Article Text

Abstract
INTRODUCTION Epidermal growth factor (EGF) is normally present as EGF1–53. A variety of C terminal truncated forms have been used in preliminary trials for treating gastrointestinal injury but their relative potency and stability when used in a clinical setting are unclear. Therefore, we compared the biological activity of recombinant EGF1–53, EGF1–52, EGF1–51, and the C terminal peptides EGF44–53 and EGF49–53.
METHODS Purity of forms was confirmed by mass spectrometry. Bioactivity of the different EGF forms was determined using [methyl-3H] thymidine incorporation into primary rat hepatocytes and their ability to reduce indomethacin (20 mg/kg subcutaneously)/restraint induced gastric injury in rats. Stability of EGF peptides was determined by serial sampling from a syringe driver system containing EGF/4% albumin in saline.
RESULTS Biological activity assays of EGF1–53, EGF1–52, and EGF1–51 gave almost identical thymidine uptake dose-response curves (maximal responses increasing baseline uptake from 4400 (600) cpm (mean (SEM)) to about 22 000 (2000) cpm when EGF was added at 1.6 nM). EGF44–53 and EGF49–53 did not stimulate 3H thymidine uptake. Control rats had 47 (4) mm2 damage/stomach, EGF1–51, EGF1–52, and EGF1–53 at 0.16 and 0.80 nmol/kg/h each reduced gastric injury by about 50% and 80%, respectively (both doses p<0.01 compared with control but no significant difference between the different forms). EGF was stable at room temperature for seven days but biological activity decreased by 35% and 40% at two and three weeks, respectively (both p<0.01). Exposure to light did not affect bioactivity.
CONCLUSION EGF1–51and EGF1–52 are as biologically active as full length EGF1–53 but the C terminal penta- and decapeptides are ineffective. Clinical trials of EGF can probably use infusion systems for at least 48 hours at room temperature and with exposure to light, without reducing biological efficacy.
- epidermal growth factor
- intestinal injury
- nutrition
Abbreviations used in this paper
- Cys
- cysteine (residues)
- EGF
- epidermal growth factor
- HPLC
- high pressure liquid chromatography
- TFA
- trifluoroacetic acid
Statistics from Altmetric.com
Epidermal growth factor (EGF) is secreted into the gastrointestinal lumen by the salivary glands and the Brunner's glands of the duodenum.1 It is initially produced as a 1207 amino acid precursor2 which is subsequently processed to the “mature” EGF1–53 form which includes three intrachain cysteine (Cys)-Cys double bonds. It is likely that the major source of EGF present in the gastric juice is derived from swallowed saliva. Although EGF1–53 is generally considered to be the “mature” form of EGF in humans, C terminal truncated forms occur naturally as a result of partial cleavage by proteases present in the gastric juice, mainly to EGF1–49 3 and in plasma to EGF1–52.4 In addition to these smaller forms of EGF, higher molecular weight forms are also found in human urine, probably derived directly from the kidney.5
EGF is a potent stimulant of proliferation and healing in vitro and in animal models in vivo. There is therefore much interest in the potential clinical applications of recombinant EGF for the treatment of gastrointestinal damage, particularly in relation to small and large intestinal disease where present therapies are suboptimal. Preliminary human trials of EGF for conditions such as neonatal necrotising enterocolitis and microvillus atrophy have provided encouraging results. However, there has been inconsistency in the form of EGF used, with various clinical trials using EGF1–48, a mixture of EGF1–51 and EGF1–52, or EGF1–53alone.6-11 As the potency of the EGF molecule is highly dependent on the C terminal residues,3 ,12 ,13 it is difficult to extrapolate the equivalent dosage (in terms of biological activity) from one trial to another.
We are aware of only one commercial product of EGF which is currently available for clinical use in an intravenous formulation (HeberBiotc SA, Havana, Cuba). This comprises a 60:40 mixture of EGF1–51 and EGF1–52. However, there are limited data on the relative biological potency of these EGF1–51 and EGF1–52 forms compared with the full length EGF1–53.
Hence we purified the separate forms of EGF and compared their biological potency with the full length molecule. In addition, as much of the biological activity of EGF appears to be dependent on the C terminal amino acids, we also examined the biological activity of synthetic peptides coding for the C terminal penta- and decapeptides of the EGF molecule.
