Article Text

Abstract
BACKGROUND The normal intestinal microflora plays an important role in host metabolism and provides a natural defence mechanism against invading pathogens. Although the microbiota in adults has been extensively studied, little is known of the changes that occur in the microflora with aging. These may have important consequences in elderly people, many of whom are receiving antibiotic therapy and who are most susceptible to intestinal dysbiosis.
AIMS To characterise the major groups of faecal bacteria in subjects of different ages using a combination of cultural, molecular, and chemotaxonomic approaches.
METHODS Comparative microbiological studies were made on four different subject groups: children (16 months to seven years, n=10), adults (21–34 years, n=7), healthy elderly subjects (67–88 years, n=5), and geriatric patients (68–73 years, n=4) diagnosed with Clostridium difficile diarrhoea. Selected faecal bacteria were investigated using viable counting procedures, 16S ribosomal RNA (rRNA) abundance measurements, and the occurrence of specific signature fatty acids in whole community fatty acid methyl ester profiles.
RESULTS The principal microbiological difference between adults and children was the occurrence of higher numbers of enterobacteria in the latter group, as determined by viable counts (p<0.05) and 16S rRNA (p<0.01) measurements. Moreover, a greater proportion of children's faecal rRNA was hybridised by the three probes (bifidobacteria, enterobacteria, bacteroides-porphyromonas-prevotella) used in the study, indicating a less developed gut microbiota. Species diversity was also markedly lower in the Clostridium difficileassociated diarrhoea group, which was characterised by high numbers of facultative anaerobes and low levels of bifidobacteria and bacteroides. Although it was a considerably less sensitive diagnostic tool, cellular fatty acid analysis correlated with viable bacterial counts and 16S rRNA measurements in a number of bacteria, including bacteroides.
CONCLUSIONS Polyphasic analysis of faecal bacteria showed that significant structural changes occur in the microbiota with aging, and this was especially evident with respect to putatively protective bifidobacteria. Reductions in these organisms in the large bowel may be related to increased disease risk in elderly people.
- intestinal bacteria
- aging
- cellular fatty acids
- ribosomal RNA
- Clostridium difficileinfection
Abbreviations used in this paper
- BPP
- Bacteroides-Porphyromonas-Prevotella
- CFA
- cellular fatty acids
- DMA
- dimethyl acyl
- rRNA ribosomal RNA
Statistics from Altmetric.com
Gut physiology and function are altered during the aging process,1 which is often accompanied by an increased incidence of gastrointestinal infections. While they can be explained by changes in environmental circumstances, and altered dietary and physiological factors, bacterial community shifts in the large bowel can also have great effects on host physiology and metabolism,2 ,3 as well as innate colonisation resistance.
The colonic microflora of infants is generally viewed as being adult-like after the age of two years, although facultative anaerobes in these children have been reported to be higher than in adults.4-6 Indeed, it is likely that the intestinal microflora does not completely resemble that of adults until much later in childhood. Once the climax microflora has become established, the major bacterial groups in the faeces of adults remain relatively constant over time.7 There are reports, however, that elderly subjects harbour fewer bifidobacteria and higher levels of fungi and enterobacteria compared with younger adults8 and it seems, therefore, that the microbiota continues to evolve throughout the lifespan of the host. Age related changes in the microbiota have been studied in animals where the intestinal microflora of young dogs was shown to comprise large numbers of bacteroides, bifidobacteria, lactobacilli, and anaerobic cocci, while older animals harboured greater numbers of clostridia and enterococci.9 In humans, the frequency of isolation of Closridium difficile is greater in the elderly. While this is partly a result of factors such as hospitalisation, clostridia in general have been found to occur in significantly higher numbers in healthy elderly volunteers compared with younger subjects.10
Immunological changes occur in the body with advancing age, as evidenced by reduced efficiency in B and T cell mediated immune responses.11-13 Other age related effects, such as decreased gastric acid secretion and increased mucosal permeability in the gut, have been linked to increases in circulating antibodies to bacteria in the microbiota in elderly subjects.14 This enables new bacterial populations to exploit novel ecological niches, thereby changing the composition and activities of the microbiota. This can be seen in the low bifidobacteria and high clostridial counts in elderly patients with colon cancer.15
Few studies have investigated bacterial succession in the colon using methods other than viable counting. However, recent investigations16 using 16S ribosomal RNA (rRNA) oligonucleotide probes to study the composition of infant faeces showed large intraindividual variations in community composition prior to weaning, with bifidobacterial 16S rRNA varying from 5% to 40% of the total bacterial 16S rRNA in a single child's faeces. After weaning, the proportion of theBacteroides-Porphyromonas-Prevotella(BPP) group increased rapidly, while bifidobacterial rRNA declined to undetectable levels. In a further set of experiments, with a larger number of subjects, these authors found that viable counting led to overestimation of bacteroides and bifidobacteria in faeces.
