Article Text

Abstract
BACKGROUND/AIM Inactivation of the p16 INK4A(p16) tumour suppressor gene by promoter region hypermethylation has been demonstrated not only in many types of tumours, including hepatocellular carcinoma (HCC), but also in early preneoplastic lesions in the lung, colon, oesophagus, and pancreas. The aim of this study was to examine the methylation status of thep16 promoter in pre- and/or non-neoplastic liver diseases.
PATIENTS/SUBJECTS/METHODS The methylation status of p16 was evaluated in 22 HCC, 17 cirrhosis, 17 chronic hepatitis, nine primary biliary cirrhosis (PBC), eight autoimmune hepatitis, seven drug induced liver disease, six fatty liver, and three normal liver tissues using methylation specific polymerase chain reaction (MSP). p16 protein expression was also examined by immunohistochemical staining.
RESULTS Methylation of the p16 promoter was detected in HCC (72.7%, 16/22) and also in cirrhosis (29.4%, 5/17) and chronic hepatitis (23.5%, 4/17), all of which were positive for hepatitis B or C virus infections. Methylation was not detected in any of the other samples. All methylation positive HCC, cirrhosis, and chronic hepatitis samples showed loss of p16 expression, and a significant correlation was found between methylation and loss of expression. Analysis of serial samples from individual patients with methylation positive HCC revealed that loss of p16 expression with promoter methylation occurred in 18 of 20 patients at the stage of chronic hepatitis without clinically detectable carcinoma.
CONCLUSIONS Our results suggest that methylation of the p16promoter and the resulting loss of p16 protein expression are early events in a subset of hepatocarcinogenesis and that their detection is useful in the follow up of patients with a high risk of developing HCC, such as those with hepatitis B or C viral infections.
- hypermethylation
- p16
- hepatocarcinogenesis
- preneoplastic diseases
- hepatitis virus infection
- methylation specific PCR
Abbreviations used in this paper
- HCC
- hepatocellular carcinoma
- HBV
- hepatitis B virus
- HCV
- hepatitis C virus
- PBC
- primary biliary cirrhosis
- PCR
- polymerase chain reaction
- MSP
- methylation specific PCR
Statistics from Altmetric.com
- hypermethylation
- p16
- hepatocarcinogenesis
- preneoplastic diseases
- hepatitis virus infection
- methylation specific PCR
Hepatocellular carcinoma (HCC) is one of the most common malignancies in the world and one of the leading causes of cancer death in Japan.1 Hepatitis B virus (HBV) and/or hepatitis C virus (HCV) infections are thought to be involved in hepatocarcinogenesis. Chronic hepatitis and cirrhosis associated with either HBV or HCV precede most HCCs. The process of chronic inflammation and cirrhosis is often accompanied by regeneration of hepatocytes, and the continuous regeneration may predispose hepatocytes to uncontrolled growth and malignant transformation as a result of disruption of cell cycle regulation, senescence, and apoptosis. HBV encodes a gene product that can interact withp53 and impair the physiological function ofp53.2 The core protein of HCV has been shown to modulate gene transcription, cell proliferation, and cell death.3 ,4 However, the mechanisms underlying virus associated hepatocarcinogenesis are still unclear.
p16, a cyclin dependent kinase inhibitor, plays an important role in cell cycle regulation5 and senescence.6p16 is one of the most frequently altered tumour suppressor genes in human cancer.7 Frequent loss of p16 expression has been reported in HCC.8 However, mutations and homozygous deletions ofp16 have been shown to be rare in HCC,8-14 although conflicting results have been reported.15,16 The reported LOH at 9p21, on whichp16 gene is located, also shows varying frequencies in HCC.9-13,15 The discrepancies in the reported frequencies in p16 alterations may be due to differences in risk factors, such as HBV, HCV, and alcohol, and differences in geographic regions.
It has been proposed that aberrant methylation of CpG islands, which are CpG dinucleotide rich areas located mainly in the promoter regions of many genes, serves as an alternative mechanism for inactivation of the tumour suppressor gene in cancer.17 ,18 In contrast with the infrequent genetic alterations, frequent methylation of the p16 promoter has been observed in HCC,11 ,13 ,14 ,19 although one study failed to detect methylation in HCC.20 The discrepancy can be explained by the fact that samples with different ethnic and aetiological backgrounds were analysed and/or that different methods were used. The discrepancy may result from different proportions of specific aetiologies, such as HBV and HCV infections, in the studies.