EGF has a short circulating half life (about eight minutes) and most clinical studies therefore use a continuous intravenous protocol (for example, see Sullivan and colleagues8). However, the stability of EGF, in terms of maintenance of biological activity while present in the infusion system (usually at room temperature), has not been fully addressed. In addition, these neonates receiving EGF for necrotising enterocolitis or congenital microvillus atrophy are also likely to be treated with phototherapy to reduce the risk of kernicterus. However, the effect of exposure to light on the biological activity of EGF has not been examined. Hence we also determined the importance of these factors on the biological activity of the EGF preparation which is available for clinical use (that is, the EGF1–51/EGF1–52 mixture).
Materials and methods
Human recombinant EGF1–51 and EGF1–52mixture, expressed in Saccharomyces cerevisiae, was obtained from Heber Biotc SA (Havana, Cuba). This product is >95% pure as assessed by high pressure liquid chromatography (HPLC). Full length EGF1–53 expressed inE coli was purchased from Promega UK Ltd (Southampton, UK) and is >95% pure, as assessed by HPLC. Cell culture media and reagents were obtained from Gibco, Life Technologies Ltd (Paisley, Scotland, UK). [Methyl-3H] thymidine was obtained from Amersham Life Science (Little Chalfont, UK). All other reagents were obtained from Sigma-Aldrich Company Ltd (Poole, Dorset, UK).
SEPARATION OF THE DIFFERENT FORMS OF EGF
EGF1–51 and EGF1–52 were purified from the commercially available recombinant mixture using reverse phase HPLC. In addition, to ensure consistency between the sample preparations, EGF1–53 was treated in a similar way.
The system used for these studies comprised the Hewlett Packard (Stockport, UK) 1100 consisting of a quaternary pump delivery system, Rheodyne-7725 sample injector, and 200 μl injection loop. Samples were collected on a Foxy-Jr fraction collector and the system was controlled by a HP pentium PC with HP Chemstation software. The column used was an analytical “Jupiter” C5, 300 Å, 5 μm (4.6×150 mm) column (Phenomenex UK Ltd) with a 0.1% trifluoroacetic acid (TFA)/acetonitrile gradient. Peptide peaks were identified by UV detection at a wavelength of 280 nm.
Purified fractions were collected, lyophilised, and protein concentration determined using a commercial, modified Lowry based, calorimetric microplate assay with human albumin as standard (Bio-Rad Laboratories Ltd, Hercules, California, USA).
The identity of each form was determined by assessment of molecular weight using the technique of matrix assisted laser desorption time of flight with a Finnigan LaserMAT mass spectrophotometer (San Jose, California, USA). Samples were mixed with 0.5 μl of alpha-cyano-4-hydroxycinnamic acid matrix (1% in 50% acetonitrile, 0.1% TFA). Standards used were oxidised insulin β chain (molecular weight 3496.9) and ubiquitin (molecular weight 8564.9). This technique has been validated previously for use in peptide sequence analyses.3-14
SYNTHESIS OF PENTA- AND DECAPEPTIDES
Peptides corresponding to the last five and 10 C terminal amino acids of human EGF were obtained from the Peptide Synthesis Laboratory (Imperial Cancer Research Fund, London, UK). They were synthesised on a 431A PE Biosystems peptide synthesiser and analysed for purity using reverse phase HPLC and mass spectrometry, as detailed above. The amino acid sequences of the two peptides were: pentapeptide—NH2 Trp-Trp-Glu-Leu-Arg COOH and decapeptide—NH2Tyr-Arg-Asp-Leu-Lys-Trp-Trp-Glu-Leu-Arg COOH.
ASSAY OF BIOLOGICAL ACTIVITY OF DIFFERENT FORMS OF EGF IN VITRO
Background
Primary rat hepatocytes provide a robust reproducible method for determining the biological activity of EGF-like molecules. We have previously used this method to determine the biological activity of other truncated forms of EGF.3 The methodology is therefore described only briefly below.
Preparation of primary rat hepatocytes
Male August rats were anaesthetised with Hypnorm (Janssen-Cilag Ltd, High Wycombe, UK) and hepatocytes were isolated by in situ collagenase perfusion. The basic protocol consists of a two step perfusion of the liver in situ via the portal vein, first with calcium free buffer followed by a calcium supplemented buffer containing collagenase.
The digested liver was removed, cells dispersed, filtered and centrifuged, and re-suspended in a plating medium. For all studies, hepatocytes were grown in Williams E medium withoutl-glutamine (Gibco BRL, Paisley, Scotland, UK) containing 5% fetal calf serum. Cell viability, determined by the ability to exclude 0.2% trypan blue, was greater than 80% in all experiments.
