Article Text

Abstract
BACKGROUND Invasion of the intestinal mucosa by leucocytes is a characteristic of intestinal inflammation but the role of the epithelium in orchestrating this recruitment has not been examined in vivo. Cultured intestinal epithelial cells secrete a wide variety of chemokines, often in response to agents present in the intestinal lumen. Macrophage inflammatory protein 2 (MIP-2) is a chemokine that attracts neutrophils, and its secretion from intestinal epithelial cells is enhanced by inflammatory stimuli such as interleukin 1β. We hypothesised that the production of MIP-2 by epithelial cells would increase leucocyte migration into the intestine.
AIM To study the effects of a chemokine secreted from intestinal epithelial cells in vivo.
METHODS MIP-2 was expressed in the mouse intestinal epithelium using an epithelial cell specific promoter from the gene encoding the intestinal fatty acid binding protein. The intestines of these transgenic mice were then analysed.
RESULTS Epithelial cells from transgenic mice expressed MIP-2 but wild-type mice did not. Neutrophil recruitment, examined by myeloperoxidase (MPO) staining and total MPO activity per unit weight of intestine, was significantly increased in transgenic mice in both the small intestine and proximal colon, and this was blocked by anti-MIP-2 antibody treatment. Both intraepithelial and lamina propria lymphocytes were also increased in transgenic mice. They showed chemotactic activity to MIP-2 in the Boyden chambers and expressed MIP-2 receptor (CXCR-2) mRNA confirmed by reverse transcription-polymerase chain reaction.
CONCLUSION These experiments are the first to show a functional role for epithelial chemokines in vivo and reveal an unexpected role for the neutrophil chemokine MIP-2 in controlling mucosal lymphocyte migration.
- chemokines
- epithelial cells
- intestinal mucosa
- leucocyte recruitment
- transgenic mouse
Abbreviations used in this paper
- CMF-HBSS
- Ca2+/Mg2+free Hanks' balanced salt solution
- Fabpi
- fatty acid binding protein of the intestine
- IEL
- intraepithelial lymphocyte
- IL
- interleukin
- LPL
- lamina propria lymphocyte
- LPS
- lipopolysaccharide
- MIP-2
- macrophage inflammatory protein 2
- MPO
- myeloperoxidase
- RT-PCR
- reverse transcription-polymerase chain reaction
- TCR
- T cell receptor
Statistics from Altmetric.com
The small intestinal epithelium regulates the absorption of nutrients and serves as a barrier to the external environment. The intestinal epithelial cell also plays a central role in mucosal immune responses. Firstly, it controls the passage of antigen from the lumen to immune cells in the lamina propria or Peyer's patches.1 Loss of barrier function through, for example, lack of E-cadherin function, results in intestinal inflammation.2 Secondly, the enterocyte is ideally placed to signal changes in the intestinal milieu through expression of immunologically active proteins.3 Candidates for signalling include chemokine secretion4-11 and antigen presentation to mucosal T cells.12-14 However, no in vivo observations have as yet demonstrated the signalling capability of intestinal epithelial cells.
In vitro experiments have repeatedly demonstrated that epithelial cell lines respond to bacterial pathogens by secreting chemokines.6-9 Moreover, changes in normal intestinal contents vary the expression of chemokines by epithelial cells. For example, butyrate, a short chain fatty acid derived from the fermentation of non-absorbed carbohydrate by intestinal bacteria, regulates expression of macrophage inflammatory protein 2 (MIP-2) and interleukin (IL)-8 in intestinal epithelial cells.10 ,11Epithelial chemokines may also play a role in Crohn's disease and ulcerative colitis.15 ,16 We hypothesised therefore that expression of chemokines by the epithelium in vivo would result in increased leucocyte recruitment into the intestinal mucosa.
MIP-2 is a member of the α chemokine family which also includes human IL-8 (which is not expressed in the mouse), human GROα, rat KC/CINC, and murine KC. MIP-2 was identified as a 6 kDa heparin binding protein secreted by the mouse macrophage cell line RAW 264.7 after stimulation with lipopolysaccharide (LPS).17 ,18 As with IL-8 in humans, MIP-2 is chemotactic for neutrophils and induces localised neutrophil infiltration when injected into the hind footpads of mice.19 MIP-2 is expressed in intestinal epithelial cells only after stimulation with LPS or proinflammatory cytokines. It is not expressed in epithelial cells isolated from healthy intestine in vivo.10 Therefore, inducing expression of MIP-2 in the epithelium enabled us to study the effect of chemokine signalling by enterocytes in vivo. In order to produce MIP-2 continuously from intestinal epithelial cells, we linked an epithelial cell specific promoter from the gene of the fatty acid binding protein of the intestine (Fabpi)20-22 to MIP-2 cDNA and established MIP-2 transgenic mice. Because these mice produce MIP-2 specifically from their intestinal epithelium, we examined the changes in the intestine and evaluated the immunological roles of epithelial cells in vivo.
