Article Text

Abstract
Background—Published data are contradictory about the importance of K-ras mutations in advanced tumours and are not available for early cancers.
Aims—To establish whether specific K-ras mutations are prognostic markers in early stage colorectal adenocarcinoma.
Methods—The presence of K-ras exon 1 mutations were correlated with tumour recurrence in two groups of patients: group 1 was a consecutive series of patients with resected colorectal adenocarcinoma at low risk of recurrence; group 2 were patients referred for chemotherapy after relapse of previously resected early stage tumours. K-ras mutations were detected by direct sequencing of whole tissue samples in all patients and in some, the leading edge and centre of the tumour were also microdissected out individually and sequenced.
Results—Mutations were present in 26 (26.5%) of 98 patients in group 1; 14 patients developed a recurrence, four (28.5%) of whom had a K-ras mutation. Seventy nine patients have not developed tumour recurrence, 22 (28%) of whom had a mutation (p=0.84). K-ras mutations were present in five of 14 patients in group 2. Microdissection did not increase the number of mutations detected.
Conclusions—Individual K-ras genotypes are distributed homogeneously throughout early stage colorectal adenocarcinomas, but detection of a mutation has no apparent prognostic value.
- Dukes’ stage
- colorectal cancer
- K-ras
- microdissection
Statistics from Altmetric.com
Between 10 and 75% of patients with colorectal cancer, depending on site and stage, develop recurrent disease despite apparently curative surgery.1 However, effective chemotherapy following surgery is becoming available.2 ,3Novel molecular treatments, such as antisense, may provide further advances. It is, therefore, becoming increasingly important to identify patients who would benefit from such adjuvant treatment. Molecular biological techniques may complement existing clinical and histological parameters, particularly as colorectal cancer is postulated to develop following the acquisition of multiple genetic changes.4Mutations of the ras gene have been observed in a number of human malignancies. The ras gene product has a key role in controlling cell growth and differentiation through its intrinsic GTPase activity.5 It also seems to interact with pathways that regulate programmed cell death,6 with other oncogenes,7 and may also play a part in drug resistance mechanisms.8 ,9 K-ras, one of three humanras genes recognised, is reported to be mutated in about half of all patients with colorectal cancers.4 The mutations are usually found at specific sites and the unique sequence of the mutated mRNA produced becomes an attractive target against which to direct antisense treatment. Indeed, targeting K-ras mutations in colon and lung tumour cell lines in vitro inhibits their growth.10 ,11 Although it has been established that mutations develop in the K-ras gene early in the progression from adenoma to carcinoma,4 their importance in prognosis of established colorectal cancer is still unclear. On the one hand, at least seven studies12-18 have suggested that the presence of a K-ras mutation has prognostic importance. Patterns of recurrence of colorectal tumours have even been attributed to specific nucleotide base mutations15-17 although, the mechanism by which such an effect may be exerted is as yet unexplained. However, at least a further four studies suggest that K-ras mutations have no prognostic importance.19-22 Similarly, studies examining the association between mutations and outcome, either after surgery to the primary tumour, in recurrent disease23 or after chemotherapy24 come to confusingly contradictory conclusions. Reports also differ as to whether an association exists between the presence of K-ras mutations and histological or demographic factors, such as age or sex.16 ,25 However, interpretation of data from other studies may be complicated by a number of factors. Firstly, all colorectal tumours have frequently been considered as a single entity. It is now recognised that the mutations in the K-ras gene may differ according to the patient’s geographic origin,26 the type of tumour27 and may be less common in tumours arising in patients with ulcerative colitis.28 In addition, many studies include only small numbers of tumours. Secondly, varying methods may also influence the frequency at which mutations have been detected. Only a few studies have used definitive, direct sequencing of the gene. In this study we looked at patients thought to have Dukes’ stage A colorectal adenocarcinoma, tumours with a generally better prognosis and a relapse rate of about 10%. Previous studies of the influence of K-ras on recurrence have included very few patients, if any, with Dukes’ stage A cancers. However, we postulated that if K-ras plays an important role in tumour progression, it’s influence would be clear in this group of patients in whom relapse is uncommon. Much of the data published to date refer to patients with relatively advanced tumours of significantly poorer prognosis. Against this background, the aims of our study were fourfold. Firstly, to establish whether the type of mutations found in a series of consecutive patients with good prognosis colorectal cancer in a British population were similar to those described for populations elsewhere. Secondly, to identify mutations which might be appropriate targets for molecular adjuvant treatments. Thirdly, to determine whether screening whole tissue samples from colorectal adenocarcinomas is a useful technique for detecting the presence of a ras mutation accurately. Finally, to attempt to clarify conflicting reports regarding the relevance of specific ras mutations as prognostic markers in primary colorectal cancer.