Assay protocol
The various purified forms of EGF, the commercial mixed EGF1–51/EGF1–52 preparation, and the C terminal peptides were added to the wells. Twelve hours later, [methyl-3H] thymidine (0.8 μM, 20 μCi/ml) was added to the wells and the plates incubated for a further 18 hours. [Methyl-3H] thymidine incorporation into TCA precipitated material was determined by liquid scintillation using a 1450 Microbeta Trilux plate reader (EG&G Wallac, Turku, Finland). Each condition was examined in at least four separate wells during each study and was also examined on three separate occasions.
As a preliminary study, dose-response curves to the different forms of EGF were performed in the presence and absence of dexamethasone 10−8M and insulin 10−7M. This was done because it has been reported that dexamethasone improves cell plating efficiency and insulin enhances the stimulatory effect of EGF.15 Although the shape of the dose-response curves were virtually identical in the presence or absence of dexamethasone/insulin, the baseline/stimulated [methyl-3H] thymidine ratios were higher when these were added. Dexamethasone and insulin were therefore always used in subsequent studies.
ASSAY OF BIOLOGICAL ACTIVITY OF DIFFERENT FORMS OF EGF IN VIVO
The ability of EGF to prevent gastric damage by indomethacin and restraint in rats was assessed using previously validated methods.3 Under light ether anaesthesia, rats (male Sprague Dawley, 225–250 g) had two subcutaneous cannula inserted into the back of the neck and were then placed in Bullman restraint cages. When the animals had recovered, a continuous subcutaneous infusion of saline or the various doses and forms of EGF or C terminal peptides was started at 1 ml/h, using a multisyringe infusion pump (Harvard Apparatus, Massachusetts, USA). EGF1–53, EGF1–52, and EGF1–51 were infused at 0.16 and 0.80 nmol/kg/h whereas much higher doses (up to 63 nmol/kg/h) were used for the short peptides. This decision was based on the fact that our in vitro studies suggested that all three long forms of EGF were likely to have similar biological efficacy and we have previously shown a good dose-response for EGF1–53 when used at these doses.3 In contrast, the in vitro studies suggested much higher doses of the penta- and decapeptides would be required to elicit any biological activity. Thirty minutes later, 20 mg/kg of indomethacin were injected subcutaneously via the second cannulae. The animals were killed by stunning and cervical dislocation three hours later and their stomachs removed and inflated with 4 ml of 10% formalin. The next day the stomachs were opened and placed in fresh formalin prior to assessment. The stomachs were randomly coded and all analyses of gastric damage were assessed blind. Total ulcerated area (mm2/stomach) was assessed using a dissecting microscope (×10) with the aid of a square grid. The stomachs were then embedded in wax and the depth of damage assessed microscopically and given a microscopic ulcer score, as previously described.16 Using this system, each stomach was given a score of 0–4 (0, no damage; 1, one small erosion (less than 0.5 mm); 2, two small or one large erosion (greater than 0.5 mm); 3, two or more large erosions; and 4, any area of ulceration extending to the muscularis mucosa).
STABILITY OF EGF SOLUTIONS IN A CLINICAL SETTING
To reproduce likely clinical conditions, the commercially available EGF1–51/EGF1–52 mixture was diluted to a concentration of 1 μg/ml in a 4.5% solution of human albumin (to minimise adhesion of peptide to the infusion system) and split into two samples. The test solutions were pumped at room temperature (24–26°C) through an infusion system comprising a syringe driver and a 50 ml syringe attached to a paediatric infusion system (“giving set”, IVAC G30402). Sample aliquots (2 ml) were taken from the distal end of the infusion system at regular intervals up to 21 days and immediately frozen and stored at −20°C until subsequent assay. One infusion system was covered in silver foil (“dark”) and the other was placed under a single 4-tube Medical Phototherapy Unit lamp (Vickers plc, UK) at a distance of 50 cm, for the duration of the experiment (“light”). This was to mimic the conditions of phototherapy treatment commonly prescribed for the jaundiced neonate. Samples were subsequently analysed for biological activity using the rat hepatocyte assay described above.
STATISTICS
Data were analysed using ANOVA. For all analyses, if a significant effect was found (p<0.05), individual comparisons were performed using t tests based on the residual and degrees of freedom obtained from the ANOVA, a method equivalent to repeated measures analysis.
Results
The different forms of EGF eluted from the column as single peaks at 39 minutes (EGF1–53), 40 minutes (EGF1–52), and 43 minutes (EGF1–51). Analyses by mass spectrometry gave molecular masses close to calculated theoretical values: EGF1–53: 6222 versus 6222 calculated; EGF1–52: 6104 versus 6109 calculated; EGF1–51: 5950 versus 5953 calculated.
