Article Text

Abstract
We report the case of a 52 year old male with diabetes mellitus and long standing evidence of hepatic iron excess. Initially considered to have haemochromatosis, this patient was reevaluated when hepatic iron stores were found to be unaffected by a prolonged course of weekly phlebotomy. The development of neurological disease prompted diagnostic consideration of aceruloplasminaemia, which we confirmed by demonstration of a novel frameshift mutation in the ceruloplasmin gene. Our inability to resolve the patient's iron overload by regular phlebotomy is consistent with recent animal studies indicating an essential role for ceruloplasmin in cellular iron efflux. Evaluation of this case underscores the clinical relevance of aceruloplasminaemia in the differential diagnosis of hepatic iron overload and provides insight into the pathogenetic mechanisms of hepatocellular iron storage and efflux.
- aceruloplasminaemia
- ceruloplasmin
- diabetes
- haemochromatosis
- iron
Statistics from Altmetric.com
Aceruloplasminaemia is an autosomal recessive disorder of iron metabolism resulting in diabetes, retinal degeneration, and neurological symptoms.1 Analysis of affected individuals reveals absent serum ceruloplasmin and evidence of excess iron accumulation in association with inherited mutations of the ceruloplasmin gene.2 ,3 Serum ferritin concentration is markedly elevated in aceruloplasminaemia and liver biopsy reveals increased hepatic iron content with abundant iron in hepatocytes and reticuloendothelial cells.4 ,5 In contrast with other forms of haemochromatosis, patients with aceruloplasminaemia do not usually present with signs or symptoms of hepatic dysfunction. Consistent with these clinical observations, liver histology from such patients reveals marked iron accumulation without evidence of fibrosis, even in advanced stages of the disease. We report here a case of aceruloplasminaemia in which the initial therapeutic approach has afforded unique insight into the mechanisms of hepatic iron accumulation in this disease.
Case report
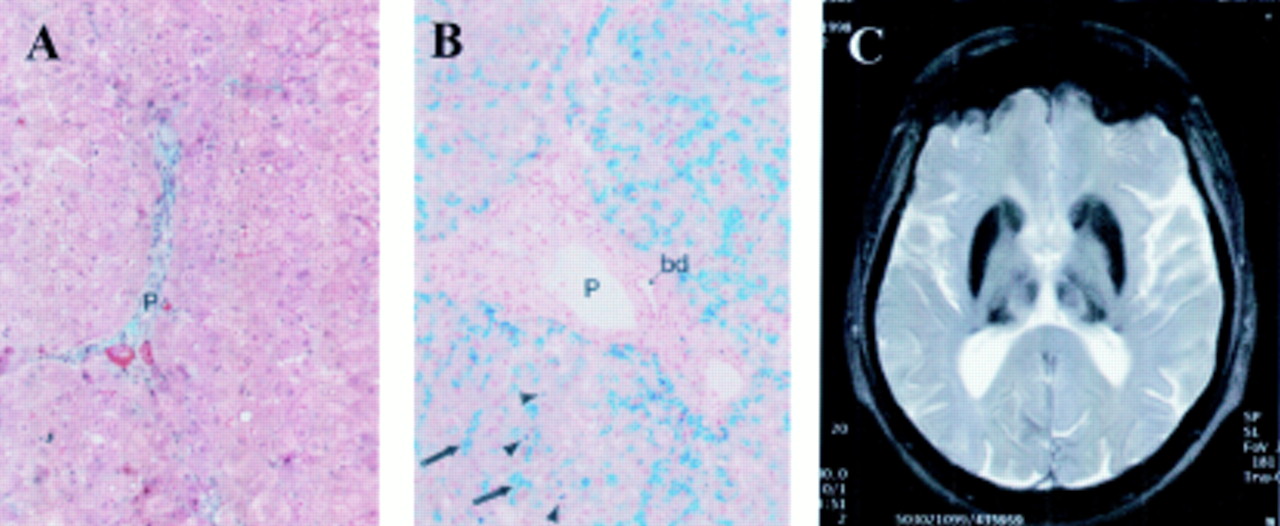
Nine years ago at the age of 43 years our patient was diagnosed with diabetes mellitus during a routine clinical visit. At this time he had no complaints with the exception of occasional joint pain attributed to his occupation as a car mechanic. Physical examination was unremarkable except for an enlarged thyroid gland. There was no history of blood transfusions and no family history of diabetes, liver dysfunction, or neurological disease. Laboratory analysis revealed elevated serum ferritin with low serum iron and a mild microcytic anaemia (table 1). Liver biopsy revealed no evidence of fibrosis or cirrhosis (fig 1A) with significant iron accumulation. Hepatic iron content was 1300 μg/g dry weight and Perl's staining demonstrated abundant haemosiderosis in hepatocytes distributed evenly within the lobule and in Kupffer cells (fig 1B). No haemosiderin pigment was found within the portal tracts or bile duct epithelia. Liver copper content was within the normal range.