Comparative analysis of 16S rRNA sequences amplified from human faeces indicated that less than 25% of the molecular species identified corresponded to known organisms, suggesting that many bacteria in the large gut have yet to be described.17 Thus classical culture based assessments of bacterial succession may not provide an accurate representation of the intestinal microbiota, and may also explain why the aetiological agent(s) of some inflammatory bowel diseases remains unknown.18
Another method of assessing bacterial community structure, without the need for cell culture, is by analysis of cellular fatty acids (CFA). This has been used to distinguish between different human faecal samples but while an altered bacterial microflora was implicated in these investigations the differences could not be related to taxa specific changes in the microbiota.19-21 Nevertheless, axenic and defined mixed culture studies have identified several CFA that might be of use as signals for the occurrence of particular bacterial groups in the bowel.22-23
The aim of this work was to compare the bacterial composition of faeces obtained from children, young adults, and elderly subjects using a polyphasic approach based on cell culture, together with molecular and chemotaxonomic methodologies. In view of the role of the microbiota in colonisation resistance against C difficile infection, the predominant bacterial populations in a group of elderly patients with C difficileassociated diarrhoea were also investigated.
Materials and methods
SUBJECTS
Fresh faecal samples were obtained from 10 children (aged 16 months to seven years), seven adults (aged 21–34 years), five elderly subjects (aged 67–88 years), and four geriatric patients diagnosed with C difficile associated diarrhoea (aged 68–73 years). The children's faecal samples were obtained from two day care centres and a local primary school in Cambridge. Faeces from young healthy adults and elderly subjects were acquired from volunteers at the Dunn Clinical Nutrition Centre and Addenbrooke's Hospital. Stool samples were obtained from C difficileassociated diarrhoea patients within 24 hours of diagnosis. These patients had commenced metronidazole therapy before faecal material was taken. None of the other subjects had a history of gastrointestinal upset or antibiotic therapy within the previous two months.. All stool specimens were processed within 60 minutes for viable bacterial counts, and samples were frozen for community CFA and 16S rRNA analysis at −20°C and −80°C, respectively.
ENUMERATION OF VIABLE BACTERIA
Viable bacterial cell counts in fresh faeces were made as described by Macfarlane and colleagues24 ,25 using the following selective media: cycloserine-cefoxitin-fructose agar (C difficile), Perfringens OPSP agar (C perfringens), Beerens agar26(bifidobacteria), Rogosa agar (lactobacilli), Blood Azide agar (enterococci), Wilkins-Chalgren agar (total anaerobes), Wilkins-Chalgren agar plus GN selective supplements (Bacteroides,Porphyromonas,Prevotella), MacConkey agar No 2 (enterobacteria), nutrient agar (total facultative anaerobes).
