Article Text

Abstract
Background—Cancer of the oesophagus has so far eluded every attempt at pharmacological treatment. The recent advent of somatic gene therapy offers a new therapeutic approach to malignant tumours.
Aim—To investigate whether and how gene transfer into the oesophagus can be achieved.
Methods—A LacZ reporter gene was used as marker and transferred into the oesophagus of rats using cationic liposomes. Gene transfer was achieved by either luminal instillation into a closed segment using a double balloon catheter, or by intramural injection through a needle. Expression of β-galactosidase was monitored in the oesophagus and various control tissues by histochemistry, polymerase chain reaction (PCR), reverse transcriptase PCR, and Southern blotting.
Results—Up to 100 days after in vivo gene transfer β-galactosidase activity could be demonstrated in the oesophagus. Following luminal application, the transgene was expressed in epithelial cells whereas intramural injection induced preferential expression in fibroblasts.
Conclusion—In vivo gene transfer into the oesophagus is feasible and safe, and the route of administration largely determines cell type specificity. This novel approach will enable in vivo studies of growth, differentiation, and malignant transformation in the oesophagus, and may open new avenues to the confinement of oesophageal malignancies.
- oesophagus
- gene transfer
- gene therapy
- liposomes
- balloon catheter
Statistics from Altmetric.com
Recent advances in recombinant DNA technology and in our understanding of eukaryotic gene regulation have resulted in the emergence of gene transfer as a potential treatment modality.1 The most advanced attempts at gene therapy have so far been made with ex vivo gene transfer techniques.2-5 This approach involves sampling of cells from the body, culture, selection, and reintroduction of genetically modified cells. Ex vivo gene transfer therefore appears well suited to the modification of circulating blood cells but represents a difficult procedure to target recombinant genes to solid organs. Furthermore, a number of cell types change their phenotype when cultured in vitro and cannot repopulate organs after propagation. Until recently, the direct in vivo introduction of recombinant genes was thought impossible. However, the development of novel gene transfer techniques now offers the possibility to target genes to specific sites in vivo.6 A number of studies have shown that either viral or non-viral vectors can be used to introduce recombinant genes into the nervous system,7 the arterial wall,6 ,8skeletal muscle,9 heart,10 ,11liver,12 ,13 lung,14-16 and into solid tumours.17-20 An alternative to viral delivery systems is the use of plasmid DNA complexed to cationic liposomes. The DNA-liposome mediated gene transfer has already been used for the delivery of recombinant genes into cells and tissues and shown to have a number of notable advantages.16 ,19-24 Because DNA is not delivered by a viral vector it is also not propagated by the transfected cell. Medical and biological safety concerns are thereby minimised. In contrast to retroviral delivery systems that only engage dividing cells, DNA-liposome mediated gene transfer does not require cell replication. Moreover, because plasmid DNA introduced with a liposome complex generally remains episomal, the potential risk of insertional mutagenesis is small compared with DNA of retroviral origin.
No successful pharmacological or chemotherapeutic treatment is currently available for oesophageal malignancies. On the other hand, the oesophagus is probably the most accessible organ for therapeutic intervention in the gastrointestinal tract. We have studied whether and how the introduction of recombinant genes into the oesophagus could be achieved by liposome mediated transfer. Our data indicate that gene transfer into the oesophagus is feasible and safe, that recombinant genes are expressed for up to 100 days, and that the route of administration largely determines target cell specificity.
Materials and Methods
CHEMICALS AND REAGENTS
The plasmid RSV-LacZ was kindly provided by Dr Gary J Nabel, University of Michigan.8 Plasmid and lipopolysaccharide (LPS) extraction kits were purchased from Quiagen. LipofectAMINE reagent, Opti-MEM, and Taq polymerase were from Gibco, BRL. A human embryonic kidney cell line, 293 cells, was purchased from ATCC (CRL 1573). X-gal chromagen (5-bromo-4-chloro-3-indolyl-β-d-galactoside) and DNAse were obtained from Boehringer Mannheim. Proteinase K was obtained from Sigma. Reverse transcriptase was purchased from Promega Biotec. Oligonucleotid primers were synthesised by Pharmacia.