Laboratory assessement of iron metabolism in 1992 and 1998

(A) Masson-Goldman trichrome stain of the patient's liver revealed no fibrosis (P, portal tract). Original magnification ×170. (B) Perl's stain of the patient's liver revealed abundant haemosiderin pigment distributed evenly throughout the hepatic lobulus. Iron accumulation can be observed in both hepatocytes (arrows) and Kupffer cells (arrowheads), but not in the portal tract (P) or bile duct epithelia (bd). Original magnification ×170. (C) T2 weighted magnetic resonance tomography revealed signal attenuation in the basal ganglia consistent with iron accumulation.
Despite the absence of increased transferrin saturation, this patient was considered to have a form of haemochromatosis and was begun on oral diabetic medication and regular phlebotomies of 250 ml/week. This treatment persisted for six years during which time serum ferritin remained elevated within the range 900–1000 μg/l. In 1998, at the age of 55 years, the patient was contacted to revisit the clinic. His diabetes was well controlled (HbA1c 7.1%) but neurological examination revealed a fine tremor of both hands, a mask-like facies, weak deep tendon reflexes bilaterally, and paraesthesias of the lower extremities. T2 weighted magnetic resonance tomography of the brain (Siemens Impact Exput) demonstrated signal attenuation in the basal ganglia consistent with iron accumulation in this region (fig 1C).
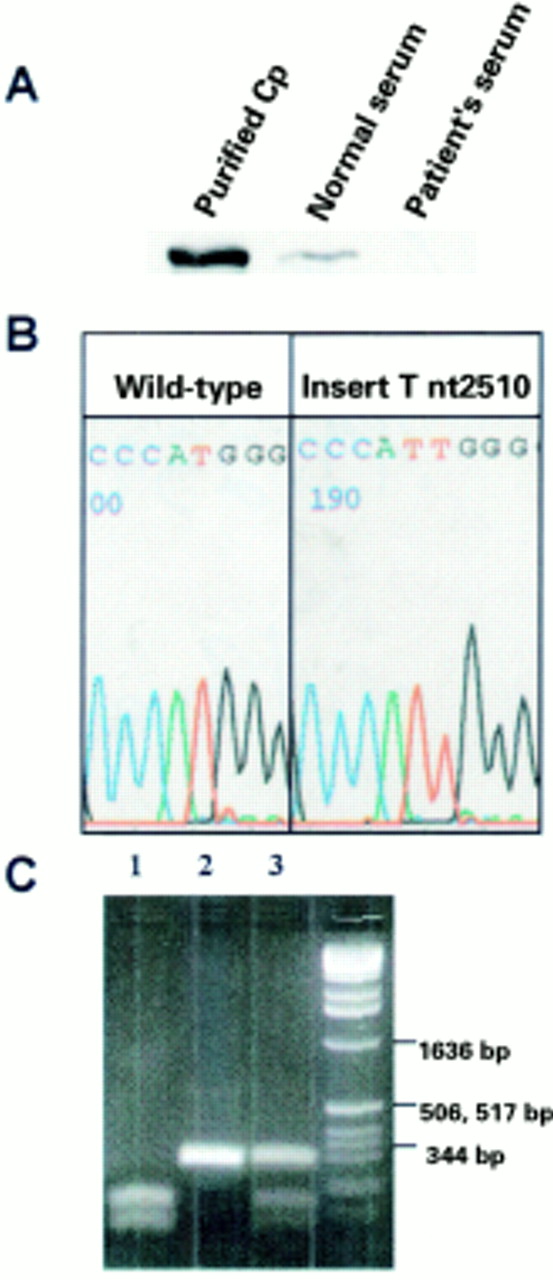
The appearance of neurological signs and symptoms in this patient together with radiographic findings and persistently elevated serum ferritin despite phlebotomy prompted evaluation for other causes of abnormal iron homeostasis. Immunoblot analysis of serum demonstrated absence of detectable ceruloplasmin (fig 2A) and consistent with this finding, sequence analysis of genomic DNA2 revealed a novel frameshift mutation in exon 14 of the ceruloplasmin gene (insert T nt 2510) (fig 2B). As this mutation results in loss of an NcoI restriction site, NcoI digestion was performed to demonstrate that the patient was homozygous for this mutation, and that the patient's asymptomatic son is an obligate heterozygote (fig 2C). Genomic sequence analysis failed to detect either of the two HFE mutations known to cause hereditary haemochromatosis (C282Y and H63D),6 or the most common mutation in the Atp7b gene found in patients with Wilson's disease (H1069Q)7 (data not shown).