In the present report, we describe for the first time that chemokine secretion by the intestinal epithelium can orchestrate leucocyte recruitment into the intestine in vivo, demonstrating the active involvement of intestinal epithelial cells in the mucosal immune system.
Materials and methods
GENERATION OF MIP-2 TRANSGENIC MICE
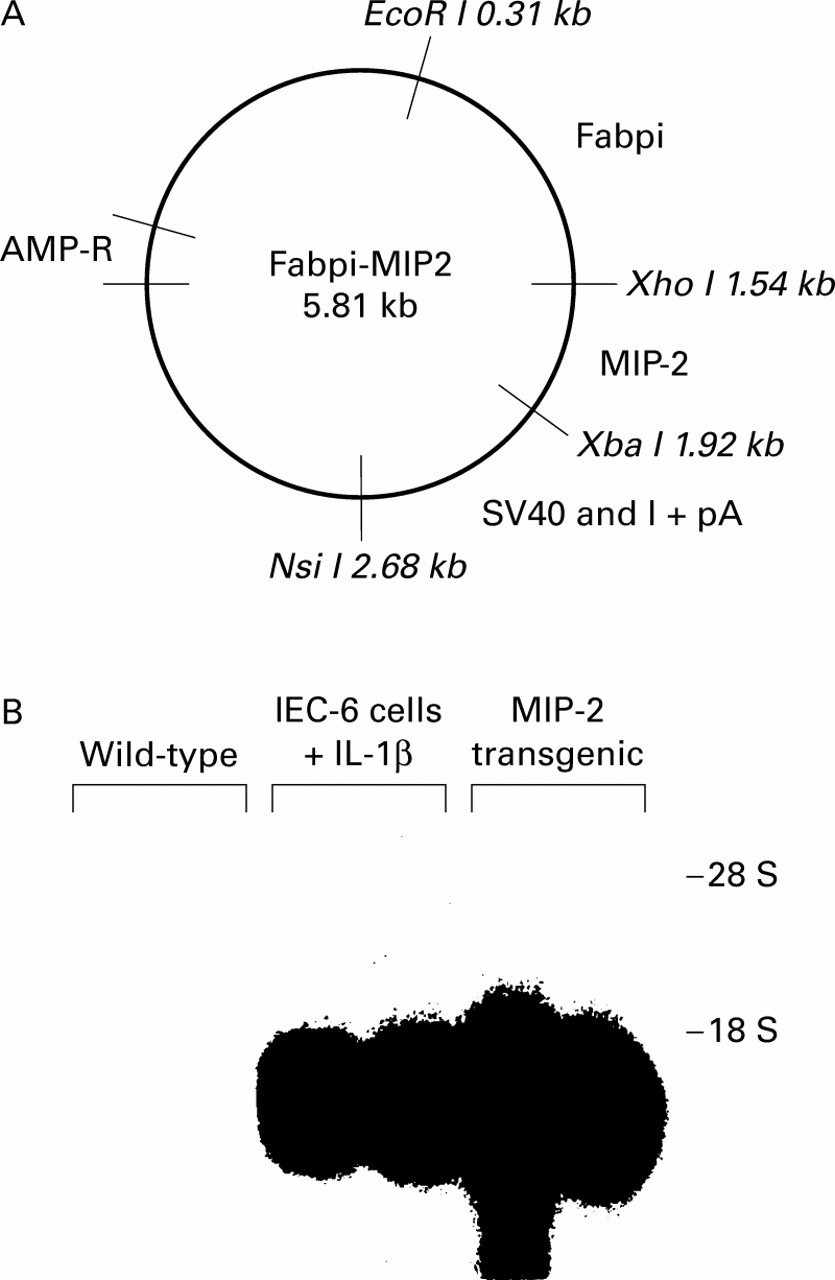
A Fabpi-MIP-2 construct was made using a JvSV plasmid containing the SV40 intron and its polyadenylation site, kindly provided by Dr E Schmidt and Dr P Leder.23 An EcoR I-BamH I fragment of the Fabpi promoter inserted into pUC13, provided by Dr J Gordon, was then cloned into it. This promoter sequence contains the 1200 bases necessary for maximal intestinal epithelial cell expression.20 We then derived a cDNA from a MIP-2 cDNA clone, provided by Chiron Co. (Emeryville, California, USA), using a polymerase chain reaction (PCR) with primers synthesised with a Xho I site (sense: 5′-CACACTCGAGCAGA CTCCAGCCACACTTCA-3′) and an Xba I site (anti-sense: 5′-TGTGTCTAGATTCTT CCGTTGAGGGACAGC-3′). The resulting Xho I-MIP-2-Xba I cDNA was inserted into the Fabpi-SV40 plasmid between the Fabpi promoter and the SV40 intron (fig 1A). The final construct was sequenced, and then the Fabpi-MIP-2-SV40 DNA was excised with EcoR I and Nsi I before oocyte injection performed by Dr A Leiter at the NIH Digestive Diseases Center Core (Boston, Massachusetts, USA). Experiments were approved by the Subcommittee on Research Animal Care of Massachusetts General Hospital, Boston, and by the Animals (scientific procedures) Act 1986, Home Office, London.

