Article Text

Abstract
BACKGROUND Inflammatory bowel disease (IBD) is a chronic inflammation of the gastrointestinal tract caused by an abnormal and uncontrolled immune response to one or more normally occurring gut constituents.
AIM Given the effects of transforming growth factor β1 (TGF-β1) on both the immune system and extracellular matrix, we postulated that alterations in TGF-β signalling in intestinal epithelial cells may play an important role in the development of IBD.
METHODS TGF-β signalling was inactivated in mouse intestine by expressing a dominant negative mutant form of the TGF-β type II receptor under the control of the mouse intestinal trefoil peptide (ITF)/TFF3 promoter. Transgenic mice (ITF-dnRII) developed spontaneous colitis presenting with diarrhoea, haematochezia, and anal prolapse when not maintained under specific pathogen free (SPF) conditions. Under SPF conditions we induced colitis by mixing dextran sodium sulphate (DSS) in drinking water to examine the significance of loss of TGF-β signalling in the pathogenesis of IBD.
RESULTS Transgenic mice showed increased susceptibility to DSS induced IBD, and elicited increased expression of major histocompatibility complex class II, generation of autoantibodies against intestinal goblet cells, and increased activity of matrix metalloproteinase in intestinal epithelial cells compared with wild-type littermates challenged with DSS.
CONCLUSIONS Deficiency of TGF-β signalling specifically in the intestine contributes to the development of IBD. Maintenance of TGF-β signalling may be important in regulating immune homeostasis in the intestine
- inflammatory bowel disease
- transforming growth factor β
- matrix metalloproteinases
- intestinal trefoil factor
- mouse
Abbreviations used in this paper
- IBD
- inflammatory bowel disease
- TGF-β
- transforming growth factor β
- MMPs
- matrix metalloproteinases
- ITF
- intestinal trefoil factor
- DSS
- dextran sodium sulphate
- MHC
- major histocompatibility complex
- SPF
- specific pathogen free
- MPO
- myeloperoxidase
- HA
- haemagglutinin
- RT-PCR
- reverse transcription-polymerase chain reaction
- HAI
- histological activity index
- TNF-α
- tumour necrosis factor α
- IL
- interleukin
- IFN-γ
- interferon γ
- TIMP
- tissue inhibitor of metalloproteinase
- IEC
- intestinal epithelial cell
- APC
- antigen presenting cell
- GABs
- anti-goblet cell autoantibody
Statistics from Altmetric.com
- inflammatory bowel disease
- transforming growth factor β
- matrix metalloproteinases
- intestinal trefoil factor
- mouse
The intestinal immune system is finely balanced, where proinflammatory and anti-inflammatory cells and molecules are carefully regulated to promote sufficient host mucosal defence capability without destruction of intestinal tissue. Once this regulatory balance is disturbed, non-specific stimulation and activation of inflammatory cells can lead to increased production and release of potent destructive immunological and inflammatory molecules.1Stimulatory molecules such as bacterial cell wall products are capable of activating macrophages and T lymphocytes to release chemotactic and proinflammatory cytokines, which can increase expression of major histocompatibility complexe (MHC) class II molecules on the surface of epithelial cells, endothelial cells, and macrophages.2Normally, these active and destructive immunological and inflammatory events are dampened through the action of suppressive cytokines such as interleukin 10 (IL-10), transforming growth factor β (TGF-β), IL-4, and IL-1 receptor antagonist. However, in inflammatory bowel disease (IBD), these downregulatory events appear to be deficient or only partially effective.1-3
In IL-10 deficient mice generated by gene targeting, most animals suffered from chronic enterocolitis, although lymphocyte development and antibody responses were normal.3 Alterations in the intestine of IL-10 null mice included extensive inflammatory reactions, aberrant expression of MHC class II molecules on the epithelium, and mucosal hyperplasia. Also, TGF-β deficient mice suffered from a more extensive autoimmune process with inflammatory infiltrates, involving multiple organs, including the intestine.4-6 Therefore, it was generally thought that IL-10 seems to be only of regional importance in preventing inflammation in contrast with the broad anti-inflammatory and immune suppressive actions of TGF-β. To date, it is open to question whether this difference in anti-inflammatory activity reflects true compartmentalisation of IL-10 function or whether TGF-β compensates for IL-10 function in IL-10 deficient mice in all organs except the intestine.1-3
Based on these findings, we hypothesised that specific loss of TGF-β signalling in the epithelia of the intestine would increase susceptibility to IBD. Germline null mutation of TGF-β1 can cause embryonic lethality and widespread inflammation in multiple organs.4 ,5 Because TGF-β1 deficient mice have a short life span and colonic epithelial cells express not only TGF-β1 but also TGF-β2 and TGF-β3, it is impossible to determine if loss of TGF-β signalling increases susceptibility to ulcerative colitis in these mice. Therefore, we overexpressed a dominant negative mutant form of TGF-β receptor II (TGF-β dnRII) with intestinal tissue specific promoter to achieve tissue restricted functional inactivation of TGF-β RII and to determine the significance of TGF-β signalling in the pathogenesis of IBD.