IN VITRO ASSAY
Each form of EGF gave virtually identical maximal responses with a sixfold increase in [methyl-3H] thymidine uptake above baseline values (all p<0.001 vbaseline values) when added at 1.6 nM. Baseline values were 4400 (600) cpm (mean (SEM)) and increased to 22 100 (4300) in response to EGF1–53, 22 100 (1000) in response to EGF1–52, 24 700 (3200) in response to EGF1–51, and 22 800 (2000) in response to the commercial mixed forms (effect of form, p=0.80 on ANOVA). Dose-response curves were also virtually superimposable (fig 1) and ANOVA showed no difference in response due to different forms (effect of form p=0.33 on ANOVA). Addition of the deca- or pentapeptide at doses up to 100 ng/ml (127 nM for pentapeptide and 68 nM for decapeptide) did not stimulate [methyl-3H] thymidine uptake above baseline values (data not shown).
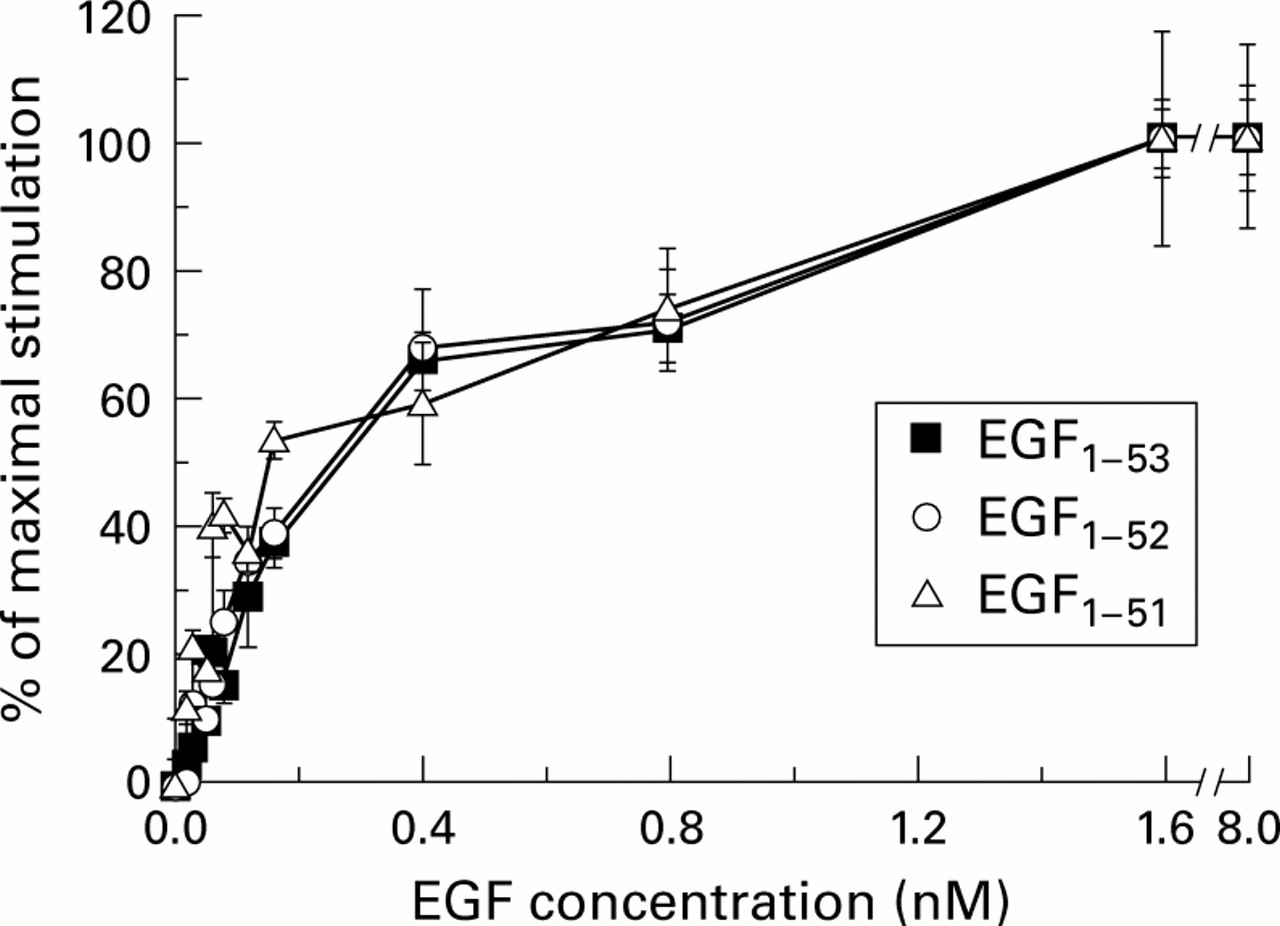
Effect of epidermal growth factors EGF1–53, EGF1–52, and EGF1–51 on [methyl-3H] thymidine uptake into primary rat hepatocytes. The dose-response curves of the various forms of EGF are virtually superimposable. Results are expressed as mean (SEM), n=4.