Methods
PATIENTS
Patients for the first part of this study were selected from the database held at the pathology departments of the Medway Hospital and the Kent and Sussex Hospital if they had undergone surgery for a Dukes’ stage A colo- rectal carcinoma between 1987 and 1994. Hospital notes of all patients were reviewed. To verify the initial staging, notes were scrutinised for evidence of metastatic spread of tumours at the time of surgery including, where available, radiological reports of chest x ray films and peri-operative liver imaging. Evidence of relapse of the disease after surgery was obtained from hospital follow up notes. All general practitioners were contacted and asked about the patients’ current status and whether there had, at any time, been evidence of relapse. If patients had died other than at the hospital where the original operation took place, cause of death was sought from the general practitioner, the hospital where they died and details of the death certificate were obtained from the Thames Cancer Registry. A second group of patients were recruited from a number of different hospitals. These patients all underwent an apparently curative resection for a Dukes’ stage A colorectal cancer before 1995. Subsequently, they all developed biopsy confirmed tumour recurrence and were all referred for palliative chemotherapy to a single oncology unit at the Royal Marsden Hospital.
HISTOLOGY
The original histological material from the resection specimens was reviewed by an experienced pathologist and graded for stage by Dukes’ classification and Astler–Coller’s modification of Dukes’ staging. Other recognised pathological risk factors for progressive disease were recorded, including tumour differentiation, mucoid subtype, vascular invasion, the degree of a host lymphocyte response, and whether the tumour margin was “pushing” or “infiltrating”. The reviewed material was also assessed for the proportion of the mucosa and the total tissue replaced by tumour in the blocks chosen for molecular analysis. The pathologist was unaware of the clinical outcome of the patients or of their K-ras status.
ANALYSIS FOR K-ras MUTATIONS
DNA extraction
All pre-PCR tissue was handled in an environment free of PCR products. All samples were coded and the investigator was blinded to all patient clinical details. For each patient, two 12 μm sections from the formalin fixed, paraffin wax representative tumour blocks were placed in a sterile tube. In all patients who relapsed and in a few long term, relapse-free survivors, where sufficient material was available, samples were also prepared for microdissection. Three 15 μm paraffin wax sections were cut from each case onto double-sided clear adhesive tape adhered to glass slides. They were lightly stained with toluene blue to permit visualisation of tissue components. Under a dissecting microscope a fine scalpel blade was used to cut around the invasive edge of the tumour defined for the purposes of this study as the 1 mm leading edge of tumour. The same area of leading edge from the three slides was peeled off and placed in a sterile tube. Using a clean scalpel blade, this same process was repeated for central tumour tissue at least 2 mm away from the leading edge. Five micron sections taken immediately before and after the 15 μm sections were stained with haematoxylin and eosin. These were used to aid the identification of the relevant areas of tumour and to ensure that lesional tissue was present on all three 15 μm sections used for microdissection. De-paraffinised tissue was recovered by a 15 minute incubation with xylene followed by centrifugation for five minutes at 14 000 rpm. This was repeated a second time. The tissue pellet was then washed twice in absolute ethanol followed by two washes in phosphate buffered saline. The pellet was incubated with 10 pellet volumes (approximately 500 μl) of lysis buffer (0.32 M sucrose, 10 mM Tris-HCl, 1% (v/v) Triton X-100) and 0.2 volumes of proteinase K (final concentration 400 μg/ml) for two to three days at 37°C. DNA was phenol-chloroform extracted and precipitated in ethanol using conventional techniques.29 The resulting DNA pellet was resuspended in 50 μl TE buffer, pH 7.4, (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, pH 8.0). DNA samples were stored at −20°C.