Materials and methods
GENERATION OF DOMINANT NEGATIVE MUTANT TGF-β RII MICE
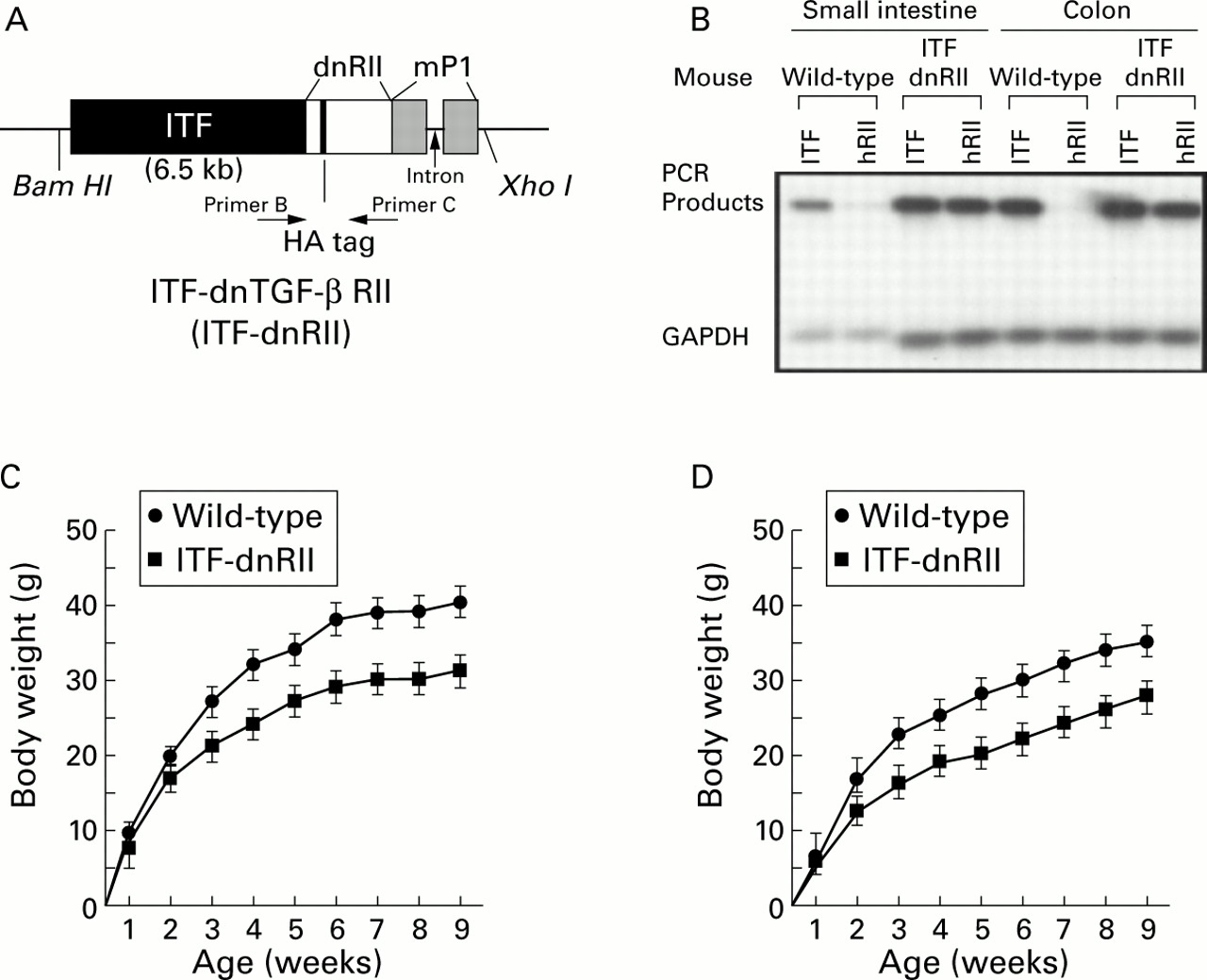
The mouse intestinal trefoil factor (ITF) fragment spans −6500 bp to +10 bp relative to the transcriptional start site.7The human TGF-β RII8 fragment spans +322 to + 911 bp, and contains a haemagglutinin (HA) tag sequence and a segment of the mouse protamine sequence that provides an intron and polyadenylation signal. After injecting the construct DNA into the male nucleus of fertilised eggs, we obtained four lines of transgenic mice designated ITF-1 to ITF-4 possessing high copy numbers of inserted TGF-β dnRII. Genotyping was performed by Southern hybridisation and polymerase chain reaction (PCR). For Southern hybridisation, DNA obtained from tails was cut with Bam H1 andXho I for ITF-dnRII, electrophoresed on 1.0% agarose gel, and transferred to a N+ membrane (BioRad, Hercules, California, USA). After hybridisation with a32P labelled dnRII probe, the presence of the 8.0 kb sized transgene for ITF-dnRII was confirmed. For PCR genotyping, the following primers were used: primer A, GGTGGTGATGTGGACAAAG; primer B, CTGCAGT-CGCTCATGCAGG. The HA sequence of TATGATGTTCCTGATTATGCTAGCCTC was inserted into the dnRII transgene construct (fig 1A).

Generation of dominant negative mutant transforming growth factor β receptor II (TGF-β RII) mice—ITF-TGF-β dnRII transgenic mice. (A) Schematic representation of the transgene. The mouse intestinal trefoil factor (ITF) fragment spans −6500 bp to +35 bp. The 0.6 kb human TGF-β RII fragment spans +322 bp to + 911 bp and contains a haemagglutinin (HA) tag sequence and a segment of the mouse protamine that provides an intron and a polyadenylation site. Transgenic mice were generated using inbred FVB/N zygotes. Of the 15 mice born, six were positive for ITF-dnRII, bred into lines, and designated ITF1 to ITF6. Pups from ITF1, ITF2, and ITF3 were used for the current experiments. (B) TGF-β dnRII expression. Using total RNA from the intestines of wild-type mice and ITF-dnRII, reverse transcription-polymerase chain reaction was performed using the specific primer sets for human TGF-β RII, mouse ITF, and mouse GAPDH. (C, D) Body weights of wild-type littermates and ITF-dnRII transgenic mice. Body weight was measured every week for nine weeks in males (C) and females (D).
ANIMAL CARE AND INDUCTION OF COLITIS
All mice were maintained in a specific pathogen free (SPF) facility and routinely monitored for absence of common mouse viruses by serology. Mice were kept in microisolator cages and provided with free access to food and water. Colitis was induced by allowing animals access to 5% (wt/vol) dextran sodium sulphate (DSS, molecular weight 47 000; ICN Biomedicals, Aurora, Ohio, USA) in their drinking water ad libitum for 10 consecutive days and were weighed every other day. This study was approved by the institutional animal care and use subcommittee of the National Cancer Institute (Bethesda, Maryland, USA).
RNA ISOLATION AND RT-PCR
Total RNA was isolated from mouse tissue using Trizol (Gibco BRL, Gaithersburg, Maryland, USA). Reverse transcription-PCR (RT-PCR) was performed according to the manufacturer's instruction using a Perkin-Elmer RT-PCR kit (Perkin-Elmer Corp., Branchburg, New Jersey, USA). The primers used were as follows: for TGF-β dnRII, 5′-ACGACATGATAGTC ACTGACAACA-3′ and 5′-TTGGGGTCA TGGCAAACTG TCTC-3′; for mouse ITF, 5′-GAAGTTTGCGTGCTGCCATGGAG-3′ and 5′-CCGCAATTAGAACAGCCTTGTG-3′. Primers for matrix metalloproteinase 9 (MMP-9) were purchased from Chemicon (Temecular, California, USA). PCR was performed with a 100 μl reaction volume. The amplification cycle (denaturation step at 94°C for one minute, an annealing step at 60°C for one minute, and an extension step at 72°C for 30 seconds) was repeated 30 times and followed by a final extension for 10 minutes at 72°C.