PLASMID DNA
A plasmid containing the Escherichia coli LacZ gene under the control of the Rous sarcoma virus long terminal repeat (LTR) using a Simian virus 40 (SV40) polyadenylation signal (RSV-LacZ) was used as a marker for gene transfer efficiency. Plasmid DNA was purified from bacterial cultures using a Quiagen extraction kit. Lysates, obtained with the alkaline lysis method, were purified by ion exchange chromatography according to the manufacturer’s protocol. Endotoxins were removed using the Quiagen LPS extraction kit. The absence of endotoxins was verified by a Limulus amebocyte assay.24
LIPOSOME PREPARATION
Approximately 15 minutes before gene transfer DNA-liposome conjugates were prepared as follows: 20 μl LipofectAMINE reagent was diluted into 100 μl Opti-MEM at room temperature, and 10 μg plasmid DNA (stock concentration greater than 1 mg/ml) was added and mixed by gentle tapping. The solution remained at room temperature for 5–10 minutes, and Opti-MEM was added to the DNA liposome solution to a final volume of 200 μl before instillation or injection, respectively.
The quality of different batches of liposomes was evaluated by transfecting 293 cells, a human embryonic kidney cell line, with RSV-LacZ. Transfections were performed as described elsewhere.25 After 48 hours cells were stained with X-gal chromagen. At least 1000 cells were counted after staining to determine percentage of positive cells.25
ANIMALS
Male Wistar rats (180–240 g body weight) were obtained from Versuchstieranstalt of the University of Ulm, Germany. Animals were kept on a 12 hour day-night cycle with free access to food (standard diet 1320, Altromin, Lage, Germany) and water. All experiments were started at 0900 hours. The fed rats were anaesthetised by intraperitoneal injection of pentobarbital (60 mg/kg body weight). Animals recovered from anaesthesia within three hours and were kept in standard laboratory conditions until they were killed. All experiments were approved by the Institutional Animal Care Committee and executed according to the National Animal Welfare Law of Germany.
GENE TRANSFER BY LUMINAL INSTILLATION
To create a fluid sealed oesophageal segment we used a double balloon catheter (a generous gift from USCI Division, CR Bard, Inc., USA). The outer diameter of the catheter was 0.8 mm (deflated balloons) and the balloons could be inflated to a diameter of 10 mm. Through the pharynx the catheter was advanced 80 mm into the rat oesophagus, which placed the distal balloon above the lower oesophageal sphincter. Inflation of both balloons in the oesophagus created a protected space between them that was used for fluid instillation (fig1). The DNA-liposome complex (200 μl) was introduced into this closed segment and allowed to incubate for 20 minutes to facilitate gene transfer. Following transfection, the balloons were deflated, and the catheter was withdrawn. The animals were allowed to wake up and resume normal activity. Liposomes alone or liposomes complexed with 10 μg Blueskript SK (Strategene) served as controls.

: Method of introducing DNA-liposome complexes with a double balloon catheter into the oesophageal epithelium.
GENE TRANSFER BY INTRAMURAL INJECTION
Under sterile conditions the abdomen was opened through a midline incision and the distal oesophagus was exposed. Freshly prepared DNA-liposome conjugate (200 μl) was injected into the oesophagus wall through a 0.5 mm needle. The abdomen was closed with running sutures of silk. Sham treated animals received an equal volume of either a DNA free liposome mixture or liposomes conjugated to Bluescript SK plasmid DNA.
HISTOLOGICAL ANALYSIS
Rats were killed three, 28, and 100 days after gene transfer. Tissue specimens from the oesophagus and various control organs were fixed for three hours in 4% paraformaldehyde/PBS solution, pH 7.4, at room temperature. The product of LacZ, β-galactosidase, was localised by incubating tissue blocks for 16 hours at 37°C in X-gal chromagen at pH 7.4. Paraffin wax sections (10 μm) were counterstained with haematoxylin and/or eosin. Cells synthesising the LacZ gene product produced a deep blue cytoplasmic precipitate.