PCR AMPLIFICATION AND DNA SEQUENCING
The first exon of K-ras-2 was amplified by PCR using primers designed to avoid amplification of the K-raspseudogene.30 ,31 The primers used were 5′-ATTATAAGGCCTGCTGAAAATG- ACTGA-3′ (upstream primer) and 5′- ATATGCATATTAAAACAAGATTTACCT- CTA -3′ (downstream primer) giving a 155 base pair product. Amplification was performed using a touchdown PCR technique32 from 63°C to 53°C over 10 cycles, followed by 30 cycles at 94°C, 53°C and 72°C. A placental DNA positive control, DNA from a colon cell line (SW480) containing an homozygously mutated valine codon 12 mutation, and a negative water control were included with each batch of samples. The cell line control was sequenced on each occasion. PCR products were electrophoresed on a 2% 1× TAE low melting point agarose gel (Gibco, Life Technologies) and extracted from the agar using Wizard PCR preps DNA purification system (Promega Corporation). PCR products were sequenced using dideoxynucleotide termination (Sequenase version 2, USB) using the downstream primer in the first instance. Sequenced samples were electrophoresed on an 8% polyacrylamide/7 M urea gel, transferred to filter paper, dried and autoradiographed for 1–7 days. If a mutation was detected, it was confirmed by amplification and sequencing of a fresh DNA sample using the upstream primer. Any sequences which proved difficult to read were re-amplified and re-sequenced. All sequencing gels were read independently by two observers who were unaware of the patients’ clinical details.
STATISTICAL METHODS
Recurrence is a time dependent variable, so to avoid bias from different lengths of follow up, survival curves were generated using the Kaplan-Meier product-limit method. The log rank test was used to evaluate differences in relapse-free survival curves. χ2tests were used for comparison of categorical data.
Results
One hundred and nine consecutive patients were identified from the database as having Dukes’ stage A colorectal cancer treated by surgical resection between 1987 and 1994 (table 1). Eleven patients were excluded using pre-determined criteria. Five patients had one or more other synchronous colonic tumours at their resection for a Dukes’ stage A tumour, one patient had had a previous gastric cancer and five patients did not have a primary colo- rectal cancer in the material provided for review. Ninety eight patients, therefore, were suitable for analysis. This included 45 men, age range 45–90 years, median 69 years, and 53 women, age range 33–89 years, median 70 years. No patient received adjuvant treatment after surgery.
Patient characteristics
HISTOLOGY
Review of the 98 patients originally believed to have Dukes’ stage A tumour, confirmed that staging in 85 patients (table 1). Twelve had a Dukes’ stage B tumour. One could not be classified accurately from the reviewed material because of insufficient architectural preservation. When classified using Astler–Coller’s modification, 22 patients were stage A, 63 stage B1 and 12 stage B2. One patient could not be classified accurately. No resection margins were involved with tumour, no lymph nodes were involved and no patient had vascular invasion.
PERI-OPERATIVE ASSESSMENT FOR DISSEMINATED DISEASE
Of the 14 patients who relapsed with disease, only one did not have a peri-operative chest x ray film. There was no evidence of disseminated disease on the other radiographs. Ten of the 14 underwent liver imaging. The results were available in nine and showed no evidence of metastases. All 14 patients were thought to have complete macroscopic clearance of disease by the surgeon. The rate of relapse in our study group concurs with rates previously reported for patients with tumours of this stage.1 Therefore, it seems fair to conclude that these patients were essentially correctly staged at presentation.
K-ras STATUS
PCR amplification and DNA sequencing enabled detection of heterozygous mutations when that mutation was present in 5% of the DNA used (fig 1). The relative amounts of non-neoplastic and neoplastic tissue within each block selected for analysis did not seem to determine whether a mutation was detected (table 2). A sequence was obtained in all 98 patients. A mutation was detected in 26 (26.5%) tumours. Twenty patients had a mutation of codon 12, six patients had a mutation of codon 13 and 72 patients had only wild type DNA (fig 2). No patient had multiple mutations. The presence of a mutation was not related to the patients’ sex, age or tumour site (p>0.05).

: Polyacrylamide sequencing gel using the downstream primer showing serial dilution of wild type K-ras DNA with DNA containing a heterozygous codon 13 guanine to adenine mutation. The arrow indicates the level of an additional band which can be seen in the lanes marked “T” when 5% or more of the DNA contained the codon 13 mutation. This extra band is not seen when 100% wild type DNA is sequenced. Wild type DNA was extracted from HT29 cells and mutated DNA from LoVo cells.