IN VIVO ASSAY
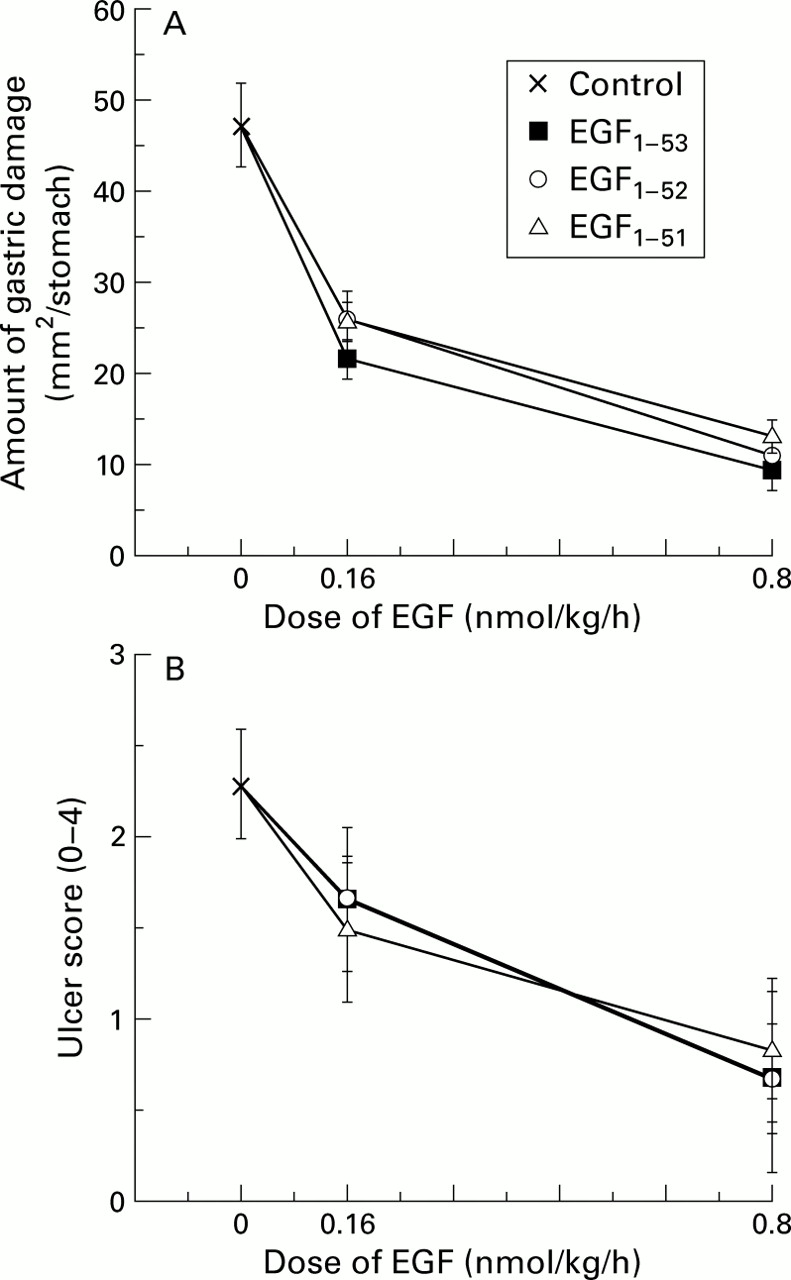
Control rats had 47 (4) mm2 damage/stomach. Administration of EGF1–51, EGF1–52, and EGF1–53, each given at 0.16 and 0.80 nmol/kg/h, reduced gastric injury by about 50% and 80%, respectively (both doses p<0.01v control but no significant difference between forms) (fig 2A). Assessment using the microscopic scoring system gave similar results (fig 2B). The penta- and decapeptides showed no protective effects when infused up to doses of 50 μg/kg/h (63 and 34 nmol/kg/h, respectively, data not shown).