16S rRNA ANALYSIS
Total rRNA was extracted from 0.5 ml aliquots of culture.16 SDS 20% (50 μl), 0.5 ml of buffer (50 mM sodium acetate, 10 mM EDTA, pH 5.1), 0.5 ml of phenol saturated with the same buffer, and 350 mg of zirconium beads (0.1 mm diameter, Biospec Products, Bartlesville, Oklahoma, USA) were added to 0.5 ml of sample in 2.0 ml screw cap vials (Sarstedt, Leicester, UK). These were subjected to two minutes of mechanical disruption on a reciprocation shaker (Mini-bead-beater, Biospec Products). Beating followed by heating at 60°C for two minutes was repeated twice. Samples were then centrifuged at 2040 g and the aqueous phase removed to a clean Eppendorf tube. Two further sequential extractions using 0.5 ml phenol:chloroform:isoamylalcohol (24:24:1, pH 5.1) and then 0.5 ml of chloroform:isoamylalcohol were carried out before total rRNA was precipitated overnight at −20°C with 7.5 M ammonium acetate (0.5 volumes) and absolute ethanol (2 volumes). Samples were washed twice in 80% ethanol and resuspended in 50 μl nuclease free water. All labware and solutions were prepared RNase free using standard protocols.26
A suite of four oligonucleotide probes (table 1) were 5′ end labelled with 32P using [γ 32P]ATP (ICN Pharmaceuticals, Basingstoke, Hants, UK) and polynucleotide kinase. Reactions contained 5 μl of probe (100 ng/ml), 2.5 μl of labelled ATP at a specific activity of 7000 Ci/mmol and a concentration of 160 mCi/ml, 3 μl of 10× kinase buffer, 1 μl of enzyme, 1.5 μl of 1% Nonidet P-40, and 17 μl of nuclease free water. Tubes were mixed gently and incubated at 37°C for 30 minutes before labelled probe was purified using an Nensorb 20 nucleic acid purification cartridge (DuPont, Brussels, Belgium). The cartridge column was rinsed sequentially with 2 ml of absolute methanol, and then 2 ml of filter sterilised 0.14% triethylamine (100 mM Tris HCl, 1 mM EDTA, pH 7.7). Triethylamine solution (470 μl) was added to the labelled probe before it was passed through the column. After sequential washes with 2 ml of triethylamine solution and 2 ml of nuclease free water, the purified probe was eluted with 0.5 ml of 50% methanol.
Sequences (listed 5′ →3′) of 16S rRNA oligonucleotide probes
Prior to blotting, extracted RNA samples were diluted to a concentration of 1–2 ng/μl. RNA in 2.5 μl of extract was denatured by addition of 30 μl of 2% glutaraldehyde in 50 mM sodium phosphate (pH 7.0) and then diluted with 7.5 μl of nuclease free water and 210 μl of dilution water (50 μg polyadenylic acid, 1 μl of 2% bromophenol blue, 50 ml of nuclease free water). A slot blot device (Minifold II, Schleicher and Schuell, Inc., Keene, New Hampshire, USA) was used to transfer 50 μl of sample in triplicate Hybond N nylon membranes (Amersham, Little Chalfont, Berks, UK), with the vacuum adjusted to filter samples in 1–2 minutes. A reference series of RNA specific for each group was blotted to each membrane for normalisation. The membranes were then air dried and baked for two hours at 80°C.
Membranes in hybridisation tubes (Robbins Scientific, Sunnyvale, California, USA) were prehybridised in 10 ml of hybridisation buffer (0.9 M NaCl, 50 mM phosphate buffer (pH 7.2), 5 mM EDTA, 10× Denhardt solution (34), 0.5% SDS, 0.05 mg/ml poly A) for two hours in a rotating hybridisation oven (Stuart Scientific, Redhill, Surry, UK) at 40°C. The hybridisation buffer was then replaced with 10 ml of fresh buffer containing labelled probe and incubated for a further 16 hours. The membranes were washed twice in 100 ml of wash solution (1% SDS, 1× SSC) for one hour at 40°C, and twice again in 300 ml of wash solution at the precise dissociation temperature for the probe. The32P signal was quantitated using an Instant Imager (Canberra Packard, Pangbourne, Berks, UK) and ImageQuant software (Molecular Dynamics, Sunnyvale, California, USA).
Abundances of specific groups of bacteria were expressed as concentrations and as percentages of total SSU rRNA in the samples, as previously described.29-31 Standard curves were calculated from reference RNA by linear regression for each probe, and this was used to account for any difference in radiosensitivity and hybridisation conditions between probes. RNA samples from the following microorganisms were used as references:Bifidobacterium longum (eubacterial domain probe and bifidobacteria genus probe), Escherichia coli (enterobacteria group probe), andBacteroides thetaiotaomicron (bacteroides group probe).