Generation of macrophage inflammatory protein 2 (MIP-2) transgenic mice. (A) Diagrammatic representation of the plasmid containing the fatty acid binding protein of the intestine (Fabpi) promoter linked to the MIP-2 gene and the SV40 intron and polyadenylation site. MIP-2 cDNA was derived from a mouse MIP-2 sequence using polymerase chain reaction with primers synthesised with a Xho I site and an Xba I site. (B) Northern blot analysis of MIP-2 mRNA accumulation in the intestinal epithelium of wild-type and transgenic mice. MIP-2 mRNA was detected using an MIP-2 cDNA probe. Epithelial cells were removed from wild-type mice (lanes 1, 2) and from transgenic mice (lanes 5, 6). MIP-2 mRNA derived from IEC-6 cells stimulated with interleukin 1β (IL-1β) was used as a positive control (lanes 3, 4).
MIP-2 SOUTHERN BLOT ANALYSIS
The presence of the transgene in genomic DNA was detected by Southern hybridisation using genomic DNA from the mouse tail. DNA samples were separated in 1% agarose gel and transferred to Gene-Screen Plus membrane (DuPont/NEN, Boston, Massachusetts, USA). Hybridisation was performed using MIP-2 cDNA labelled with [32P] dCTP (DuPont/NEN). All experiments were performed on mice aged 3–6 months.
CELL PREPARATIONS
Each intestinal sample was removed and flushed with Ca2+/Mg2+ free Hanks' balanced salt solution (CMF-HBSS; Sigma Chemical Co., St Louis, Missouri, USA), and all Peyer's patches were removed, prior to isolation of epithelial cells, intraepithelial lymphocytes (IELs), and lamina propria lymphocytes (LPLs). The intestine was then opened longitudinally, washed gently with CMF-HBSS, and cut laterally into small pieces. Each segment was incubated in CMF-HBSS containing 1 mM EDTA and stirred for 20 minutes four times to remove epithelium.24 Cell suspensions were filtered through nylon mesh and centrifuged. The cell pellet, consisting of epithelial cells and IELs, was suspended in RPMI 1640 medium (Sigma) containing 10% fetal calf serum, 10 mMl-glutamine, 0.05 mM 2-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin. For isolating LPLs, the remaining intestine was transferred to flasks containing RPMI 1640 with 90 U/ml collagenase (Sigma) and 2.5 U/ml protease (Sigma), and stirred for 60 minutes in a 37°C water bath. Cell suspensions, containing LPLs, were filtered through nylon mesh and centrifuged. Epithelial cells, IELs, and LPLs were purified using a 45–70% discontinuous Percoll (Sigma) gradient as previously described.24 Cells at the surface were collected as epithelial cells. Contamination with underlying lamina propria cells in epithelial cells was negligible using this technique. Those cells at the interface were collected as IELs and LPLs, and suspended in RPMI. For IELs and LPLs, cell viability was assessed by trypan blue dye exclusion. IELs and LPLs were greater than 85% viable and the stained preparations of IELs and LPLs displayed the characteristic morphology of lymphocytes with minimal epithelial cell contamination.
For the migration assay, as a control, systemic lymphocytes were isolated from the spleen by Ficoll density gradient centrifugation.
MIP-2 mRNA TRANSFER BLOT ANALYSIS, mRNA RT-PCR, AND WESTERN BLOT ANALYSIS
Hybridisation experiments were performed using a MIP-2 cDNA after mRNA from epithelial cells was separated by electrophoresis and transferred onto nylon membranes, as described previously.10 In brief, epithelial cells were transferred to guanidinium isothiocyanate buffer and homogenised. mRNA was extracted by phenol extraction. The samples were separated in 4-morpholinepropanesulphonic acid-formaldehyde agarose gel and transferred to Gene-Screen Plus membrane (DuPont/NEN). Inserts were labelled with [32P] dCTP (DuPont/NEN) and blots were hybridised. Washed blots were autoradiographed at −70°C. mRNA derived from IEC-6 cells stimulated with IL-1β (1 ng/ml) was used as a positive control. To measure mRNA by reverse transcription (RT)-PCR, amplification was performed by specific MIP-2 primers, as previously described.10
MIP-2 protein was identified in homogenates by western blotting as previously reported.10
MYELOPEROXIDASE MEASUREMENTS
CMF-HBSS (25 ml) was perfused transcardially to flush all blood components from the intestinal vasculature. Intestine was then removed and washed gently with CMF-HBSS and snap frozen in liquid N2. The neutrophil infiltration of the intestinal mucosa was examined by staining frozen sections (4 μm) for myeloperoxidase (MPO) utilising the Hanker Yates' reaction.25 Sections were incubated with Hanker Yates' reagent (Sigma): 1 mg/ml in 10 ml of 0.1 M Tris buffer (pH 7.6) containing 1 μl/ml of 3% H2O2 for 30 minutes and counterstained with haematoxylin before examination by light microscopy.