Spectrum of K-ras mutations detected and number of patients with mutations who relapsed
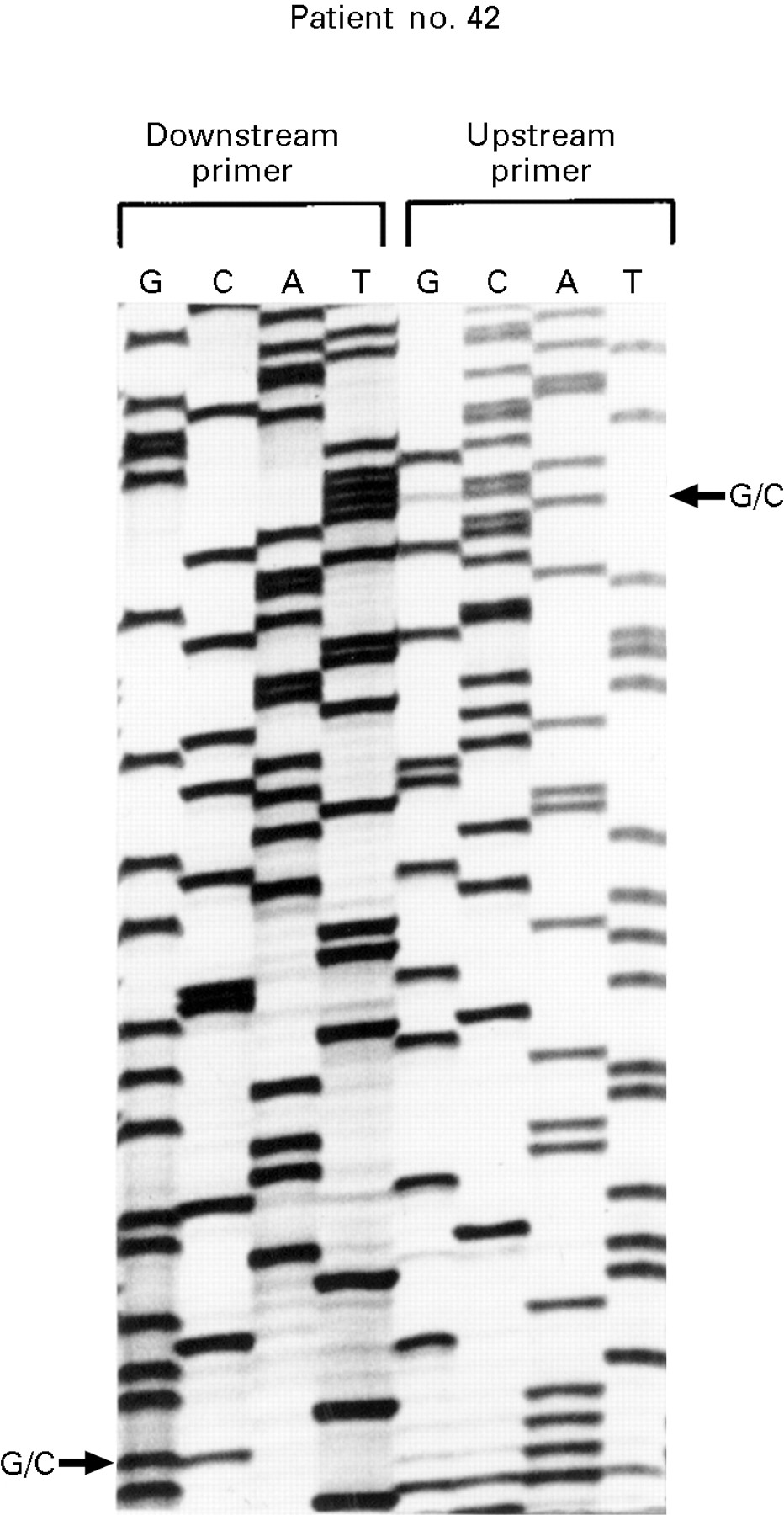
: Polyacrylamide sequencing gel showing K-ras exon 1 gene sequence from patient 42. The DNA has been sequenced using both the upstream and downstream primers. An arrow indicates a G to C mutation at position 2 of codon 12 giving an amino acid change from glycine to alanine.
K-ras AND HISTOLOGY
The presence of any mutation was not associated with Dukes’ staging or Astler–Coller’s modification (table 3). Neither was the presence of a mutation associated with the other histological parameters recorded, degree of differentiation of the tumour, mucoid subtype, degree of host lymphocyte response, or whether the tumour margins were “pushing” or “infiltrating” (p>0.05).