Comparison of the biological activity of various forms of epidermal growth factor (EGF) using attenuation of gastric damage as a bioassay. Rats were restrained and given a continuous subcutaneous infusion of saline (control), EGF1–53, EGF1–52, or EGF1–51. Thirty minutes later, all animals also received indomethacin (20 mg/kg subcutaneously). Three hours after administration of indomethacin, all animals were killed and the amount of gastric injury assessed. (A) All forms of EGF reduced the amount of total ulcerated area (mm2/stomach) (all p<0.01 v control) but there were no differences in potency between the forms. (B) Assessment using a microscopic damage score, where each stomach was given a score of 0–4 (0, no damage; 1, one small erosion (less than 0.5 mm); 2, two small or one large erosion (greater than 0.5 mm); 3, two or more large erosions; and 4, any area of ulceration extending to the muscularis mucosa). Results are expressed as mean (SEM), n=6 animals per datum point.
STABILITY OF INFUSION SOLUTION OF EGF
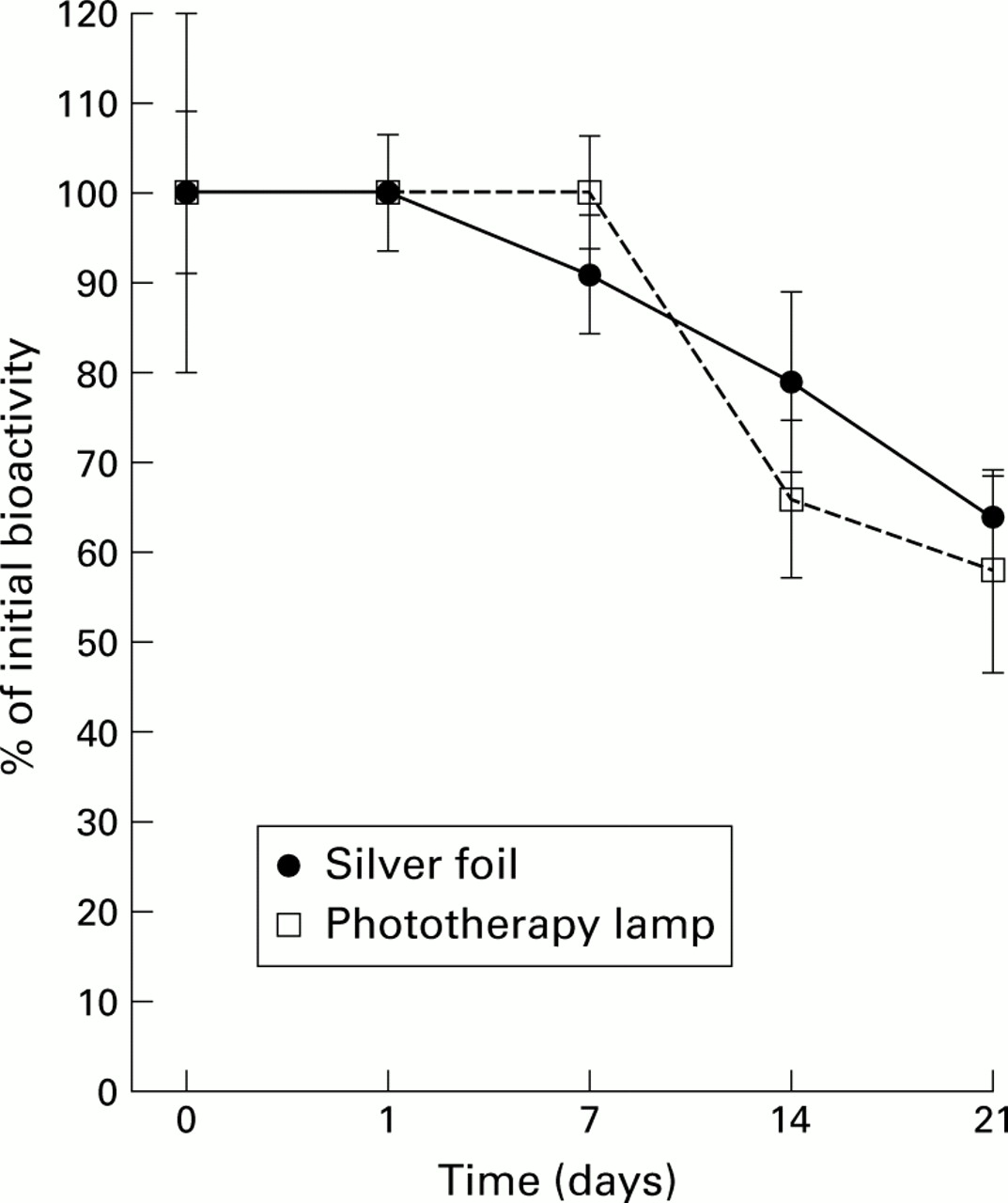
Under both “light” and “dark” conditions, EGF was stable at room temperature for seven days but biological activity decreased by about 20–30% at two weeks and by 35–40% at three weeks (both p<0.05 v initial levels) (fig 3). Exposure to light did not affect bioactivity (p=0.06 on ANOVA).