Recently, aberrant methylation of the p16promoter has also been reported in early preneoplastic lesions in the lung,21 colon,22 oesophagus,23and pancreas.24 These findings suggest that loss of p16 function, often due to promoter methylation, may be an early event in the pathogenesis of various types of tumours.25 In this regard, it is of interest that weak amplification of methylated DNA has been seen in a small number of non-tumour liver tissues in which strong signals of methylated DNA were detected in the corresponding tumour tissues.13 Although these findings suggest that de novo methylation of the p16 promoter could occur in premalignant or early subcellular malignant changes in hepatocarcinogenesis, the functional significance of these observations has not been elucidated. In this study, we analysed 5′ CpG island methylation of p16 and correlated the results with immunohistochemical expression of p16 protein in 89 specimens from HCCs, pre- and/or non-neoplastic liver diseases, with or without hepatitis virus infections, and normal livers.
Materials and methods
TISSUE SPECIMENS
Twenty two HCC (mean age 59.2 (SD 9.3) years; 19 males and three females; nine HBV positive and 13 HCV positive), 17 cirrhosis (58.2 (9.7) years; 15 males and two females; seven HBV positive and 10 HCV positive), 17 chronic hepatitis (44.0 (13.7) years; nine males and eight females; seven HBV positive and 10 HCV positive), nine primary biliary cirrhosis (PBC) (55.9 (11.4) years; one male and eight females), eight autoimmune hepatitis (61.9 (4.5) years; one male and seven females), seven drug induced liver disorder (45.1 (16.2) years; two males and five females), six fatty liver (46.7 (15.3) years; two males and four females), and three normal liver (29.0 (14.0) years; two males and one female) tissue specimens were obtained surgically by needle biopsy or at autopsy. HBV and HCV infections were diagnosed by HBs antigen (LPIA-200; Diatron laboratories, Tokyo, Japan) and anti-HCV antibody (Immunocheck-HCV Ab; International Reagent, Kobe, Japan), respectively. All specimens were diagnosed pathologically and frozen at −80°C.
BISULPHITE MODIFICATION
The principles of the method have been described previously.25 In the chemical modification of cytosine to uracil by bisulphite treatment, all cytosines are converted to uracil, but those that are methylated are resistant to this modification and remain as cytosine. One can design polymerase chain reaction (PCR) primers to distinguish methylated from unmethylated DNA in bisulphite modified DNA, taking advantage of the sequence differences resulting from bisulphite modification.25 Genomic DNA (2 μg) extracted from frozen tissues by the standard phenol/chloroform procedure was denatured in 0.2 M NaOH for 15 minutes. Sodium bisulphite (Sigma Chemical, St Louis, Missouri, USA) was added to a final concentration of 3.1 M, and hydroquinone (Sigma Chemical) was added to a final concentration of 0.5 mM. The reaction was performed at 50°C for 16 hours. Modified DNA was then purified using Wizard DNA purification resin (Promega, Madison, Wisconsin, USA) according to the manufacturer's protocol and eluted with 50 μl of water. DNA was treated with NaOH (final concentration 0.3 M) for five minutes at room temperature, followed by ethanol precipitation. Modified DNA was resuspended in water and used immediately or stored at −20°C.
METHYLATION SPECIFIC PCR (MSP)
Bisulphite modified DNA was subjected to MSP using primers specific for unmethylated p16(5′-TTATTAGAGGGTGGGGTGGAT TGT-3′ and 5′-CAACCCCAAACCACAAC CATAA-3′) or methylated p16(5′-TTAT TAGAGGGTGGGGCGGATCGC-3′ and 5′-GACCCCGAACCGCGACCGTAA-3′).25PCR reactions were performed in a volume of 50 μl containing 1×PCR buffer, 0.25 mM dNTP, 1 mM of each primer, and 2.5 U of Taq polymerase. PCR conditions were as follows: one cycle of 95°C for five minutes; 35 cycles of 95°C for 30 seconds, 60°C (unmethylated) or 65°C (methylated) for 30 seconds, and 72°C for 30 seconds; and one cycle of 72°C for four minutes.25 PCR samples were then electrophoresed on a 3% agarose gel.
IMMUNOHISTOCHEMISTRY
Tissue sections (4–5 μm) were deparaffinised and incubated in target retrieval solution (Dako, Carpinteria, California, USA) at the recommended dilution and heated by steam for 15 minutes. Slides were incubated overnight at 4°C with a mouse monoclonal anti-p16 antibody (Neomarkers, Union City, California, USA) diluted to a final concentration of 4 μg/ml. Negative control slides were treated with normal rabbit immunoglobulin under similar conditions. Slides were then rinsed with phosphate buffered saline and incubated with the secondary antibody using the Dako LSAB 2 System (Dako), developed with diaminobenzidine (Kanto Chemical, Tokyo) for five minutes, and counterstained with haematoxylin for one minute.