IMMUNOHISTOCHEMICAL STAINING
Immunohistochemical stainings with HA tag (LCRC, National Cancer Institute) and MHC class II (OX-3; Serotec, Raleigh, North Carolina, USA) antibodies were performed as previously described.9
IN SITU HYBRIDISATION
The human TGF-β dominant negative RII complementary DNA coding region (198 bp) and MMP-9 were synthesised by RT-PCR using the primer sets described above and inserted into pBluescrip SK(−) vector. Each DNA probe was labelled with biotin using the BioNick labelling system (Gibco-BRL). In situ hybridisation was performed as described previously.9
SCORING OF DEGREE OF ULCERATIVE COLITIS
Tissue samples, fixed in formalin and routinely processed, were assessed histologically by light microscopy, performed in a blinded fashion. Three variables were considered, each scored on a 0–4 scale according to the severity of the induced damage: extent of ulceration; mucosal submucosal inflammatory infiltration; and wall thickness. The criteria for scoring are defined in the legend to fig3.
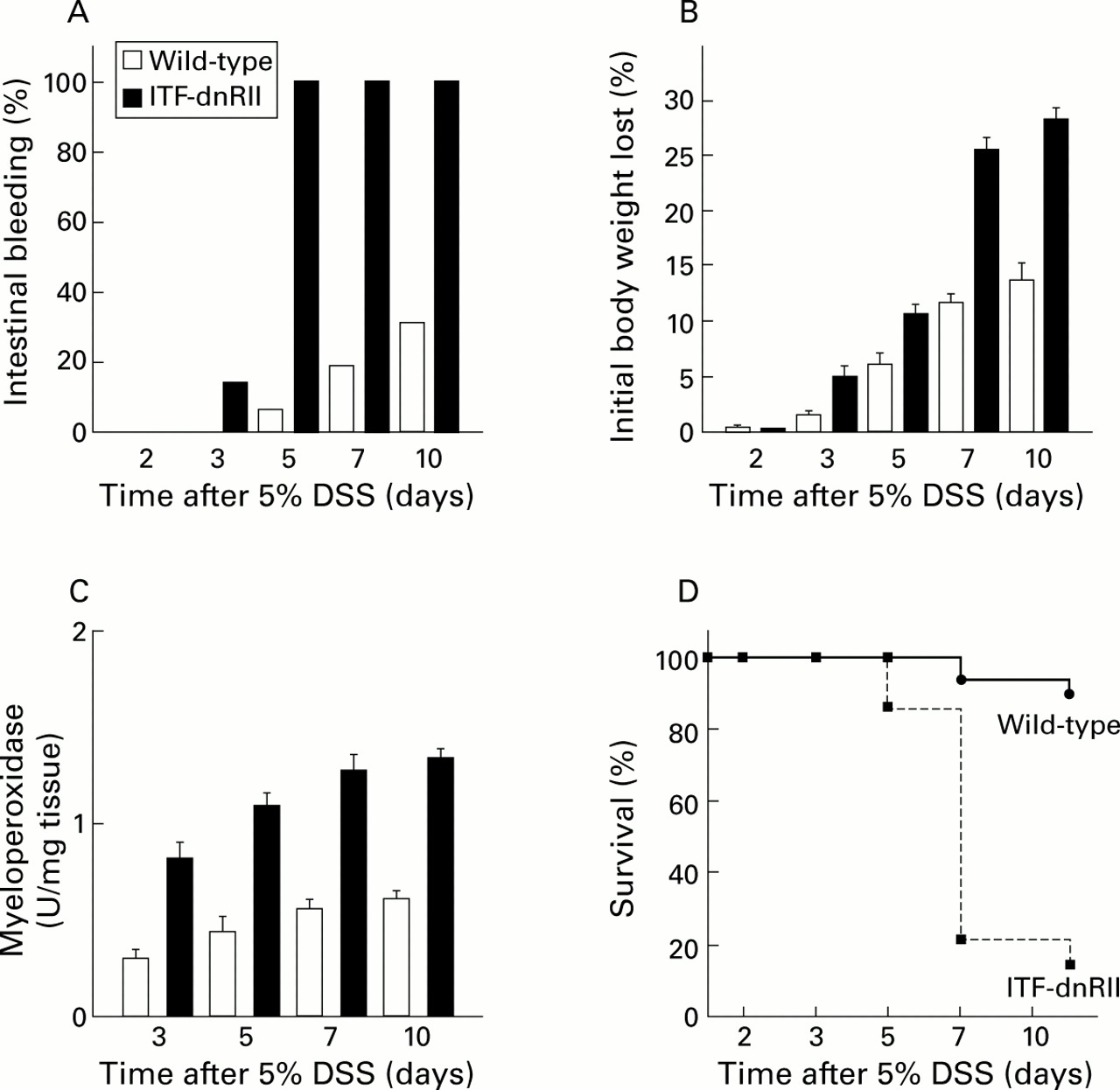
Dextran sodium sulphate (DSS) administration resulted in an increased incidence of intestinal bleeding, weight loss, colitis, and death in ITF-dnRII transgenic mice. (A) Prevalence of haematochezia. Intestinal bleeding (% incidence) was more common in ITF-dnRII mice. Earlier and more severe anal bleeding developed in mice lacking TGF-β signalling after administration of DSS (p<0.001). (B) Changes in body weight. Weight loss (expressed as per cent of initial body weight) was more prominent in ITF-dnRII mice than in wild-type mice. (C) Mucosal myeloperoxidase (MPO) activity. Changes in mucosal MPO activity in the colon of wild-type littermates and ITF-dnRII mice. ITF-dnRII mice showed significantly higher levels of mucosal MPO than wild-type mice (p<0.01). (D) Survival. Survival was significantly decreased in ITF-dnRII mice compared wild-type littermates using Kaplan-Meyer analysis. (ITF-dnRII transgenic mice were from ITF1–3 lines.)
AUTOANTIBODIES AGAINST INTESTINAL CELLS
Autoantibodies were detected by indirect immunofluorescence methods using cryopreserved slides of monkey ileum cells (Biochips; Euroimmun, Lubeck, Germany). For immunofluorescence, sera were diluted 1:20 in phosphate buffered saline containing 2% bovine serum albumin. Positive or negative control sera (20 μl) were used on two of the 10 well defined areas of each glass slide containing monkey ileum or pancreas. On the remaining eight areas, 20 μl of diluted serum were applied. After incubation in a humid chamber for 30 minutes at room temperature and incubation for another 30 minutes with a 1:50 dilution of fluorescein isothiocyanate labelled Fab′(2) goat antimouse immunoglobulin antibodies (Sigma Chemical, St Louis, Missouri, USA), the slides were washed again in phosphate buffered saline for five minutes and then mounted.