For measurement of functional MPO activity, tissue specimens were weighed and homogenised in 5 ml of 50 mM phosphate buffer (pH 6.0) at 4°C, and centrifuged at 30 000 g for 30 minutes at 4°C. MPO was extracted with 0.5% hexadecyltrimethylammonium bromide (HTAB; Sigma) in 50 mM phosphate buffer (pH 6.0) at 25°C.26 Samples were frozen on dry ice, thawed, and sonicated three times. These were then centrifuged at 30 000 g for 30 minutes. Supernatants were reacted with o-dianisidine dihydrochloride (Sigma) containing 1 μl/ml of 3% H2O2, and MPO activity was assayed spectrophotometrically. Results are expressed per unit weight of intestine. The thickness of the small intestine is greater than in the colon due mainly to greater amounts of muscle and connective tissue. Expression of MPO activity as a proportion of unit weight of whole tissue appears therefore to be greater in colon samples than in the small intestine. Nevertheless, this does not invalidate comparisons of similar organs between transgenic and wild-type mice.
TREATMENT WITH ANTI-MIP-2 ANTIBODY
Goat antimouse MIP-2 antibody (Sigma) was injected intravenously into mice to block the effect of MIP-2 secreted from epithelial cells. On days 1–3, 1 μg/g body weight of antibody was infused into the mouse tail vein as a bolus. Mice were then sacrificed on day 7 and neutrophil recruitment was examined. As a control, an identical volume of phosphate buffered saline was injected into mice as was used to inject antibody in the experimental mice.
IMMUNOHISTOCHEMICAL ANALYSIS
Jejunal mucosa was rapidly frozen in OCT embedding medium using liquid nitrogen cooled isopentane and stored at −70°C until processing. Frozen sections (4 μm in thickness) were cut and air dried. Sections were fixed with cold acetone for 10 minutes and incubated with monoclonal hamster antimouse CD3 antibody (145-2C11). After washing, sections were incubated with biotin conjugated goat antihamster IgG antibody (Vector Laboratories, Peterborough, UK) containing 4% normal mouse serum. Sections were then incubated with avidin peroxidase (Sigma). Peroxidase activity was detected with 3,3′-diaminobenzenzidine-tetra-hydrochloride (Sigma) in Tris HCl containing 0.01% H2O2. Every section was counterstained with haematoxylin before examining by light microscopy. Non-specific staining was evaluated on sections stained without the primary antibody.
To evaluate IEL and LPL numbers in the intestine, at least 10 villi were examined in each section as previously described.24The number of IELs per 100 epithelial cells was counted for each specimen. The number of LPLs per 1 mm2 was determined by image analysis.
ANALYSIS OF LYMPHOCYTE SUBPOPULATIONS
Immunofluorescence staining was performed on IELs and LPLs to examine their subpopulations with monoclonal rat antimouse T cell receptor (TCR)-αβ, and TCR-γδ antibodies (PharMingen, San Diego, California, USA). Positively stained cells were detected with FITC conjugated antirat IgG (Dako, High Wycombe, UK). These antibodies were used at recommended dilutions according to the manufacture's instructions. Single colour flow cytometric analysis was performed using a FACScan (Becton Dickinson, San Jose, California, USA). Lymphocytes were gated on light scatter characteristics, and the percentage of positive cells was calculated in comparison with a negative control consisting of cells stained with FITC conjugated antirat IgG alone.
LYMPHOCYTE MIGRATION ASSAY
Twenty four well Boyden chemotaxis chambers (Becton Dickinson) were used to measure lymphocyte migration in response to MIP-2 compared with that of IL-10, which is known to be chemotactic for IEL.27 The upper and lower chambers were separated by a polyvinylpyrrolidone free polycarbonate membrane with 3 μm pores. IELs and splenocytes (2×105/0.2 ml) from wild-type mice were placed in the upper chambers in the RPMI 1640 media containing 10 U/ml of mouse recombinant IL-2: 0, 0.1, and 1 μg/ml of purified recombinant mouse IL-10 (Sigma) and MIP-2 (Sigma) with or without 10 μg/ml of anti-MIP-2 antibody (Sigma) were added to the lower chambers. All experiments were done in duplicate. After four hours of incubation at 37°C, the number of lymphocytes in the lower chamber was counted.