STATISTICAL ANALYSIS
Methylation of the p16 promoter was assessed for associations with clinicopathological characteristics using the χ2 or Fisher's exact test. A p value <0.05 was considered statistically significant.
Results
METHYLATION OF THE p16 PROMOTER IN VARIOUS LIVER DISEASES
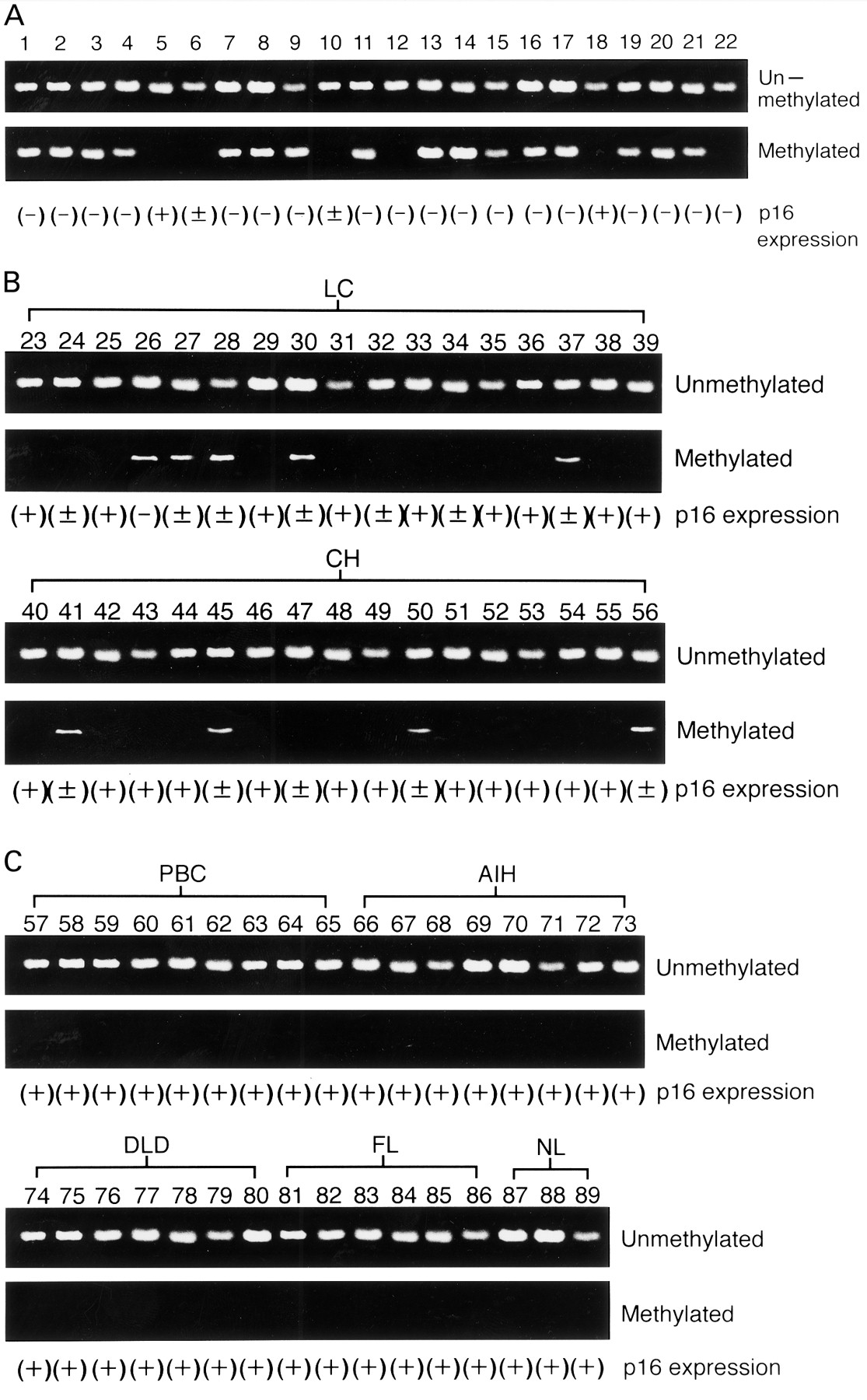
Methylation of the p16 promoter was detected in 72.7% (16/22) of HCC (fig 1A), 29.4% (5/17) of cirrhosis (fig 1B), and 23.5% (4/17) of chronic hepatitis tissue samples (fig1B, fig 2, table 1). In contrast, none of the nine PBC, eight autoimmune hepatitis, seven drug induced liver disease, six fatty liver, or three normal liver tissues showedp16 promoter methylation (fig 1C, fig 2). All methylation positive tissue samples were positive for either HBV or HCV (fig 2, table 1). Positivity for p16promoter methylation was not different between HBV and HCV positive patients in HCC (7/9 v 9/13), cirrhosis (2/7v 3/10), or chronic hepatitis (1/7v 3/10). Unmethylated bands were also detected in all methylation positive cases (fig 1A, 1B). There was no significant correlation between methylation of thep16 promoter and age, type of viral infection, tumour cell differentiation, tumour size, or histological findings in HCC (table 1).