CHANGES IN INTESTINAL MMPs
Intestinal tissues were homogenised, mixed with sample buffer, and directly applied, without prior heating or reduction, to 4–16% acrylamide gels containing 1 mg/ml of gelatin. After removal of sodium dodecyl sulphate from the gel by incubation in 2.5% (v/v) Triton X-100 for one hour, the gels were incubated at 37°C for 16 hours in 50 mM Tris HCl, pH 7.6, containing 0.2 M NaCl, 5 mM CaCl2, and 0.02% (w/v) Brij-35. The gels were stained for three hours in 40% methanol/10% glacial acetic acid containing 0.2% Coomassie brilliant blue G-250 and de-stained in the same solution without dye. Each MMP was evident as a clear band against the blue background of stained gelatin. For western blot analysis, protein extractions were performed with lysis buffers containing sodium orthovanadate and sodium dodecyl sulphate. Proteins were separated by electrophoresis on 4–20% polyacrylamide gels under reducing conditions and transferred to nylon membranes. After treatment with a blocking solution, the nylon membranes were incubated overnight at 4°C with mixture of antibodies to MMP-2 (R&D, Minneapolis, Minnesota, USA) and MMP-3 (Chemicon, Temecula, California, USA) diluted 1:200 in Tris buffered saline containing 5% non-fat milk. The nylon membranes were washed and incubated with peroxidase conjugated goat antimouse IgG antibody. After washing the blot in Tris buffered saline containing 0.1% Tween 20, proteins were detected using a chemiluminescence procedure (Pierce Chemical Co., Rockford, Illinois, USA).
Results
GENERATION OF DOMINANT NEGATIVE MUTANT MICE OF TGF-β RECEPTOR II USING MOUSE ITF PROMOTER
We generated a construct in which expression of the human TGF-β dnRII spanning nucleotides +322 to +911 and encoding the extracellular and transmembrane domains of TGF-β RII8 ,9 was regulated by the ITF promoter7 (fig 1A). The presence of human dnRII was detected by RT-PCR (fig 1B). All three lines of ITF-dnRII transgenic mice (ITF1–ITF3) developed spontaneous colitis when not maintained under SPF conditions. In both males and females, there was no difference in body weight until the first week after birth but after the second week body weight was approximately 20–30% lower in ITF-dnRII mice compared with those of wild-type mice (fig 1C, D). Some ITF-dnRII transgenic mice spontaneously developed severe diarrhoea and haematochezia accompanied by weight loss and a shaggy appearance. A few transgenic mice died due to weight loss probably caused by the development of spontaneous ulcerative colitis, and some showed rectal prolapse. However, these effects rarely occurred when mice were maintained under SPF conditions.
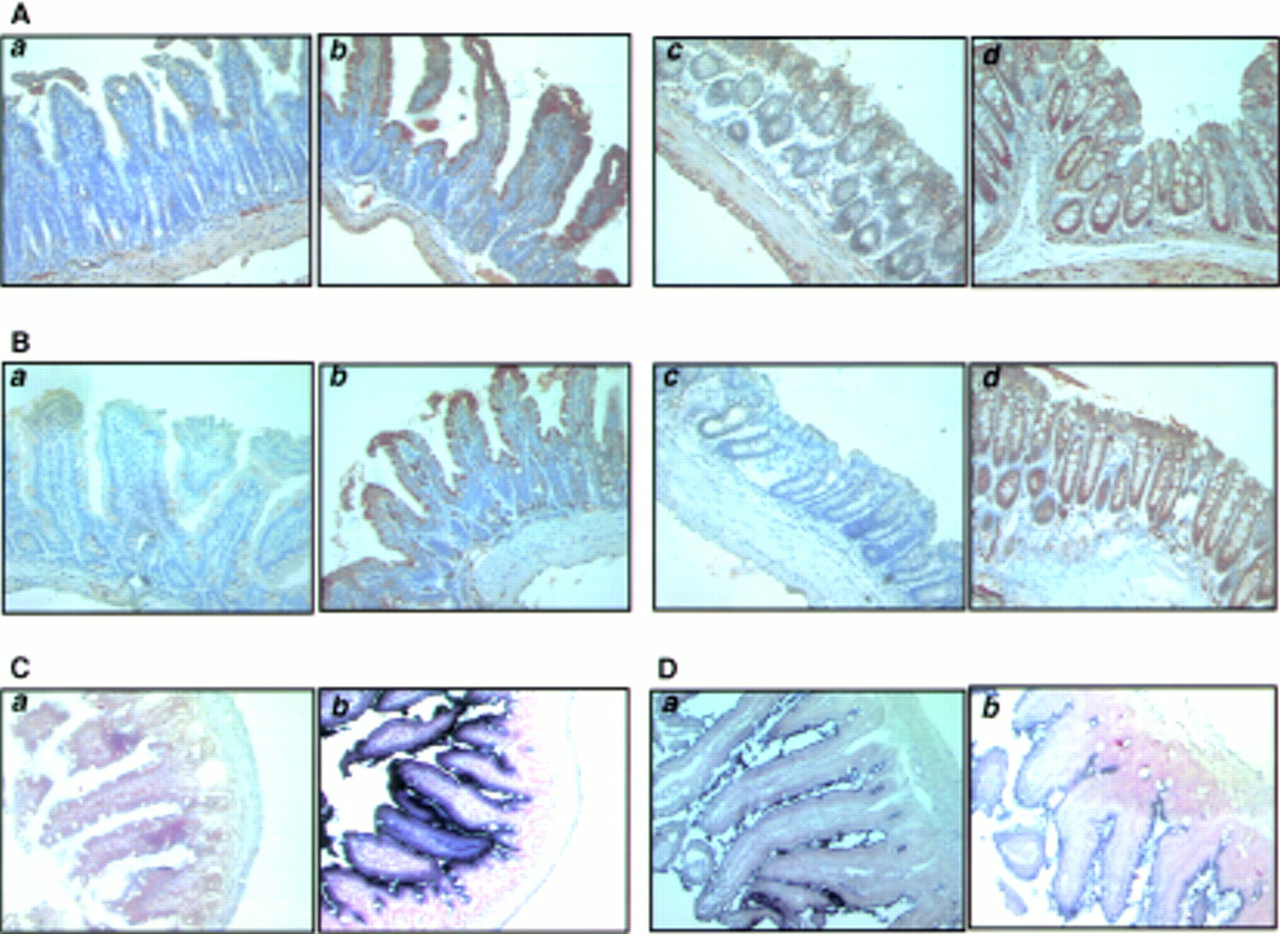
We performed immunohistochemical staining using TGF-β RII antibody which detects both mouse and human TGF-β RII. In the intestine of wild-type mice, TGF-β RII expression was noted on the epithelial cells of the luminal surface, muscularis mucosa, and proper muscle layers. However, in the intestine of ITF-dnRII transgenic mice, positive staining was markedly increased throughout the entire epithelial and muscular layers (fig 2A(b), 2A(d)). These findings were reconfirmed using in situ hybridisation of human TGF-β RII mRNA (fig2C). Purple coloured positive hybridised signals of TGF-β RII were only seen in the intestines of ITF-dnRII transgenic mice because transgenic mice have human TGF-β type II receptor. As we inserted the HA sequence in dnRII transgene construct, this allowed expression of the ITF-dnRII transgene to be visualised by immunohistochemical staining with a HA epitope antibody. The distribution of positive dnRII staining paralleled expression of HA tag antigens. While staining of HA tag was not detectable in intestines of wild-type littermates it was strongly expressed in the intestines of ITF-dnRII transgenic mice (fig2B). There was no significant change in expression of ITF mRNA between wild-type littermates and ITF-dnRII transgenic mice (fig 2D)