Correlation of the presence of a ras mutation with clinical and histological findings
K-ras, RELAPSE AND FOLLOW UP
Five patients died in the peri-operative period from complications of their surgery. Four patients died from unrelated causes 8.5, 28, 32, and 50 months after surgery. Fourteen patients relapsed with recurrent tumours. The remaining 79 patients have no evidence of recurrence after a median 44 months of follow up. Of the 14 patients who developed recurrence, four (28.5%) had a K-ras mutation. In three patients, this was in codon 12, two aspartate and one serine. One was an aspartate mutation in codon 13. Eleven of the 14 died of their recurrent bowel cancer. These deaths occurred between 8 and 66.5 months from the date of the original surgery (median 31.5 months). Three other patients developed a recurrence which was treated surgically at 40, 47 and 50 months after their first operation. These three patients had wild type K-ras DNA. There was no significant difference in the median time to relapse in the four patients with a K-rasmutation—24 months (range 8–66.5 months) compared with those without a mutation, 30 months, (range 11–61 months). Twenty two (28%) of 79 patients also had a K-ras mutation but remain free of recurrent disease (the five patients who died peri-operatively are not included). For the whole cohort, one and three year relapse-free survival was 92% and 87.1%, respectively, for patients with a K-ras mutation and 95.5% and 86.5% for patients without a mutation. So, there was no detectable differences in relapse-free survival between patients with or without a K-ras mutation (p=0.84, log rank test).
SECOND GROUP OF PATIENTS
In view of the small numbers of patients who relapsed in the consecutive cohort of 98 patients, the finding that K-rasstatus is not important could be due to the inability to detect a real difference. Therefore, 14 more tumours from patients who originally had apparent complete resection of histologically verified Dukes’ stage A tumours but who later developed a histologically proved recurrence, were obtained. The Astler–Coller’s stage of the tumours was A in four patients, B1 in eight and B2 in two. Patients relapsed between 8 and 55 months after their original surgery (median 34 months). Sequencing whole tissue samples from these 14 patients detected wild type genotypes in 10 patients, one patient with an aspartate and another with an arginine codon 12 mutation and two patients with an aspartate codon 13 mutation. Thus, the rate of mutation in this second group of patients who relapsed was similar to that in the group of patients who relapsed in the consecutive series.
RESULTS OF MICRODISSECTION
Despite the sensitivity of the PCR sequencing technique used (fig1), the possibility of error due to failure to dissect away surrounding stroma and hence mask the presence of a mutation with non-neoplastic cells carrying a wild type genotype was considered. Therefore, in 18 patients who developed recurrence and in five long term survivors without evidence of recurrence, the leading edge of the tumour was dissected out and sequenced separately. Similarly, the central core of the tumour was dissected out and sequenced separately (fig 3). A sequence was obtained in all 23 whole tissue samples (five mutated, 18 wild type) (table 4). A sequence was also obtained in 22 of the microdissected leading tumour edges and 20 of the microdissected samples from the tumour centres. In every patient, the same genotype was found in both microdissected samples, those taken from the leading edge or from the centre. In no case did a mutation appear in microdissected samples which was not detected in the whole tissue sample. These findings suggest, firstly, that colorectal tumours are homogeneous for K-ras genotype, and, secondly, that using PCR amplification followed by direct sequencing of whole tissue samples is adequate and that microdissection of tumours is not necessary. In one patient, a mutation was detected that was present in the whole tissue sample, but was not present in the microdissected samples for that tumour. The presence of K-ras mutations have been described in normal epithelium around tumours and shown not to correlate with the presence of mutations within tumours.33This is probably the case in this patient. This effect, however, seems to be rare and it seems unlikely that the detection of occasional extra mutations from the epithelium around the tumour would have affected our overall result significantly.

{kind=link}
{kind=link}
{kind=link}
: Toluene blue stained 15 μm paraffin wax section of a Dukes’ stage A, Astler–Coller’s modification B1 colorectal adenocarcinoma with the leading edge (1) and the core (2) of the tumour removed by microdissection.
Summary of microdissection results in 23 patients
Discussion
This study has looked at a group of patients with colorectal cancer of good prognosis. It used the definitive technique of direct sequencing to identify the presence of a mutation. We have shown that there does not seem to be any relation between the presence of a K-ras mutation and clinical or histological parameters or cancer recurrence. We have also shown that microdissection of tumours is not necessary when our technique of direct sequencing is used. In addition, our results suggest that cells within colorectal adenocarcinomas are essentially homogeneous for the K-rasgenotype. Our results cannot support those of previous studies which have suggested that the likelihood of development of K-rasmutations depends on the tumour site within the lower gastrointestinal tract, is sex specific or is related to a patient’s age. This study also failed to support earlier suggestions that specific mutations of K-ras predict biological behaviour of tumours and are of prognostic importance. However, G to A or T nucleotide changes, particularly at the second base of the codon, are the most common in this population, as seen elsewhere in patients with more advanced tumours. The striking feature of previously published data on K-ras in colorectal cancer is the remarkably contradictory conclusions reached. There are several possible reasons why we too might have reached different conclusions to other studies. The first area of potential bias is our patient selection and follow up. Our patient groups represented individuals thought to have a low risk of recurrence. Initial staging may have missed metastatic disease at surgery in some individuals, but staging as performed in these patients represents currently accepted practice in the United Kingdom. As for recurrence, this study has been able to identify outcome of patients from this group with confidence because of the co-operation of well informed general practitioners.