Methylation specific polymerase chain reaction (PCR) analysis and expression of p16 gene. (A) Hepatocellular carcinoma (HCC) samples. (B) Liver cirrhosis (LC) and chronic hepatitis (CH) samples. (C) Primary biliary cirrhosis (PBC), autoimmune hepatitis (AIH), drug induced liver disease (DLD), fatty liver (FL), and normal liver (NL) samples. Unmethylated and methylated indicate methylation specific PCR analysis specific for unmethylated and methylated promoter of p16, respectively. (+), normal expression of p16; (±), partial loss of p16 expression; (−), complete loss of p16 expression.

Summary of the frequencies of p16 promoter methylation. The frequency of the p16 promoter methylation is expressed as a percentage. The number of positive cases per total number of cases is also shown.
Summary of methylation and expression of p16 in hepatocellular carcinoma (HCC) tissues
p16 EXPRESSION
All normal liver tissues (fig 3A) and those with non-viral liver diseases (fig 3B–E) showed positive nuclear staining for p16. In contrast, complete or partial loss of p16 expression was observed in 90.9% (20/22) of HCC, 47.1% (8/17) of cirrhosis, and 29.4% (5/17) of chronic hepatitis cases, all of which were associated with hepatitis virus infections (fig 3F–I). Partial loss of p16 expression (fig 3F,3G) was observed in four of the five methylation positive chronic hepatitis tissues but in only one of the 13 methylation negative chronic hepatitis tissues (p=0.002) (fig 1B). One complete (fig3H) and four partial losses of p16 expression were observed in the five methylation positive cirrhosis tissues while only three of 12 methylation negative cirrhosis tissues showed partial loss of p16 expression (p=0.009) (fig 1B). Complete (fig 3I) or partial loss of p16 nuclear staining was observed in 81.8% (18/22; 8/9 HBV positive and 10/13 HCV positive) and 9.1% (2/22; both HCV positive) of HCCs, respectively. All of the 16 methylation positive cases showed complete loss of p16 expression (fig 1A, table 1), and a significant correlation was found between methylation and complete loss of expression (p=0.002).

{kind=link}
{kind=link}
{kind=link}
Immunohistochemistry of p16 using the mouse anti-p16 monoclonal antibody. Strong nuclear immunostaining was detected in normal liver (A), fatty liver (B), drug induced liver disease (C), autoimmune hepatitis (D), and primary biliary cirrhosis (E) samples. (F) and (G) Partial loss of p16 expression in chronic hepatitis samples. Complete loss of p16 expression in cirrhosis (H) and hepatocellular carcinoma (I) samples.
Analysis of serial samples from individual patients with methylation positive HCC revealed that loss of p16 expression with promoter methylation occurred in 18 of 20 patients at the stage of chronic hepatitis without clinically detectable carcinoma (data not shown).
Discussion
In this study, we analysed the methylation status of the 5′ CpG island of the p16 gene promoter in a series of HCC and pre- and/or non-neoplastic liver diseases with or without hepatitis virus infections. Methylation of thep16 promoter was detected in 72.7% of HCCs, 29.4% of cirrhosis, and 23.5% of chronic hepatitis tissues, all of which were positive for HBV or HCV infections. In contrast, methylation was not detected in any of the tissue samples from non-viral liver diseases, including PBC, autoimmune hepatitis, drug induced liver disease, and fatty liver, suggesting that this epigenetic change may be related to hepatitis virus infections.