ANALYSIS OF CXCR-2 EXPRESSION ON LYMPHOCYTES
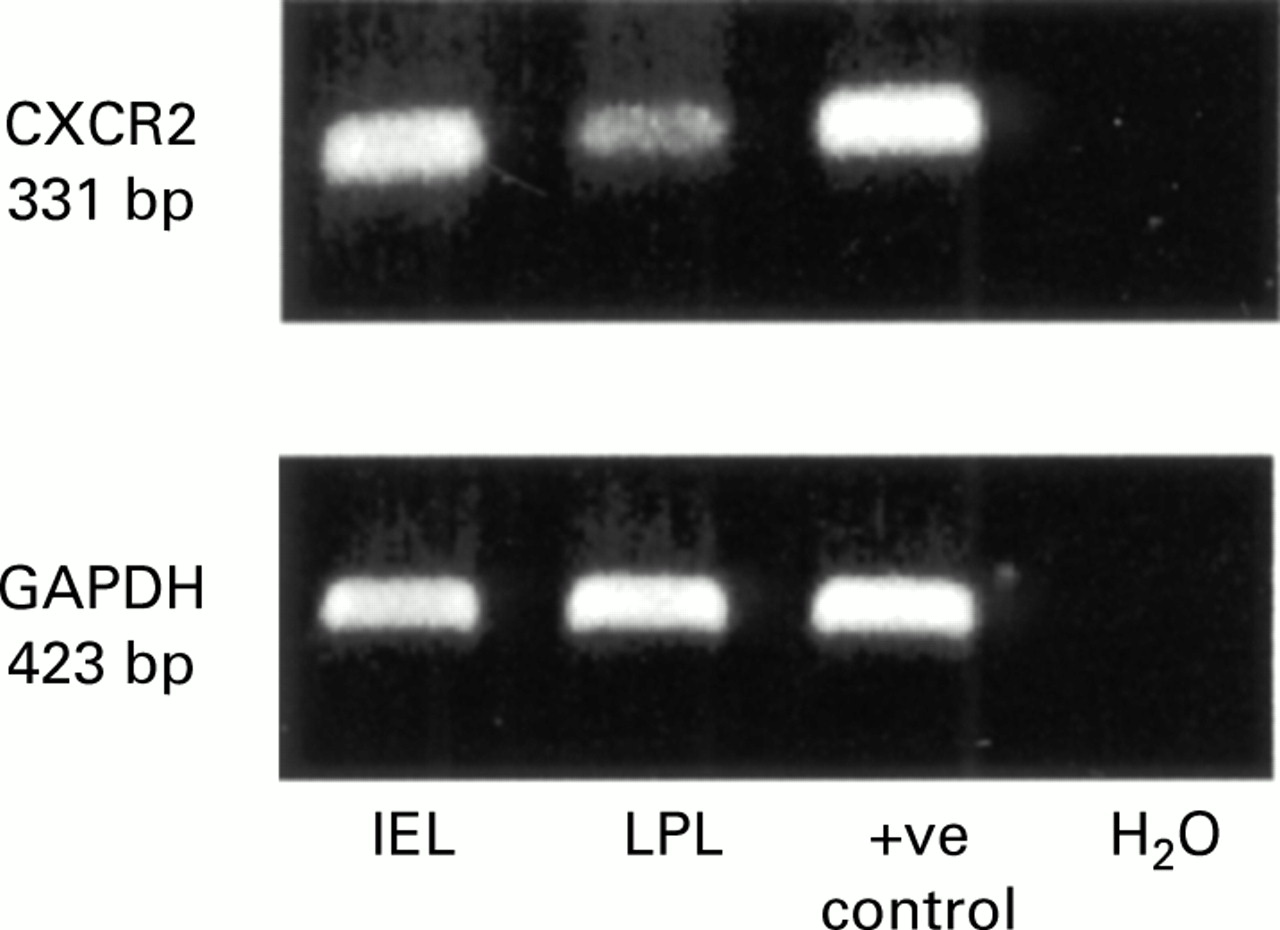
IELs and LPLs from wild-type mice were pooled and homogenised in a solution of guanidinium isothiocyanate buffer and phenol, and then mixed with chloroform, followed by isopropanol precipitation. cDNA was synthesised by RT at 42°C for 30 minutes, from 0.5 μg of total cellular RNA using the GeneAmp RNA PCR kit (Perkin-Elmer, Branchburg, New Jersey, USA) according to the manufacturer's protocol. The thermal cycle was programmed with a hot start at 95°C for five minutes followed by 30 cycle at 95°C for one minute, annealing at 62°C for one minute, followed by extension at 72°C for one minute. PCR primers for mouse CXCR-2 and GAPDH were as follows: CXCR-2 sense: 5′-CT CCTTGGTGATGCTGGTCA-3′; antisense: 5′-AGAATTAAGATGGGCAGGGC-3′, resulting in a 331 bp PCR product. GAPDH sense: 5′-CTGGAAAGCTGTGGCGTGAT-3′; antisense: 5′-CAGGGTTTCTTACTCCTTGG- 3′, which produce a 423 bp PCR product. RT-PCR products were electrophoresed in 1% agarose gel stained with ethidium bromide and visualised through a UV light. RNA extracted from whole spleen was used as a positive control and PCR mixture without RNA but distilled H2O was used as a negative control.