{kind=link}
{kind=link}
{kind=link}
Stability of epidermal growth factor (EGF) when placed in a clinical setting. A 60:40 mixture of EGF1–51and EGF1–52 is commercially available in an intravenous formulation. To determine its stability under likely clinical conditions, the EGF1–51/EGF1–52 mixture was diluted to 1 μg/ml in a 4.5% solution of human albumin and divided into two samples. The test solutions were pumped at room temperature (24–26°C) through an infusion system. One infusion system was covered in silver foil and the other was placed under a phototherapy lamp to mimic the conditions of phototherapy treatment commonly prescribed for the jaundiced neonate. Samples were subsequently analysed for biological activity using the rat hepatocyte assay. Results are expressed as mean (SEM), n=4.
Discussion
We used two well validated in vitro and in vivo models to compare directly the biological activity of EGF derivatives. In both systems, EGF1–51, EGF1–52, and EGF1–53had similar potencies whereas the C terminal penta- and decapeptides were inactive. Stability studies showed that solutions of EGF were stable for at least 48 hours at room temperature and with exposure to light, without any reduction in biological efficacy.
For the in vitro studies, we used the rat hepatocyte system as it provides a robust, reproducible model for examining growth factor activity and we have previous experience of its reliability in assessing the biological activity of other EGF derivatives.3 It provides a highly reproducible dose-response curve and is of particular value for examining growth factor activity in intestinal juice as hepatocytes are not adversely affected by normal small intestinal luminal contents.17This assay system has the advantages of using primary cells of gastrointestinal origin and providing a steep dose-response curve. This latter point is particularly relevant if comparing molecules which have similar potency. It cannot be used, however, to assess changes in cell number as under the conditions used, hepatocytes complete cell DNA synthesis but cell division occurs infrequently and no net increase in cell number is seen.15 Other assays which allow assessment of change in cell number have the limitation of poor dose-response curves and use malignant cell lines or cells of limited relevance to the gastrointestinal tract (for example, 3T3 fibroblasts).
EGF has many effects, including stimulation of cytoprotection, mucus production, and cell migration. Some workers have used inhibition of acid secretion as a marker of biological activity of C truncated EGF derivatives. The physiological relevance of luminal EGF in controlling acid secretion is, however, unclear as it is unlikely to reach its receptor under non-damaged circumstances.18 In addition, there is little consistency in results from studies examining the effect of C terminal truncation of EGF on acid secretion; Hollenberg and Gregory found that EGF1–47 had no loss of biological activity compared with EGF1–53 in terms of acid inhibitory activity, despite a 10-fold loss of ability to incorporate3H thymidine into fibroblasts.19 In contrast, Gregory and coworkers found that EGF1–47 had only an eighth of the activity of the intact molecule in a similar assay.12 We therefore decided to use a cytoprotection assay for the in vivo studies.
There are many well validated acute models of gastric injury. We chose the indomethacin/restraint model as our in vivo assay system because peptic ulceration due to non-steroidal anti-inflammatory drugs is a major source of morbidity and mortality in humans20 and we have previously used it to examine the influence of peptic digestion on the EGF molecule.3 Results from the in vitro and in vivo models gave consistent results in that all three forms of EGF gave similar dose-responses whereas the C terminal peptides were without biological effect.