Tissue distribution of transforming growth factor β receptor II (TGF-β RII), intestinal trefoil factor (ITF), and dnRII. (A) Immunohistochemical staining of TGF-β RII using anti-TGF-β RII antibody and (B) TGF-β dnRII using anti-haemagglutinin (HA) tag antibody in the small intestine of wild-type littermates (a), small intestine of ITF-dnRII mice (b), colon of wild-type littermates (c), and colon of ITF-dnRII transgenic mice (d). Slightly increased expression of TGF-β type II was observed in the intestine of ITF-dnRII mice compared with wild-type littermates. Because we inserted the HA sequence in transgene construct, the HA tag was identified only in the intestine of ITF-dnRII mice by immunohistochemical staining with HA antibody; none was stained in the intestine of wild-type littermates. (C) In situ hybridisation was performed for the presence of human TGF-β RII mRNA using a biotinylated TGF-β RII probe (206 bp) in the colon of wild-type littermates (a) and the colon of ITF-dnRII transgenic mice (b). (D) In situ hybridisation of mouse ITF mRNA. Expression pattern of mouse ITF gene was similarly expressed in transgenic mice (a) and wild-type littermates (b) (×200 magnification)
LOSS OF TGF-β SIGNALLING INCREASES SUSCEPTIBILITY TO ULCERATIVE COLITIS
To see if the intestine of mice lacking TGF-β signalling is more prone to IBD, we administered DSS in drinking water, which is known to produce both acute and chronic ulcerative colitis with features similar to the symptomatic and histological findings in humans.10 ,11 After preliminary experiments to determine the effects of various concentrations of DSS in wild-type mice, a group of 16 wild-type littermates (FVB/N background) and 28 ITF-dnRII mice, each about 30 g in weight, were treated with 5% DSS in their drinking water for designated periods of time. ITF-dnRII mice appeared highly susceptible to DSS induced ulcerative colitis; nearly all developed frankly bloody diarrhoea with occasional rectal prolapse within five days after DSS administration and most died by about day 7. Some mice showed severe haematochezia even on day 3 after DSS administration, and earlier mortality was noted on day 5 after DSS administration. The FVB/N strain was somewhat less susceptible to DSS induced colitis compared with other strains of mice, including ICR, C57BL/6, and Balb/c, based on preliminary observations. Only 25% of wild-type mice treated with DSS exhibited melena and occasional bloody diarrhoea on day 10 (fig 3A). Severe wasting was more prevalent in ITF-dnRII mice than in wild-type mice (fig 3B). We measured mucosal myeloperoxidase (MPO) activity, an index of neutrophil infiltration in tissue,12 and found that mucosal MPO activity of ITF-dnRII mice was significantly elevated compared with that of wild-type mice in the colon (fig 3C). The difference in survival between the two groups was statistically significant (p<0.005) (fig 3D). On gross examination of the intestine from ITF-dnRII mice after DSS treatment, multiple linear ulcerations were detected in the small intestine and colon. Extensive haemorrhage had occurred in the small and large intestine, most severely in the rectal area. Microscopic examination revealed severe mucosal ulcerations with prominent inflammatory cell infiltrations. DSS treated wild-type mice showed only scattered erosions and haemorrhagic spots on days 7 and 10.
Pathological scores, measured using the histological activity index (HAI), of the ITF-dnRII mice were significantly higher than those of wild-type mice (fig 4A, B). Even at three days after 5% DSS administration, HAI was significantly increased in ITF-dnRII mice. Prominent differences in HAI scores were also noted in the extent of mucosal ulcerations (fig 4C). These differences in HAI scores came from the observation that most of the ITF-dnRII mice showed focally deep to extensive ulceration with moderate to severe inflammatory infiltrates. This finding correlated with the observations that transgenic mice showed higher mortality due to intestinal bleeding and increased MPO activity. Therefore, these results suggest that TGF-β signalling is important either in maintaining normal intestinal mucosal integrity or in modulating intestinal immune homeostasis.