STATISTICAL ANALYSIS
All data were analysed by performing one tailed unpaired Student's t tests; p<0.05 was considered significant.
Results
INTESTINAL FATTY ACID BINDING PROTEIN GENE INDUCES MIP-2 TRANSCRIPTION IN EPITHELIAL CELLS OF TRANSGENIC MICE
An epithelial cell specific promoter from the gene of the Fabpi20 was linked to the cDNA of the MIP-2 gene with an SV40 intron and its polyadenylation site (fig 1A). This DNA construct was injected into C57BL/6 oocytes to generate transgenic mice. Two founder pups containing the transgene were detected by probing genomic DNA from mouse tails with the SV40 polyadenylation sequence. Heterozygous F1 mice were bred from one founder and their offspring were genotyped. MIP-2 transgenic mice developed normally, with no diarrhoea, and revealed no significant differences in body weight compared with wild-type.
Epithelial cells derived from wild-type mice showed no MIP-2 mRNA by either northern blots (fig 1B) or RT-PCR (data not shown). However, both methods detected MIP-2 transcripts in the intestinal epithelium of MIP-2 transgenic mice. MIP-2 protein was present in cytoplasmic lysates from epithelial cells examined in western blots. Again, the production of MIP-2 was observed in transgenic mice but not in wild-type mice (data not shown).
MIP-2 EXPRESSION BY EPITHELIAL CELLS RESULTS IN NEUTROPHIL RECRUITMENT INTO THE SMALL AND LARGE INTESTINE
Because MIP-2 is known as a chemoattractant for neutrophils,19 we examined whether MIP-2 secreted by intestinal epithelial cells increased neutrophil recruitment in vivo. Histological analysis demonstrated that MPO positive neutrophils were increased in the lamina propria of MIP-2 transgenic mice (fig 2A). There were no crypt abscesses in either wild-type or MIP-2 transgenic mice.


Neutrophil recruitment increases in macrophage inflammatory protein 2 (MIP-2) transgenic mice. (A) Neutrophils in the mid jejunum were stained by myeloperoxidase (MPO) using the Hanker Yates' reaction. Magnification ×100. (B) Neutrophil recruitment is demonstrated by total MPO activity. MPO was extracted with 0.5% hexadecyltrimethylammonium bromide. Total MPO activity is expressed per unit weight of tissue derived from wild-type MIP-2 transgenic mice. Expression of MPO activity as a proportion of unit per weight of whole tissue appeared to be smaller in the small intestine than in the colon due to the heavier weight per unit length of the small intestine. Data are presented as mean (SD) of eight animals for each group. **p<0.01 versus wild-type.
We also quantified invasion of neutrophils by measuring MPO activity in full thickness intestine (fig 2B). Total MPO activity per unit weight of intestine was significantly elevated in MIP-2 transgenic mice in the small intestine, caecum, and proximal colon (p<0.01). Total MPO activity was not increased in tissues where the Fabpi promoter was inactive: MPO activity in the distal colon, liver, and spleen of transgenic mice was not significantly different from wild-type mice. To confirm that neutrophil recruitment in transgenic mice was due to MIP-2 production, mice were treated with an anti-MIP-2 antibody and neutrophil recruitment was examined. The increase in neutrophils into the intestine was prevented by anti-MIP-2 antibody treatment in transgenic mice (fig3).

Neutrophil recruitment due to macrophage inflammatory protein 2 (MIP-2) secretion was blocked with an anti-MIP-2 antibody. Anti-MIP-2 antibody was injected intravenously seven days and four days before examining the mice. Neutrophil recruitment was examined by measuring myeloperoxidase (MPO) activity per unit weight of tissue samples derived from wild-type and MIP-2 transgenic mice. Ab (−), mice received phosphate buffered saline only; Ab (+), mice received anti-MIP-2 antibody treatment. Data are presented as mean (SD) of eight animals for each group. **p<0.01.
MIP-2 EXPRESSION BY EPITHELIAL CELLS INDUCES AN INCREASE IN INTRAEPITHELIAL AND LAMINA PROPRIA LYMPHOCYTES
Lymphocytes were counted by staining CD3 positive cells in intestinal sections. Greater numbers of CD3 positive cells were present in the intestinal samples taken from MIP-2 transgenic mice (fig 4A). Numbers of IELs and LPLs were both significantly increased in MIP-2 transgenic mice compared with age matched wild-type mice (p<0.01 and p<0.05, respectively) (fig 4B).