CELLULAR FATTY ACID ANALYSIS
Bacterial community fatty acid structure was analysed by extraction of CFA from bacterial cell mass in a 10% faecal slurry in sterile distilled water. Duplicate aliquots (1 ml) of the fresh slurry were centrifuged (13600 g, 10 minutes) in an Eppendorf bench centrifuge and the resultant pellets washed in an equal volume of 0.7% MgSO4 (w/v). CFA were extracted as described previously32 using a model 5898A Microbial Identification System (Microbial ID Inc., Newark, Delaware, USA) which consisted of a Hewlett-Packard Model 6890 gas chromatograph fitted with dual 5% phenyl-methyl silicone capillary columns (0.2 mm×25 m), flame ionisation detectors, a Hewlett-Packard Model 7637A automatic sampler, and a Hewlett-Packard Vectra XM computer (Hewlett-Packard, Palo Alto, California, USA). Samples (2 μl) were injected onto the column with a temperature gradient from 170 to 270°C, injection port temperature 300°C, and ultra high purity hydrogen as the carrier gas. Peaks were automatically integrated and fatty acids identified using MIS Library Generation software.
CHEMICALS
Unless otherwise stated, all fine chemicals were obtained from Sigma Chemical Co (St Louis, Missouri, USA). Bacteriological culture media and associated antibiotic selective supplements were purchased from Oxoid (Basingstoke, UK).
STATISTICAL ANALYSIS
Analysis of variance (ANOVA) between each group of subjects was calculated with Data Desk (version 6.0) software (Data Description Inc., Ithaca, New York, USA).
ETHICAL PERMISSION
Subject consent was obtained for the investigations, and the study was approved by both the Dunn Clinical Nutrition Centre and Addenbrooke's local research ethics committees.
Results
BACTERIOLOGICAL ANALYSES
Results in figs 1 and 2 show that although total anaerobe counts were comparable in all four groups, bacterial compositions at the genus level varied markedly. BPP group organisms were significantly lower inC difficile associated diarrhoea patients compared with children (p⩽0.0001), young adult (p⩽0.0001), and healthy elderly groups (p<0.001). Overall, bifidobacterial numbers were reduced in the elderly where two of four subjects had no detectable bifidobacteria, and one had low counts ofBif angulatum (log10 7.5 g/faeces). In contrast, however, the remaining subject in this group had very high numbers of these organisms in faeces (Bif angulatum 9.8; Bif adolescentis 9.6; Bif longum 9.2). Bacterial diversity was notably lower in the C difficile associated diarrhoea subjects, with only one bacteroides (Bacteroides species BL) and one bifidobacterium (Bif adolescentis) being detected in this group (results not shown). Altogether, the most prevalent bifidobacterial species were Bif angulatum and Bif adolescentis, while the range of bacteroides species was more diverse. In contrast with bacteroides and bifidobacteria, species variations in clostridia and lactobacilli did not decline in the C difficile associated diarrhoea group, with the total counts of clostridia and lactobacilli being significantly higher in theC difficile associated diarrhoea group compared with young adults (p<0.05) and elderly subjects (p<0.01), respectively (fig 2).

Viable counts of bacteroides, bifidobacteria, enterobacteria, and total anaerobes in faeces of different age groups. Results are mean (SEM) in children (n=5), adults (n=7), elderly subjects (n=4), and in patients with Clostridium difficile associated diarrhoea (CDAD, n=4). BPP, Bacteroides-Porphyromonas-Prevotella.

Viable counts of clostridia, lactobacilli, and enterococci in faeces obtained from children (n=5), adults (n=7), elderly subjects (n=4), and patients with Clostridium difficile associated diarrhoeal (CDAD, n=4).
Facultative anaerobes were lowest in the young adult group, with the highest populations occurring in C difficileassociated diarrhoea patients, although the only statistically significant difference was the increased prevalence of enterobacteria in children relative to adults (p<0.05). However, little interindividual species variation was observed with respect to these organisms.
