studies are needed, however, to evaluate these potential mechanisms.
Acknowledgments
This work was supported in part by a grant from Otsuka America Pharmaceutical Company.
References
Footnotes
- Abbreviations used in this paper:
- ROM
- reactive oxygen metabolites
- IBD
- inflammatory bowel disease
- EGF
- epidermal growth factor
- TGF-α
- transforming growth factor α
- DMEM
- Dulbecco's minimum essential medium
- LSCM
- laser scanning fluorescent microscopy
- FSA
- fluorescein sulphonic acid
- DNP
- dinitrophenylhydrazine