Intraepithelial lymphocyte (IEL) and lamina propria lymphocyte (LPL) numbers were increased in the small intestine of macrophage inflammatory protein 2 (MIP-2) transgenic mice. (A) Lymphocytes were detected with an anti-CD3 Ab (145-2C11). Magnification ×100. (B) IELs were expressed as number of CD3 positive cells in the epithelium per 100 epithelial cells. LPLs were expressed as CD3 positive cells in the lamina propria per mm2. Data are presented as mean (SD) of six animals for each group. *p<0.05, **p<0.01.
Because infiltration of IELs and LPLs were observed in MIP-2 transgenic mice, we examined the TCR subpopulations of these lymphocytes using FACScan (table 1). In MIP-2 transgenic mice, the subpopulation of TCR-αβ was significantly increased in IELs (p<0.05). Although there was no significant increase in TCR-αβ positive cells in LPLs, the subpopulation of TCR-γδ positive cells was significantly decreased in LPLs (p<0.05).
T cell receptor (TCR)-αβ positive T cells were infiltrated into the mucosa in MIP-2 transgenic mice
MIP-2 CHEMOATTRACTS MUCOSAL LYMPHOCYTES IN VITRO
To determine if MIP-2 induced lymphocyte infiltration, a migration assay was performed in vitro. The responses of mouse IELs and splenocytes to IL-10 (as a positive control) corresponded to those observed in human lymphocytes.27 Approximately 4×104 IELs and splenocytes migrated to a lower chamber spontaneously. However, this increased twofold with 1 μg/ml of MIP-2 (fig 5). The migration response of IELs and splenocytes to both MIP-2 and IL-10 depended on the cytokine concentration. Anti-MIP-2 antibody blocked IEL and splenocyte migration responses to MIP-2.

Macrophage inflammatory protein 2 (MIP-2) attracts lymphocytes, including intraepithelial lymphocytes (IEL) in vitro. Lymphocyte migration in response to interleukin 10 (IL-10) and MIP-2 in the absence or presence of anti-MIP-2 antibody (MIP-2/Ab) was studied using the Boyden chamber. Lymphocytes (2×105/0.2 ml) were placed in the upper chambers, and migrated cells were counted after four hours of incubation at 37°C. Each experiment was done in duplicate. Data are presented as mean (SEM) of six animals for each group. Significant differences were evaluated in assays between supernatants with or without anti-MIP-2 antibody. **p<0.01.
Since mucosal lymphocytes were attracted by MIP-2 in vitro, we examined expression of the MIP-2 receptor, CXCR-2, on these cells by RT-PCR. CXCR-2 mRNA was confirmed in IELs and LPLs, and expression of CXCR-2 was relatively higher in IELs than in LPLs (fig 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of CXCR-2 expression on lymphocytes from wild-type mice. Agarose gel stained with ethidium bromide showing reverse transcription-polymerase chain reaction (RT-PCR) products for CXCR-2 (331 bp) and GAPDH (423 bp). Expression of CXCR-2 and GAPDH mRNA was confirmed in intraepithelial lymphocytes (IEL) and lamina propria lymphocytes (LPL) isolated from wild-type mice. RNA extracted from whole spleen was used as a positive control and PCR mixture without RNA but distilled H2O was used as a negative control. Representative of three independent experiments.
Discussion
The primary function of the intestinal epithelial cells is to absorb nutrients while providing a physical and immunological barrier to the external world. Nevertheless, epithelial cells are in direct contact with the intestinal milieu and they produce several chemokines influenced by the luminal contents that include bacteria, bacterial products, and chemical stimuli.6-11 These findings suggest that intestinal epithelial cells may actively alter mucosal immune responses. Previous experiments in lung have shown neutrophils recruited by chemokines, possibly released by epithelial cells using an external stimulus, α-quartz.28 Although careful experiments demonstrate chemokine specificity by blocking with antibodies, these experiments do not exclude the possibility that the same external stimulus also activated non-epithelial cells in the tissue.
We therefore established transgenic mice whose epithelial cells secrete MIP-2 by using an epithelial cell specific promoter, Fabpi. Because this promoter is active only in epithelial cells,20-22the technique allowed us to examine the effect of epithelial cells directly. In this study, we demonstrated that chemokine expression from intestinal epithelial cells alters the mucosal immune system in vivo. Invasion of neutrophils into the lamina propria was significantly increased in MIP-2 transgenic mice compared with wild-type mice. The increase in neutrophils was greater in the small intestine and proximal colon than in the distal colon or rectum. This observation corresponds to the known activity of the Fabpi promoter that we used to regulate expression of MIP-2 in transgenic mice.20 The Fabpi promoter is strongest in the small intestinal epithelium and becomes weaker from the proximal to the distal colon; the distribution of neutrophil recruitment corresponded to this expression. Similarly, MPO activities in the liver and spleen of transgenic mice (where the Fabpi promoter is inactive) were not significantly different from wild-type mice. We also demonstrated that the increased neutrophil infiltration into the intestine was blocked by an anti-MIP-2 antibody (fig 3). This demonstrated that the increased neutrophil infiltration into the intestine in the MIP-2 transgenic mice was directly due to MIP-2 and not to an unrelated effect of establishing a strain of transgenic mice.