16S rRNA
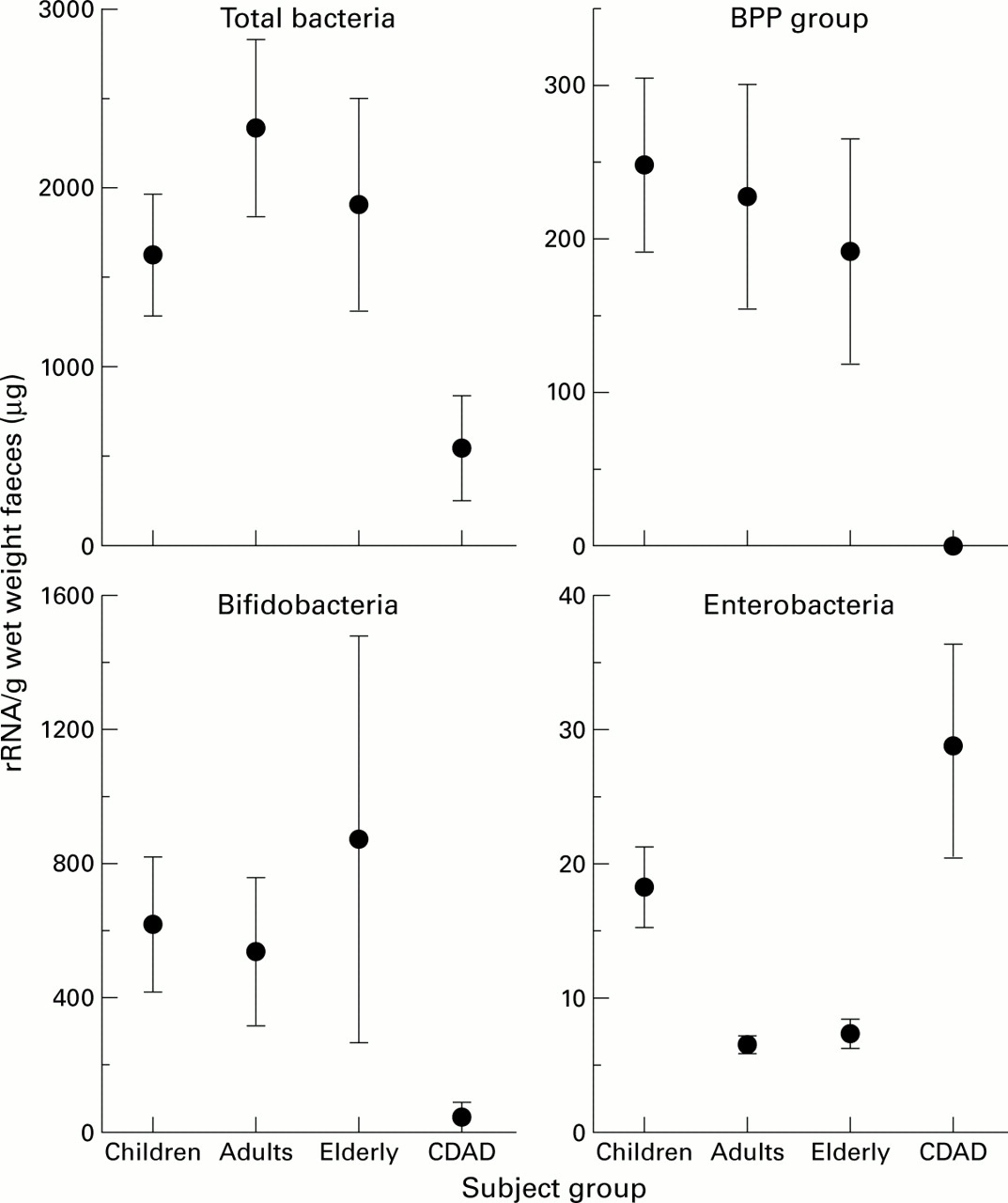
Figure 3 shows that total bacterial 16S rRNA peaked in young adults whereas concentrations in C difficileassociated diarrhoea patients were significantly lower than in all other groups. The BPP probe showed similar levels of this rRNA in each subject group, except for the C difficileassociated diarrhoea group where none was detected. Bifidobacterial rRNA concentrations were twice as high as the BPP group, and again very small amounts were present in the faeces of C difficile associated diarrhoea patients. Stools from healthy elderly subjects contained either negligible or high amounts of bifidobacterial rRNA, resulting in a high standard error in this group. Low concentrations of enterobacterial 16S rRNA were found in all subjects in the study, with children's faeces containing twice the amount found in adults (p<0.01) and elderly subjects (p<0.01). The highest levels of enterobacterial rRNA were detected inC difficile associated diarrhoea patients.