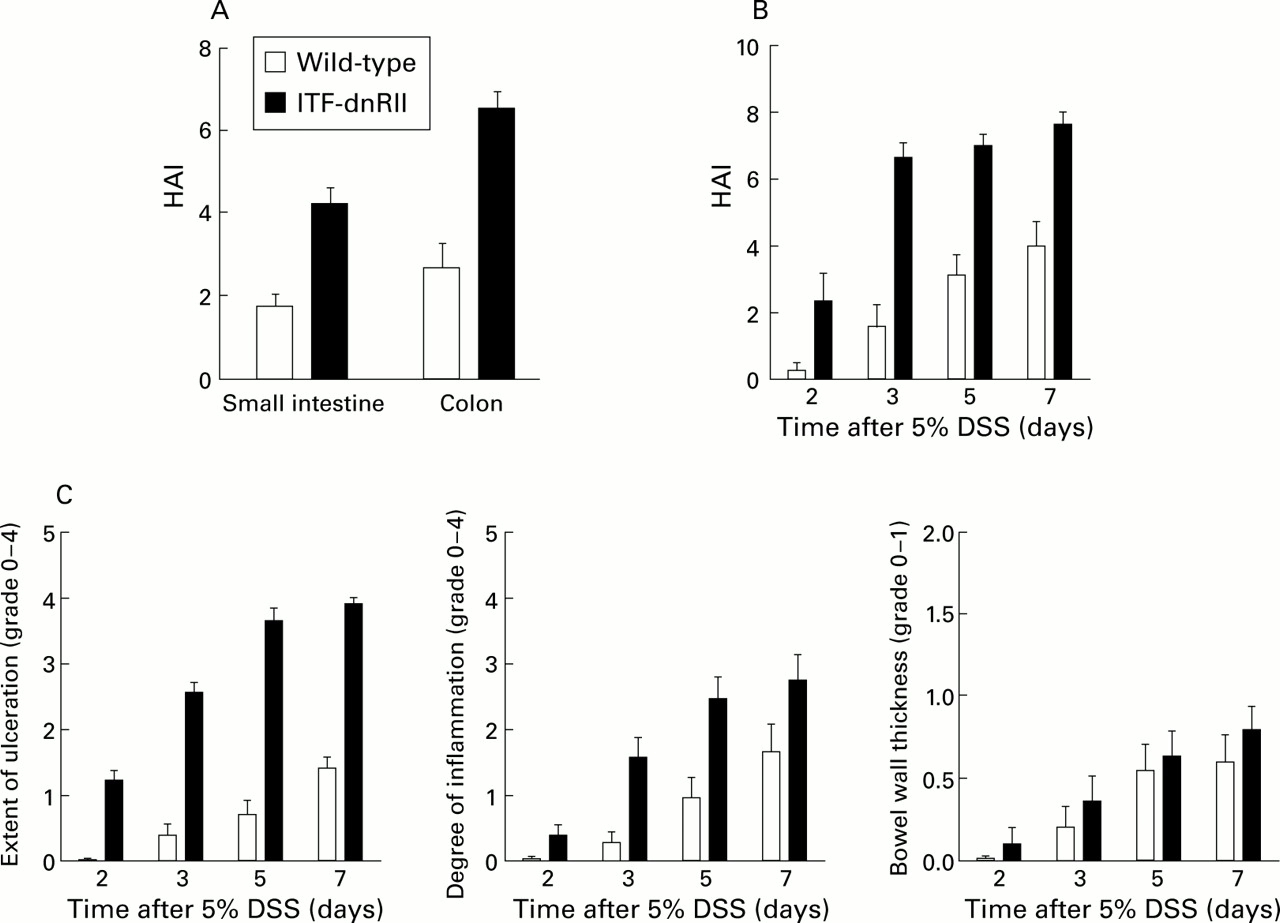
ITF-dnRII transgenic mice showed increased pathological scores Score criteria for extent of ulceration are as follows: 0, no architectural change; 1, focal superficial ulceration; 2, diffuse superficial ulceration; 3, focal gland dropout or focal deep ulceration; 4, extensive glandular dropout or deep ulceration. Scores for inflammation are as follows: 0, no increased inflammatory infiltrates; 1, focal mild inflammation; 2, diffuse mild inflammation; 3, cryptic abscess formation; 4, diffuse dense inflammation. Presence of oedema: 0, no significant oedema; 1, broad zone of oedema. Based on these criteria, comparison of histological activity index (HAI) is shown according to site of inflammation (A), days after 5% dextran sodium sulphate (DSS) administration (B), and criteria of each score (C). The extent of mucosal ulceration was significantly different between transgenic mice and wild-type littermates (p<0.001). (ITF-dnRII transgenic mice were from ITF1–3 lines.)
INCREASED MHC CLASS II ANTIGENS AND AUTOANTIBODIES FOLLOWING LOSS OF TGF-β SIGNALLING
We studied expression of MHC II in the intestines of ITF-dnRII mice and wild-type littermates as TGF-β null mice showed increased synthesis of MHC class II.6 Immunohistochemical staining and western blot with MHC class II antibody showed increased expression in the intestinal epithelium of transgenic mice whereas immunostaining and immunodetection of MHC class II was less common in the intestinal epithelial cells of wild-type mice (fig 5A, B). MHC class II molecules are normally expressed on limited cell types involved in presenting antigen to T helper cells. These cell types commonly include B lymphocytes, activated T lymphocytes and macrophages, dendritic cells, glial cells, and thymic epithelial cells.13 This elevated expression of MHC class II in epithelial cells of the intestine was seen even prior to any evidence of inflammatory infiltrates and became more prominent after provoking colitis in transgenic mice (fig5A).
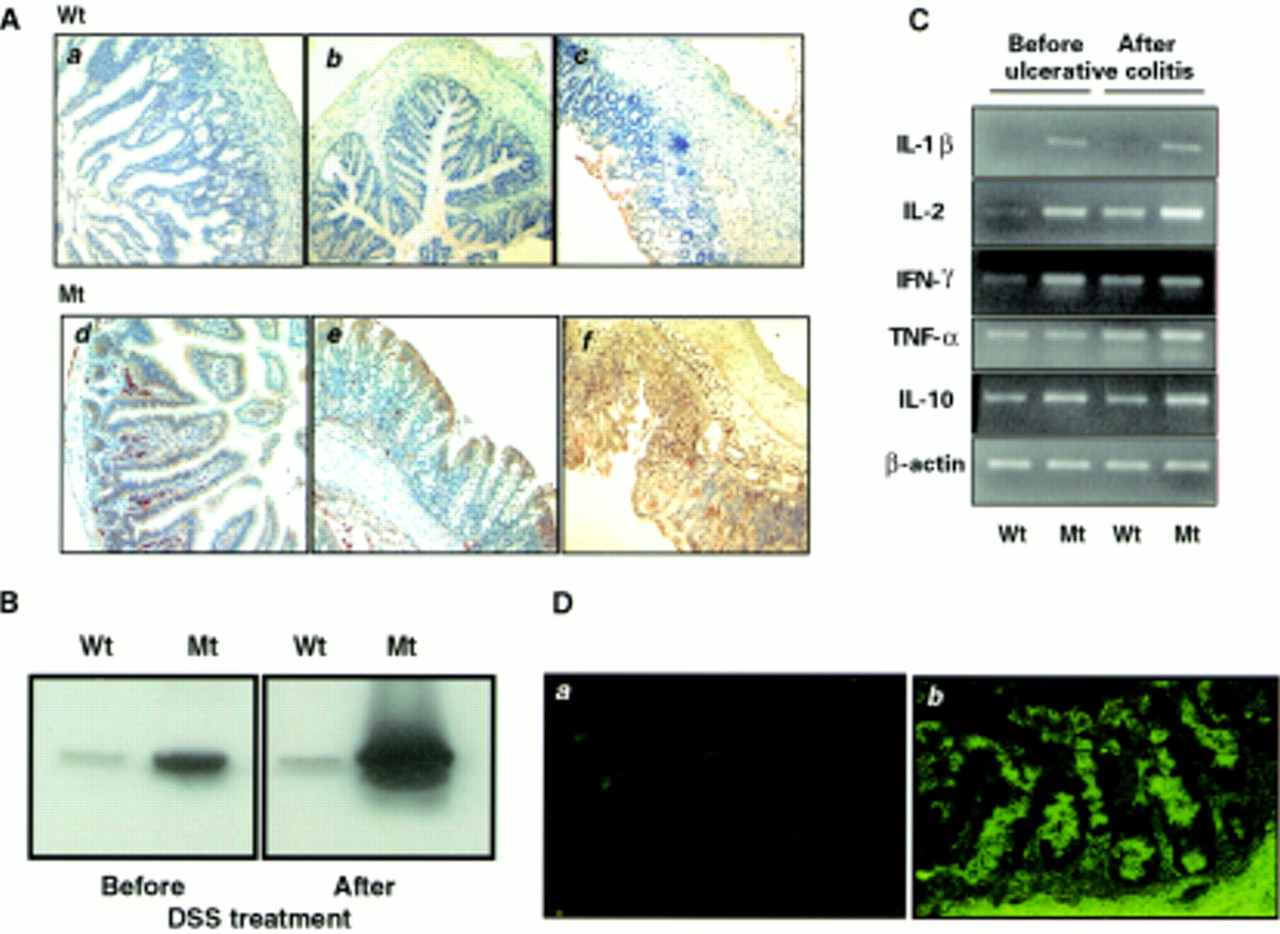
Increased expression of major histocompatibility complex (MHC) class II, cytokines, and autoantibodies in the intestine of ITF-dnRII transgenic mice. (A) Immunohistochemical staining of MHC class II. Scant MHC class II expression was observed in epithelial cells of the small intestine (a) and colon (b) of wild-type (Wt) FVB/N mice but increased expression of MHC class II was observed in epithelial cells of the small intestine (d) and colon (e) of ITF-dnRII transgenic mice (Mt). After induction of inflammatory bowel disease (IBD), MHC class II antigen expression was more markedly increased in transgenic mice (f) compared with wild-type mice (c) (×100 magnification). (B) Western blotting of MHC class II. Western blotting showed similar findings, with ITF-dnRII mice (Mt) showing increased expression of MHC class II antigens in intestine compared with wild-type littermates (Wt) (DSS, dextran sodium sulphate). (C) Changes in cytokines. Interleukin (IL)-1β, IL-2, interferon γ (IFN-γ), and IL-10 were all expressed at higher levels in the colons of transgenic mice (Mt) compared with those of wild-type mice (Wt). Expression of tumour necrosis factor α (TNF-α) was increased after induction of ulcerative colitis. (D) Autoantibodies in serum of ITF-dnRII transgenic mice. Autoantibodies were identified by indirect immunofluorescence using monkey ileum sections and FITC coupled rat antimouse immunoglobulin polyclonal antibody. Autoantibodies were detected in the serum of ITF-dnRII mice (b) whereas none was seen after applying sera obtained from wild-type littermates (a). Positive sera containing IgG autoantibodies showed blurry, drop-like staining (×200 magnification).