EGF is produced as a 1207 amino acid precursor2 and is subsequently processed to the mature 53 amino acid form. In addition to the salivary glands, gastrointestinal EGF is also produced by the Brunner's glands of the duodenum and the ulcer associated cell lineage—a recently identified glandular structure induced at sites of injury. Its concentration in gastric juice is about 500 ng/l3 ,21 with urine concentrations being about one tenth of those found in gastric juice.22 Circulating levels of EGF are extremely low and probably consist mainly of the EGF1–52 form4 with much of this circulating EGF being bound to platelets. Early studies on the stability of EGF within the gastrointestinal lumen reported that EGF1–53was stable in acid and pepsin, but we have subsequently shown that it is cleaved to EGF1–49 (and to a lesser extent EGF1–46) in acidic gastric juice in vitro and in vivo.3 EGF is also susceptible to digestion by pancreatic proteases within the small intestinal lumen.17 The major site of EGF production outside of the gastrointestinal tract is the kidney where much larger forms of EGF are produced (molecular weight approximately 30 000) and excreted into urine.5
It is now generally accepted that the C terminal amino acids of the EGF molecule play a key role in mediating much of its biological activity. Loss of the C terminal seven (EGF1–46) amino acids results in at least 80–90% reduction in the potency of the ligand to bind in receptor binding studies,12 with site directed mutagenesis studies showing that the leucine residue at position 47 is of particular importance in maintaining biological activity.23 We have previously shown that loss of the terminal five (EGF1–48) or four (EGF1–49) amino acids also has a marked effect on biological activity, their potency being only about 25–30% of EGF1–53 as assessed using in vitro and in vivo systems.3 ,13 Although less well studied, EGF1–50 also has reduced biological activity, having about 50% of the activity of EGF1–53, as assessed by [3H] thymidine uptake into 3T3 fibroblasts.12
There have been only limited studies examining the importance of the final two C terminal amino acids of the EGF molecule in mediating biological activity. Araki and coworkers reported a 4% and 10% reduction in the binding affinities of EGF1–51 and EGF1–52 to KB cells.4 However, in vivo studies were not performed. Our results therefore support the idea that the terminal two amino acids do not play a key role in mediating the biological effects of EGF and extend previous findings by showing that this is also true when a pathophysiologically relevant in vivo model is used. This is an important point as differences in functional biological activity due to factors such as the systemic handling of the various peptides (for example, circulating t1/2) would not have been apparent from in vitro studies. Conversely, the results of the in vitro assay strongly suggest that the lack of activity of the penta- and decapeptides was not due to rapid excretion in vivo. Formal pharmacokinetic studies would be of interest but are complicated by the fact that the immunoreactivity of the different forms of EGF varies3 and that standard size exclusion column separation techniques of radiolabelled EGF are too insensitive to detect minor clipping of the EGF molecule.3 Analysis of the circulating form(s) and bioactivity of EGF in vivo is therefore difficult to achieve.
As stated previously, the importance of the C terminal seven amino acids in mediating biological responses is well demonstrated by the fact that most of the biological activity of EGF is lost if EGF1–46 is used.12 However, it is also apparent that the intrinsic biological activity of the EGF molecule requires additional areas of sequence, as shown by our findings that the penta- and decapeptides were inactive. Identification of which residues outside of the C terminal region of the EGF molecule are vital for biological activity is difficult and complex.24 Part of this complexity results from the fact that, in common with many other biologically active peptides, Cys-Cys disulphide bridging within the EGF molecule brings amino acid residues which appear distant on the primary sequence into close proximity in its correctly folded tertiary state.
Recombinant peptides are being increasingly used for a variety of clinical conditions. Examples include recombinant human insulin for the treatment of diabetes, erythropoietin for renal failure induced anaemia, and interferon for viral hepatitis. The use of recombinant peptides for “hollow organ” gastrointestinal conditions is at a more preliminary stage although early reports appear encouraging.25 EGF has been advocated for a wide variety of gastrointestinal conditions, including necrotising enterocolitis, congenital microvillus atrophy, and inflammatory bowel disease. For all of these conditions, data are only available from limited clinical studies involving very few patients, single case reports, or animal experiments.6-9 ,26 Formal double blind, randomised, controlled trials are planned by several groups (personal communications) and it is therefore important that researchers appreciate the importance of consistency regarding the forms/dosage of EGF to be used (at least in terms of biological equivalence). Our studies have shown that EGF is a robust molecule, in terms of preservation of biological activity, when used in a typical clinical setting. Provided standard aseptic techniques are used, infusions of EGF (with a carrier protein such as albumin) can be continued for 24–48 hours in the light without significant loss of activity, minimising the time (and cost) required for its usage.
Acknowledgments
We thank H Hansen for performing the mass spectrometry, Drs N O'Reilly and D Gadhia, ICRF, London, for synthesising the deca- and pentapeptides, and Mr D Floyd and Mrs K Witty for manuscript preparation. Grant support was from the Wellcome Trust and MRC.
Abbreviations used in this paper
- Cys
- cysteine (residues)
- EGF
- epidermal growth factor
- HPLC
- high pressure liquid chromatography
- TFA
- trifluoroacetic acid