{kind=link}
{kind=link}
{kind=link}
16S rRNA concentrations in faeces. Results are mean (SEM) in children (n=10), adults (n=7), elderly subjects (n=5), and in patients with C difficile associated diarrhoea (CDAD, n=4). BPP, Bacteroides-Porphyromonas-Prevotella.
Expressed as a percentage of total bacterial rRNA, the BPP group was still significantly lower in the C difficileassociated diarrhoea patients (table 2). Despite retaining a similar pattern to results expressed as a concentration, variation in the percentage of bifidobacterial rRNA within groups was seen to increase, particularly in patients with C difficileassociated diarrhoea which, as a consequence, was no longer significantly different from the other groups. With respect to the percentage of enterobacterial rRNA, the infant faecal samples were found to contain significantly higher amounts than the young adults (p<0.01) or healthy aging subjects (p<0.05), although all accounted for less than 2% of total rRNA. The proportion of enterobacterial rRNA in the C difficile associated diarrhoea group was markedly higher, averaging about 10% of the total, which despite the high degree of variation within this group of subjects was found to be highly significant.
Percentage of specific 16S rRNA in the faeces of the different age groups
CELLULAR FATTY ACID MEASUREMENTS
Whole community CFA analysis showed that faecal profiles differed considerably between the four groups (table 3) The straight chain saturated fatty acids 16:0 and 18:0 always predominated. Amounts of 12:0, 14:0, and 15:0 were lower in patients with C difficile associated diarrhoea, while the longer fatty acid 20:0 was significantly greater in this group. Of the unsaturated straight chain CFA, the absence of 16:1 cis9 was a characteristic of children, while relatively high amounts of the long chain 20:1 cis11 were only seen in theC difficile associated diarrhoea patients. Faeces in the latter group also contained a greater percentage of 18:1cis9 although this was only found to be significantly higher than in children.
Bacterial community cellular fatty acid (CFA) composition in the faeces of the different age groups
The highest levels of branched CFA were seen in young adults with the total averaging almost 5% whereas these fatty acids comprised only 0.1% of the total CFA in stools of C difficile associated diarrhoea patients. Deficiency of 15:0 anteiso and 15:0 iso CFA moieties was a characteristic of theC difficile associated diarrhoea group and, although only minor amounts of 14:0 iso were detected in adults, its presence distinguished these subjects from children. Elevated 17:0 anteiso CFA were also found in this adult group, although this was not significantly different from the other subjects.
Dimethyl acyl (DMA) molecules were often present in lower proportions in the faeces of children and C difficileassociated diarrhoea patients, although the total values were not significantly different. However, when viewed individually, certain DMA analogues were indicative of different subject groups. Relatively high percentages of 18:1 cis11 DMA and 14:0 DMA distinguished adults from children and those with C difficile associated diarrhoea, and 15:0 anteiso DMA was only detected in samples from the adult group. Furthermore, children's stools were unique in their lack of 18:0 DMA.
The cyclopropane fatty acid 19cyc9,10 and its DMA analogue, together with the hydroxyl CFA 18:0 12OH, were not detected in any stool samples. The shorter hydroxyl fatty acid 16:0 3OH was only found in faeces from young adults, although its presence was not consistent or statistically significant.
Discussion
In addition to bacterial culture, in situ hybridisation with rRNA targeted oligonucleotide probes and CFA analysis can be used to monitor bacterial populations in complex microbiotas. rRNA synthesis is affected by cell growth rate and the nutritional environment, and therefore the amount of bound 16S rRNA probe also provides a measure of the metabolic status of individual bacterial populations.