We performed RT-PCR to compare mRNA levels of inflammatory or anti-inflammatory cytokines, including IL-2, IL-1β, tumour necrosis factor α (TNF-α), interferon γ (IFN-γ), and IL-10 in intestinal tissues of transgenic mice and wild-type littermates. Even prior to inducing ulcerative colitis, steady state levels of IL-1β, IL-2, IFN-γ, and IL-10 mRNAs were higher in intestinal tissue from ITF-dnRII mice than those from wild-type littermates. After DSS treatment, levels of IL-1β, IL-2, TNF-α, and IL-10 transcripts were further induced. IL-4 mRNA was slightly increased in intestinal tissue from transgenic mice compared with levels in wild-type mice, and the level of TGF-β1 expression was also higher in intestinal tissues from transgenic mice (fig 5C). Further evidence for an autoimmune mechanism to explain why loss of TGF-β signalling in ITF-dnRII mice resulted in increased susceptibility to ulcerative colitis was supplied by the presence of autoantibodies against intestinal cells.14 We identified autoantibodies against intestinal cells in the serum of ITF-dnRII mice (fig 5D). Autoantibodies were detected in 88% (21 of 24 tested) of ITF-dnRII mice while none was detected in the sera of wild-type littermates (0 of 20). There was a significant correlation between the titre of autoantibodies and HAI (r=0.77, p<0.01). All mice who showed positive autoantibodies in 1:100 diluted sera also showed a high HAI.
DISRUPTED MUCOSAL INTEGRITY BY LOSS OF TGF-β SIGNALLING
We saw a significant difference in pathological scoring between wild-type littermates and ITF-dnRII mice, suggesting that normal TGF-β signalling in the intestine may be important in maintaining mucosal integrity. As TGF-β is known to be an important regulator of MMP and tissue inhibitor of metalloproteinase (TIMP),15 we studied changes in MMP. Even before inducing ulcerative colitis, MMP gelatinolytic activity was elevated in tissue homogenates from ITF-dnRII mice compared with those from wild-type littermates. MMP gelatinolytic activities were more significantly increased after induction of ulcerative colitis (fig 6A). Despite loading only one tenth of the amount of protein compared with wild-type littermates onto the gelatin zymography gels, MMP gelatinolytic bands were prominently increased in ITF-dnRII mice. These MMPs were further confirmed by western blot analysis using a mixture of antibodies against MMP-2, MMP-3 (fig 6B), and in situ hybridisation of MMP-9 mRNA (fig 6C). Increased MMPs in the colons of ITF-dnRII mice may contribute to the increased mucosal ulcerations after DSS administration.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Matrix metalloproteinase (MMP) expression in mucosal homogenates of wild-type and transgenic mice. (A) Even before induction of ulcerative colitis, colon extracts of transgenic mice showed increased MMP gelatinolytic bands (52, 72, and 92 kDa) which become more prominent after induction of ulcerative colitis. Even applying only one tenth of the amount of protein compared with wild-type (Wt) littermates onto gelatin zymography gel, MMP activity was significantly increased in ITF-dnRII (Mt) mice. (B) MMP expression was confirmed by western blot analysis using a mixture of MMP-2, MMP-3, and MMP-9 antibodies. (C) In situ hybridisation of MMP-9 showed increased mRNA expression in small intestine (c) and colon (d) of ITF-dnRII transgenic mice (Mt) compared with wild-type littermates (Wt) (a, b) (×200 magnification).
Discussion
In this study, we have shown that loss of TGF-β signalling in the intestinal epithelium increases susceptibility to ulcerative colitis in transgenic mice through combinations of increased MHC class II molecule expression, increased intestinal MMP activities, and increased autoantibody production, all of which cooperatively led to aggravated mucosal destruction.