All of the HCC, cirrhosis, and chronic hepatitis samples with promoter methylation showed loss of expression, and a significant correlation between methylation and loss of expression was observed. Therefore, methylation of the p16 promoter detected in this study appears to have a functional significance. Furthermore, it is notable that the majority of patients with methylation positive HCC had loss of p16 expression with promoter methylation at the stage of chronic hepatitis without clinically detectable carcinoma. Our results suggest that methylation of the p16 promoter is one of the earliest events in a subset of hepatitis virus associated hepatocarcinogenesis. Our results are consistent with the findings of several recent studies indicating that inactivation ofp16 by hypermethylation is an early event in cancer development of the lung,21 colon,22oesophagus,23 and pancreas.24 Thus our results confirm recently reported observations of frequent methylation of the p16 promoter in HCC and extend the observations to chronic liver diseases associated with hepatitis virus infections. Further analysis, including a number of non-virus associated cases, is required to clarify the possible intriguing relationship between methylation of the p16promoter and chronic hepatitis virus infections.
In all of the methylation positive cases, unmethylated bands were also detected. One possible explanation is that these unmethylated bands were due to contaminated normal tissues in the tumour, such as inflammatory cells and normal liver cells. Considering the loss of expression, both alleles appear to be inactivated by biallelic methylation or methylation in conjunction with genetic alterations, such as mutation, LOH, and homozygous deletion, as shown in a previous study.13 Several HCC, cirrhosis, and chronic hepatitis samples without methylation also showed loss of p16 expression, suggesting that the above mentioned genetic alterations play a role in inactivation of the p16 gene. In contrast with our results, several previous studies have failed to detect methylation of the p16 promoter in chronic non-tumour liver diseases, including those of viral origin.11 ,14 These conflicting observations may be due to differences in the methods for detection of methylation and/or in the ethnic and aetiological backgrounds of individuals. However, these factors in our study were similar to those in the study of Matsuda and colleagues.14 Nevertheless, our observation is unlikely to be simply overestimation of methylation as we found a good correlation between methylation and loss of expression.
All methylation positive HCC showed complete loss of p16 expression while the majority of methylation positive cirrhosis and chronic hepatitis samples showed partial loss of expression. It has been suggested that accumulation of methylated cytosine in the 5′ CpG island of p16 may silence this gene in a dose dependent manner.26 It has been reported that partial loss of p16 expression was associated with lower levels of methylation than complete loss, suggesting that methylation, and loss of expression, of p16 occurs in a progressive manner.14 ,27 Gradual loss of p16 expression associated with progressivep16 promoter methylation has also been reported in the escape of mammary epithelial cells from senescence.28 Therefore, partial loss of p16 expression in cirrhosis and chronic hepatitis may be due to the heterogeneity of methylation in hepatocytes, and the population of cells with methylated alleles may increase during progression to HCC. It seems intriguing to speculate that cells with loss of p16 expression are more likely to progress to tumorous cells than are those with normal p16 function.
The precise mechanisms leading to aberrant methylation of CpG islands in neoplastic cells are not known. Moreover, little is known of the link between viral infections and methylation machinery. Recently, human immunodeficiency virus has been shown to induce methylation of interferon γ through increased DNA methyltransferase activity.29 Interestingly, Sun et al have reported increased levels of DNA methyltransferase mRNA in liver tissues from chronic hepatitis and cirrhosis compared with those in normal liver tissues.30 However, several lines of experimental evidence suggest that there is no correlation between DNA methyltransferase activity and aberrant methylation of endogenous genes.31 Therefore, aberrant methylation of thep16 promoter may not be caused directly by elevated DNA methyltransferase activity in hepatocarcinogenesis. Other mechanisms such as loss of protection against de novo methylation may underlie aberrant methylation of p16 during hepatocarcinogenesis.17 ,18 Our results warrant further investigation.
Although further study is necessary to clarify the link between hepatitis virus infections and methylation ofp16 in hepatocarcinogenesis, detection ofp16 promoter methylation could be a useful molecular marker to follow up patients with a high risk of developing HCC, such as those with HBV or HCV infections. It would be of interest to prospectively examine whether patients withp16 methylation positive cirrhosis or chronic hepatitis have a higher risk of developing HCC compared with those without p16 methylation.
Acknowledgments
This work was supported by grants in aid from the Ministry of Education, Science, Sports, and Culture (FI and KI) and from the Ministry of Health and Welfare (FI and KI), Japan.
Abbreviations used in this paper
- HCC
- hepatocellular carcinoma
- HBV
- hepatitis B virus
- HCV
- hepatitis C virus
- PBC
- primary biliary cirrhosis
- PCR
- polymerase chain reaction
- MSP
- methylation specific PCR