Secondly, it is inevitable that because of the expected low relapse rate, the small numbers with a higher stage of tumour and varying lengths of follow up there is a risk of a type II statistical error. In addition, data from a group where few events occur, of necessity, produce wide confidence intervals. Some of these problems can be compensated for by using the log rank test to analyse the data, as we have done. In addition, the validity of our findings can be assessed by comparing our results with data from other studies. Large studies have shown that patients with Dukes’ stage A tumours have a five year event free survival of 90%1 and Dukes’ stage B tumours a three year event free survival of 79%3 and a five year overall survival of around 75%.34 Results from our series are entirely in keeping with these figures. However, we did not only rely on these statistical devices. We sought out more Dukes’ stage A tumours in patients who after their resection had developed a histologically proved tumour recurrence. We found K-rasmutations in these patients with an identical frequency to that in our consecutive cohort.
Thirdly, it is also unlikely that the molecular screening methods used were insufficiently sensitive to find an association. The PCR based sequencing method detects the presence of an heterozygous mutation clearly when as little as 5% of the genomic DNA contains mutated K-ras (fig 1). We are able to detect mutations when less than 25% of the block is replaced by tumour. Our data show the same proportion of mutations is detected from these blocks as from blocks where all tissue is replaced by tumour. However, to confirm the validity of the sequences obtained from whole tissue samples, we dissected out specific areas in about one quarter of this series as genotypic heterogeneity within the tumour could be a reason for failing to detect an association between K-ras mutation and relapse. However, as it is generally accepted that K-ras mutations are acquired early in the progression from adenoma to carcinoma, it has always seemed improbable that heterogeneity is important and K-ras status in tumours should be homogeneous. But, to the best of our knowledge, this has not been examined before. As we expected, with these data, we have shown that not only are tumours homogeneous for the K-ras genotype, but also that microdissection is not necessary.
This study raises intriguing questions. In particular, why have published results conflicted to such a degree? Some of the reasons for this are no doubt methodological. Complete surgical excision of high risk tumours which present early may also contribute. But it may be that as yet, investigators are not asking the right questions. For, it may be that the presence of a mutation in itself is insufficient and it may only be a marker of exposure to a carcinogen. The biological behaviour of that tumour may depend on whether that mutated allele is expressed in preference to the wild type allele. No large studies have looked at this. In addition, what is happening in tumours which progress where no mutated K-ras allele is detected? We did not look for for the presence of K-ras codon 61 or N-ras mutations which together provide about 10% ofras mutations in colorectal cancer.4 Even had we done so, it is likely that 16 or 17 patients who relapsed would have had only wild type ras. So, in these patients was there no contribution at all from the ras pathway to disease progression or were other genes upstream or downstream within the pathway altered by mutation thereby effectively upregulating K-ras function? Finally, what is the mechanism whereby the K-ras gene is so consistently mutated at exactly the same points and so rarely elsewhere within the genome?
In summary, we have shown that early colo- rectal adenocarcinomas are homogeneous for K-ras genotype. We found that only 27.6% of tumours contained a mutation which is lower than in some studies of more advanced tumours. However, in these patients we could not detect any prognostic significance from the presence of a mutation in codon 12 or 13 of K-ras. Although gene mutation analysis will undoubtedly contribute to patient management in the future, at present K-ras status per se is not helpful in this respect for early colo- rectal adenocarcinomas.
Acknowledgments
We would like to thank Jacqui Oates and Theresa Oliver for help with entering our data into the computer database and updating our follow up records; Dr R Lindlay and Messrs D I Beeby, C M Butler, R Hoile, O Khan, and P Webb (Medway Hospital) and Dr G Russell and Messrs P G Bentley, A J Cook, T F Ford, J L Lewis, and T G Williams (Kent and Sussex Hospital) for allowing us to use tissue from their patients and giving us access to their notes; and The British Digestive Foundation who fund Dr Andreyev with a Research Fellowship.