Viable counts of the predominant bacteria in the faeces of adults and children highlighted differences in microbiota composition, such as higher bifidobacteria and clostridia populations in children. More significantly, enterobacterial carriage was 100-fold greater in these subjects (fig 1). Moreover, 16S rRNA abundance measurements demonstrated that enterobacterial, bifidobacterial, and BPP group organisms were proportionally high in children, showing that their microbiotas were less complex bacteriologically than those of the adult microflora (fig 3). Other workers have reported that while the overall structure of the microbiota in infants may be adult-like, facultative anaerobes often remain at elevated levels,5 and our results show that this extends beyond infancy into childhood. The eventual decline in enterobacteria as the microbiota develops is due to proliferation of anaerobic species and increasing competition for nutrients from other microorganisms.
Viable counts and estimations of 16S rRNA abundance in this study are in agreement with early investigations8 ,9 that reported declining anaerobe populations and increased enterobacterial numbers in elderly people. However, bifidobacterial RNA results from elderly subjects were highly skewed because while these individuals, as a group, had reduced levels of culturable faecal bifidobacteria, one subject, who was a 67 year old unusually health and fitness conscious male, had very high counts of these organisms, belonging to several species. It was these bacteria that were responsible for the high mean 16S rRNA concentrations shown in fig 3.
The percentage of bifidobacterial 16S rRNA in adults was greater than that found in other investigations that have examined 16S rRNA/DNA by fluorescent in situ hybridisation and random sequencing.17 ,33 ,34 However, in our work, population abundance was expressed as both total quantities of SSU rRNA per gram of faeces and as a proportion of the total. This was made possible by careful quantitation of the procedure used to extract rRNA from faeces. Expressing the abundance of a specific microbial population as total quantities allows more confident expression of the contribution made by a bacterial population to overall community composition. Changes in population abundance, measured and expressed as a proportion of total rRNA, may represent shifts in total community structure and ribosomal abundance, and not changes in absolute amounts.
Discrepancies between methods used for quantitation of bifidobacteria in faeces have been reported by other workers. For example, fluorescent in situ hybridisation gives a lower estimate of bifidobacteria compared with plate counts.35-37 Although these observations reflect inherent differences in experimental methodology, it would be useful to be able to compare the origins and transit times of the faecal samples used, since after thriving in the caecum the suboptimal environmental conditions encountered by bacteria in the distal bowel or faeces, such as depletion of preferred energy substrates, would result in lower rRNA yields.
Despite the low numbers of C difficileassociated diarrhoea patients, the faecal microflora in this group was markedly different from all other samples (figs 1-3). Only one bacteroides was isolated from these subjects, and the absence of specific 16S rRNA confirmed their low prevalence. Bifidobacteria were also minor constituents of the faecal microflora, although this was in part a reflection of low bacterial species diversity and reduced cell numbers in the microbiota as a whole. The sharp reduction in anaerobes was primarily related to metronidazole treatment, even though faecal samples were obtained from C difficileassociated diarrhoea patients on the day of diagnosis. This was because a combination of severe diarrhoea and incapacity impeded stool collection, and the first dose of metronidazole was often administered before a suitable specimen could be obtained. The increase in facultative anaerobes in these subjects was further indicative of metronidazole therapy, which is selective against obligate anaerobes. By far the greatest enterobacterial activity was detected in theC difficile associated diarrhoea group (9.3% of total rRNA) reflecting severe disturbances in the gut microbiota in these patients. Nevertheless, viable counts showed that anaerobic species such as clostridia and lactobacilli were present in high numbers, indicating that the altered state of the microflora was not entirely antibiotic related.