Comparisons of MPO activity are valid when comparing wild-type and transgenic animals over the same area of intestine. However, it is not possible to extrapolate variations in neutrophil recruitment from MPO activity per weight of tissue between different parts of the intestine. This is because the amount of serosal tissue varies considerably between different sections. The weight of upper small intestine per unit length is much greater than the large intestine. Moreover, different preparations of large intestine preserve different amounts of serosal tissue per unit length. Thus differences occur when comparing whole large intestine with individual segments.
Increases in IEL numbers occur in disease states, including coeliac disease and cows' milk sensitive enteropathy.29 ,30 A number of factors have been shown to regulate IEL populations, including αEβ7 expression on the lymphocyte surface and MAdCAM-1 expression on endothelial cells.31 In vitro studies have shown human IELs to be attracted by various cytokines, including IL-10 and the chemokines IL-8, RANTES, MCP-1, and MIP-1.27 The present study showed that MIP-2 attracts lymphocytes, particularly TCR-αβ positive cells, into the intestinal mucosa. Since most peripheral T cells are TCR-αβ, we suspect that these T cells have migrated from peripheral blood rather than proliferating within intestinal epithelium. We confirmed in vitro that MIP-2 was chemotactic for mucosal lymphocytes because it increased the populations of IELs in the Boyden chamber in a dose dependent manner. Thus not only can epithelial cells orchestrate neutrophil invasion through chemokine secretion but they can also regulate lymphocyte populations. The only receptor in the mouse described to date that interacts with MIP-2 is the CXCR-2. Expression of this receptor was confirmed on IELs and LPLs by RT-PCR (fig 6). Interestingly, expression of CXCR-2 mRNA was relatively higher in IELs than in LPLs. A recent study suggested that interferon γ maintains expression of CXCR-2 on lymphocytes.32 It is possible that interferon γ influences lymphocytes in the epithelial compartment more than in the lamina propria, resulting in higher expression of CXCR-2 mRNA in IELs (fig 6).
These experiments were designed to test the hypothesis that epithelial cell gene expression markedly affects the mucosal immune system. The work was not intended to examine when a model of inflammatory bowel disease could be developed from unrestricted chemokine secretion by the epithelium. While the epithelial cell may play a role in generation of inflammation in such conditions it seems unlikely that it is the only important cell in its initiation. Tissue remodelling is an important component of inflammatory bowel disease. Such changes are induced by factors that include transforming growth factor β, tumour necrosis factor α, and matrix metalloproteinases. No direct attempt was made to alter expression of these proteins. Recruitment of leucocytes could in theory indirectly alter the concentration of some of these proteins but without significant T cell activation such effects would be small.
The transgenic mice described in this report exhibited neutrophil and lymphocyte infiltration into the mucosa due to a single chemokine expressed by the epithelial cells. Mice showed no symptoms of diarrhoea or weight loss. We would not expect that a single chemokine secreted from the epithelium would be sufficient to cause a disease-like status. In inflammation, many different chemokines are expressed by the intestine at different times following epithelial cell stimulation.33 Such a phenomenon would increase the complexity of signals on leucocytes. Nevertheless, this article shows for the first time in vivo that a chemokine secreted by epithelial cells directly alters the mucosal immune system. Epithelial cells can therefore regulate inflammatory cells in the intestine and may do so in response to stimuli from the intestinal lumen by secreting chemokines.
Acknowledgments
We thank Drs E. Schmidt and P Leder for the JvSV plasmid; Dr J Gordon for Fabpi promoter; and Dr A Leiter for oocyte injection. We are grateful to Professor TT MacDonald for his comments on the manuscript. This work was supported by the following grants from the NIH: AI43472, DK47753, DK43351, DK40561; the Joint Research Board of St Bartholomew's Hospital, London, UK; the Uehara Memorial Foundation, Tokyo, Japan; and the Medical Research Council.
Abbreviations used in this paper
- CMF-HBSS
- Ca2+/Mg2+free Hanks' balanced salt solution
- Fabpi
- fatty acid binding protein of the intestine
- IEL
- intraepithelial lymphocyte
- IL
- interleukin
- LPL
- lamina propria lymphocyte
- LPS
- lipopolysaccharide
- MIP-2
- macrophage inflammatory protein 2
- MPO
- myeloperoxidase
- RT-PCR
- reverse transcription-polymerase chain reaction
- TCR
- T cell receptor