To understand why selective loss of TGF-β signalling in the intestinal epithelial cells triggered increased susceptibility to IBD, a brief description of immune regulation in the intestine is required. Recognition that immune responses in the intestine differ from those seen systemically has led to a search for a novel pathway involved in mucosal immunoregulation. One cell type that has surfaced as a prime candidate for such a regulatory role is the intestinal epithelial cell (IEC). The IECs sample luminal antigens and present these to primed T cells. Therefore, the IEC acts as a key regulator of the immunological state of the intestine.16 But IECs have been grouped into a category of non-professional antigen presenting cells (APCs) because non-professional APCs do not constitutively express MHC class II molecules and do not activate T cells in a conventional manner as they do not express specific co-stimulatory molecules.17Therefore, IECs can present antigens to primed T cells.18However, processing of intact protein antigens may be limited and the ability to generate potent immune responses would be reduced. In normal individuals, IECs present antigens to T cells and, in so doing, preferentially stimulate proliferation of CD8+ T cells. As CD8+ cells are associated with suppressor effects, it has been proposed that IEC antigen presentation is a component of an immunoregulatory mechanism that leads to downregulation of mucosal immune responses.19 However, in patients with IBD, IEC antigen presentation leads primarily to stimulation of CD4+T cells which are associated with helper activity. In this case, IEC antigen presentation can lead to upregulation of mucosal immune responses, and consequently aggravated chronic inflammation.20 Therefore, selective loss of TGF-β signalling in the intestine rendered affected IECs APCs through increased expression of MHC class II, which was shown in current ITF-dnRII transgenic mice. Conversely, intact TGF-β signalling in colonic epithelium seems to be critical in maintaining normal mucosal immunoregulation.
TGF-β is a multifunctional growth factor and has important chemotactic and anabolic actions on fibroblasts involved in tissue repair. It stimulates their production of critical components of the extracellular matrix, such as collagen and fibronectin, and proteoglycans. It inhibits the action of proteolytic enzymes that destroy newly formed connective tissues. TGF-β promotes collagen deposition by smooth muscle cells isolated from Crohn's disease tissue, a process that may contribute to fibrosis and stricture formation in Crohn's disease.21 TGF-β is also known to inhibit intestinal epithelial cell proliferation, a process that may inhibit re-epithelialisation after surface injury. Babyatsky and colleagues22 showed that TGF-β may play a role in promoting healing of the overlying epithelium by modulating epithelial cell restitution, which serves to re-establish surface continuity after mucosal injury. A new finding discovered in this study was that loss of TGF-β signalling in the intestine increases susceptibility to ulcerative colitis, suggesting that TGF-β signalling is a critical determinant of tissue injury in inflammatory disease. Although there were some reports on TGF-β RII mutation in relation to ulcerative colitis associated neoplasia,23 ,24 it may be interesting to determine the level of TGF-β receptors in human colonic mucosa in patients with ulcerative colitis.
IBD is a classic autoimmune disease involving either a reaction against a self constituent or a gut constituent that cross reacts with a self constituent. These autoantibodies then interact with non-specific cytotoxic cells to cause tissue injury via an antibody dependent cell mediated cytotoxic mechanism.25 In autoimmune disease, B cells secreting pathogenic autoreactive antibodies are supported by TH cells, implying defects in the regulation of both types of cells. Defective central or peripheral tolerance, inadequate suppression of autoreactive cells, and/or unusual activation of T and B cells may underlie the persistence of autoantibodies that characterise autoimmune disease.26 The autoantibodies of interest in IBD include antineutrophilic cytoplasmic antibodies, VH3–15 gene product, colonic 40 kDa protein, anti-goblet cell autoantibody (GAB), antiendothelial cell antibody, and anticardiolipin antibody.27 GAB reacts with phenol water extracts of fetal colonic tissue and mucopolysaccharides derived from colonic goblet cells. Folwaczny and colleagues28 detected significantly higher prevalence of IBD in relatives of GAB positive patients than in relatives of GAB negative patients. Thus GABs are prevalent in a cohort of healthy individuals with an increased risk of developing IBD, and these autoantibodies may be a marker of susceptibility to chronic IBD. In patients with infectious enteritis or colitis, or in patients with diverticulitis or other non-autoimmune diseases of the colon, GABs were not found. Therefore, development of GABs is unlikely to result from a non-specific immunoreactivity during inflammation of the intestinal mucosa.29
Egger and colleagues30 showed that mice lacking TGF-α have an increased susceptibility to DSS induced colitis but their findings were due to lower levels of ITF in intestinal epithelial cells because TGF-α has been shown to stimulate ITF synthesis and release from goblet cells. Thus it can be hypothesised that diminished ITF production and release in TGF-α null mice leads to increased susceptibility to intraluminal DSS. In contrast, our ITF-dnRII transgenic mice showed unaltered ITF mRNA by in situ hybridisation and RT-PCR. Therefore, increased susceptibility to DSS induced ulcerative colitis shown in our transgenic mice does not seem to be due to loss of ITF.
TGF-β has been known to upregulate TIMP mRNA and downregulate MMP. TGF-β suppresses expression of MMP mRNA by binding to an inhibitory element of the MMP promoter, known as the TGF-β inhibitory element.31 ,32 In situ hybridisation studies showed interstitial collagenase and stromelysin-1 mRNA in granulation tissue, which is associated with Crohn's disease, ulcerative colitis, and gastric ulcers.33 Uncontrolled production of MMPs could have devastating consequences similar to those in the intestine of our transgenic mice. MMP expression was much higher in our transgenic mice compared with wild-type littermates even before induction of ulcerative colitis. Although we did not measure MMP activity in tissues, extensive mucosal ulceration seen in the ITF-dnRII mice may have resulted, at least in part, from markedly increased MMP expression, implicating TGF-β signalling in the maintenance of mucosal integrity of the intestinal tract. The aetiology and exact pathogenesis of human ulcerative colitis, a major subgroup of IBD, are still unknown. Although a number of animal models of IBD have been described, including T cell receptor α deficient, IL-2, and IL-10 deficient mice, and rats transgenic for HLA-B27, these models explain only one abnormal aspect of mucosal immunoregulation.3 ,34 ,35 Our study showed that TGF-β signalling may be the critical determinant either in the underlying pathophysiology of all of these reported IBD animal models or in maintaining mucosal immune homeostasis and epithelial integrity.
Acknowledgments
We thank Drs R Chinery and HM Cox for mouse ITF gene, Drs AB Roberts and W Strober for critically reviewing the manuscript, and C Lee for preparation of the manuscript.
Abbreviations used in this paper
- IBD
- inflammatory bowel disease
- TGF-β
- transforming growth factor β
- MMPs
- matrix metalloproteinases
- ITF
- intestinal trefoil factor
- DSS
- dextran sodium sulphate
- MHC
- major histocompatibility complex
- SPF
- specific pathogen free
- MPO
- myeloperoxidase
- HA
- haemagglutinin
- RT-PCR
- reverse transcription-polymerase chain reaction
- HAI
- histological activity index
- TNF-α
- tumour necrosis factor α
- IL
- interleukin
- IFN-γ
- interferon γ
- TIMP
- tissue inhibitor of metalloproteinase
- IEC
- intestinal epithelial cell
- APC
- antigen presenting cell
- GABs
- anti-goblet cell autoantibody
References
Linked Articles
- Commentary