The absence of bifidobacteria, or their low numbers in the elderly, may have metabolic and health consequences for the host because they play an important role in the body affecting immune system reactivity and a multiplicity of other physiological functions.38Bifidobacteria are also involved in colonisation resistance in the bowel,39-41 for example, oral administration ofBif breve has been shown to eradicateCampylobacter jejuni from stools of children with enteritis.42
The scarcity of bifidobacteria, together with low levels of bifidobacterial rRNA in some elderly subjects, suggests that treatment with probiotics or prebiotics may be beneficial in these individuals.30 ,43 ,44 Due to low bifidobacterial species diversity, probiotics would provide an immediate increase in bifidobacteria in the bowel while prebiotic feeding could be used to facilitate expansion of indigenous bifidobacteria populations and maintain the probiotics. However, bifidobacteria have been shown to exhibit strong carbohydrate substrate preferences,45demonstrating the need to identify the species specificities associated with individual prebiotics if they are to be used effectively.
CFA analysis has been used in several studies on bacterial composition and biomass estimations in a variety of environments.19 ,20 ,21 ,46 ,47 In the current work, whole community CFA analysis confirmed that this technology could distinguish between the faeces of different age groups. This was achieved through cataloguing specific signature fatty acids that could be used to identify particular groups of bacteria. Thus branched chain CFA have been linked to Gram positive microorganisms while cyclopropane CFA are associated with anaerobic species, although neither are exclusive to these groups.48 A potential confounding factor in using CFA profiles in this way is that fatty acid synthesis is strongly affected by environmental conditions, which due to variations in substrate availability in the bowel and differences in gut transit times could affect faecal analysis.49 However, branched chain fatty acids form an intrinsically high proportion of CFA in bacteroides, even during growth in diverse nutritional and environmental conditions,23 and it is noteworthy that these molecules were not present in C difficile associated diarrhoea stools. Since levels of 15:0 anteiso CFA exhibited a degree of correlation with 16S rRNA abundance and viable counts for BPP group organisms, this fatty acid appears to be a useful marker for these bacteria in faeces. TheC difficile associated diarrhoea group was also characterised by high levels of 20:1cis11 in their stools. While not present inC difficile, this CFA is a minor cell membrane component in other intestinal clostridia. Pure culture experiments with C septicum have shown that 20:1 cis11 is associated with high specific growth rates23 and so its detection inC difficile associated diarrhoea patients is likely to be related to more rapid gut transit times. The notion that 20:1 cis11 indicated elevated clostridial populations in faeces was supported by viable count data (fig 2), although its close relationship with bacterial growth renders it unsuitable as a signal molecule for faecal clostridia.
DMA analogues were minor CFA in the C difficile associated diarrhoea group compared with young adults and elderly subjects. The reduction in 18:1cis9 DMA, and to a lesser extent 18:1cis11 DMA, which have previously been linked with bifidobacteria,22 could be taken as evidence of their low numbers. Despite this, the association of DMA synthesis with low specific growth rates23 indicates these reductions can also be attributed to rapid colonic transit times associated with diarrhoea. Conversely, 18:0 DMA CFA were mainly detected inC difficile associated diarrhoea subjects due to their high levels of clostridia and enterococci.22The proportion of 18:0 DMA was significantly lower in the children's group compared with all other groups, as indeed were the majority of DMA moieties. This was probably as a result of low enterococcal carriage in these subjects (fig 2).
CFA measurements do not discriminate between bacterial and mammalian fatty acids, and faecal CFA profiles may have been affected by residual dietary lipids or exfoliated colonic epithelial cells because they contain some fatty acids that occur in bacteria, such as palmitate (16:0).50 However, as faeces largely consist of bacterial cell mass,2 these factors are unlikely to have had significant quantitative or qualitative effects on the analyses made in this investigation.
In summary, polyphasic analysis of the faecal microflora demonstrated major changes in bacterial community structure associated with disease and aging in the host. In general, viable counts of gut microorganisms correlated with rRNA hybridisation data. While CFA analysis was useful in distinguishing between stools provided by different subject groups, and 15:0 anteiso CFA related to the occurrence of bacteroides in faeces, fatty acids identified as putative indicators of Gram positive species such as bifidobacteria, enterococci, and clostridia were less promising in that no one signal molecule was exclusive to these groups.
Abbreviations used in this paper
- BPP
- Bacteroides-Porphyromonas-Prevotella
- CFA
- cellular fatty acids
- DMA
- dimethyl acyl
- rRNA ribosomal RNA