{kind=link}
{kind=link}
(A) Immunoblot demonstrating absence of serum ceruloplasmin (Cp) in the patient compared with serum from a normal patient and purified ceruloplasmin (Vital Products Inc., St Louis, Missouri, USA). (B) Demonstration of the frameshift mutation insert T nucleotide 2510 in exon 14 of the ceruloplasmin gene by sequence analysis with an ABI Prism sequencer (PE Biosystems, Foster City, California, USA). (C) Restriction analysis of patient DNA demonstrates homozygosity of the insert T nucleotide 2510 mutation. As this frameshift mutation results in the removal of an NcoI restriction site, NcoI digestion of the ceruloplasmin exon 14 polymerase chain reaction product was performed on a control individual (homozygous WT, lane 1), patient (homozygous frameshift mutation, lane 2), and the patient's son (obligate heterozygote, lane 3). DNA size markers are indicated at the far right.
Discussion
Taken together, the results of this analysis are consistent with a diagnosis of aceruloplasminaemia. Nevertheless, the initial consideration of haemochromatosis in this case is understandable given that aceruloplasminaemia was not even described as a distinct clinical entity at the time when he initially presented. Importantly, these disorders share specific laboratory features, including diabetes mellitus secondary to pancreatic iron accumulation and marked elevation of serum ferritin reflecting increased hepatic iron stores.8 ,9 Although the hepatic iron index in this case was normal, hepatic iron content was well within the range observed in hereditary haemochromatosis. As it is now recognised that up to 15% of patients homozygous for the C282Y mutation in the haemochromatosis gene have hepatic iron indexes less than 1.9, the initial clinical impression in this case would seem reasonable.10 The normal biopsy appearance in this patient did not mitigate against hereditary haemochromatosis, as fibrosis is often not observed early in the course of this disease and this picture would be consistent with anticipation of a reasonable response in reduction of iron overload with phlebotomy.11
In the past few years it has become increasingly apparent that complete absence of serum ceruloplasmin in combination with clinical and radiographic evidence of central nervous system iron accumulation is consistent with a diagnosis of aceruloplasminaemia. Demonstration of a novel frameshift mutation in the ceruloplasmin gene in this patient confirms this diagnosis and adds to the growing list of mutations reported in this disease. In all cases thus far, including this one, such mutations would be anticipated to interrupt the ceruloplasmin open reading frame resulting in a molecule devoid of the trinuclear copper cluster essential for ferroxidase activity.9 Although the presence of neurological disease and decreased serum ceruloplasmin concentration can lead to diagnostic confusion with Wilson's disease, the normal hepatic copper content as well as the marked increase in hepatic iron content in aceruloplasminaemia permits diagnostic differentiation between these two disorders.12
The therapeutic rationale behind phlebotomy in this case was that the erythropoietic demand created by removal of red blood cells would result in continued iron efflux from mobilisable storage sites in the liver.8 ,11 Such an approach is effective in haemochromatosis, as monitored by serum ferritin concentration, a gauge of intracellular iron stores which decreases with increasing red blood cell removal. While phlebotomy is quite successful in removing iron stores in patients with haemochromatosis, this case demonstrates that such an approach is of no benefit in aceruloplasminaemia and suggests that the pathological accumulation of hepatic iron is qualitatively different in these two diseases. The inability to mobilise hepatic iron stores in our patient despite induction of a marked erythropoietic demand is consistent with experimental observations in aceruloplasminaemic mice and supports the proposed concept that ceruloplasmin functions as a plasma ferroxidase with an essential role in the mobilisation of iron from tissue stores.13 In the absence of ceruloplasmin the increased need for iron brought about by phlebotomy in this patient was not able to be met by increased hepatocyte or RE cell efflux and therefore no substantial decrease in hepatic iron content occurred.
Beyond emphasising the importance of considering aceruloplasminaemia in the differential diagnosis of disorders causing hepatic iron overload, this case reveals that despite equivalent amounts of excess hepatic iron the pathogenetic mechanisms of iron accumulation and efflux differ among these disorders. Ceruloplasmin should now be considered as a protein which plays an essential role in iron metabolism and, as illustrated by this case, knowledge of the mechanisms by which ceruloplasmin functions is essential in guiding therapy. Further understanding of the mechanism by which ceruloplasmin regulates cellular iron efflux will be of importance not only for development of effective therapy in this disease but also in the design of novel therapeutic approaches in other disorders of iron overload where compartmentalisation or detoxification of iron is a desirable clinical consequence.14
Acknowledgments
NH and MS contributed equally to this study. Supported by funds from NIH grant HL41536 (JDG).