IMMUNOHISTOCHEMISTRY
Specimens of rat oesophagus tissue were fixed in 4% neutral buffered formalin at 37°C for 72 hours, incubated in X-gal chromagen as described above, and embedded in paraffin wax. Sections of 5 μm were blocked with a 30% H2O2 solution, deparaffinised in an ascendant ethanol succession, and washed in PBS buffer at pH 7.4. After preincubation with rabbit serum, sections were incubated with a monoclonal anti-β-galactosidase antibody at a 1/100 dilution in 0.5% bovine serum albumin-Tris (Sigma, St Louis, USA) for 30 minutes. The secondary antibody, rabbit antimouse (Dako, Glostrup, Denmark) was used as conjugate with biotin in a 1/600 dilution for 30 minutes. Antibody binding was visualised with a streptavidin-peroxidase complex (Dako, Glostrup, Denmark) diluted 1/1000 in PBS followed by the addition of 1 mg/ml DAB (Dako) in 0.02% H2O2for 20 minutes. Cells positive for β-galactosidase showed a brown staining pattern throughout the entire cytoplasm. Within the same cells β-galactosidase activity was detected as a deep blue precipitate using X-gal. As negative controls, tissue sections were treated similarly but without adding the primary antibody.
ANALYSIS OF GENE TRANSFER BY POLYMERASE CHAIN REACTION
Tissue from transfected, sham transfected oesophageal segments, and control tissues were incubated in 600 μl lysis buffer containing 100 mmol/l NaCl, 100 mmol/l EDTA, 50 mmol/l Tris pH 8, 1% sodium dodecyl sulphate (SDS), and 0.6 mg/ml proteinase K at 55°C overnight. Phenol/chloroform (1:1) (600 μl) was added and vigorously vortexed. This extraction was repeated twice followed by a chloroform extraction and ethanol precipitation. Dried DNA was resuspended in TE and subjected to polymerase chain reaction (PCR) analysis.
Oligonucleotide primers were synthesised to E coli LacZ (sense 5′-GCC AGC GCG GAT CAT CGG TCA GAC G-3′; antisense 5′-GCC TGC GAT GTC GGT TTC CGC GAG G-3′). The primers gave rise to a 318 bp product. The PCR reactions were performed in 100 μl containing 20 mmol/l Tris pH 8.4, 50 mmol/l KCl, 1.5 mmol/l MgCl2, 100 ng genomic DNA, and 10 mmol of each primer. The reaction mixture was heated to 95°C for five minutes and subsequently 2.5 units of Taq polymerase was added. The tubes were transferred to an automated DNA thermal cycler (Strategene) for 35 cycles of denaturing (94°C, one minute), annealing (60°C, two minutes), and polymerisation (72°C, two minutes). A portion of the reaction mixture (20 μl) was electrophoresed in a 1.6% NuSieve agarose gel (FMC Bioproducts), stained with ethidium bromide to visualise the amplification product, and recorded on Polaroid film. Positive controls were 1 ng of the RSV-LacZ plasmid or 293 cells transfected with 5 μg RSV-LacZ.
ANALYSIS OF β-GALACTOSIDASE EXPRESSION BY POLYMERASE CHAIN REACTION
Recombinant LacZ mRNA expression was analysed by reverse transcriptase PCR. Oesophageal samples were obtained, and total cellular RNA was prepared using acid guanidinium thiocyanate/phenol/chloroform as previously described26with the modification that the acid phenol extraction was performed twice and the final ethanol precipitation was performed in the presence of 0.3 M sodium acetate, pH 5.2. RNA integrity was confirmed by gel electrophoresis.26 Nucleic acids were extracted and treated with RNAse free DNAse at 37°C for 30 minutes to eliminate DNA contaminations.
After a phenol/chloroform extraction, complementary DNA (cDNA) was prepared from 1 μg unfractionated RNA in 20 μl reverse transcription buffer containing 20 mmol/l Tris, pH 8.4, 50 mmol/l KCl, 5 mmol/l MgCl2, 1 mmol/l each of deoxyadenosine triphosphate, deoxycytidine triphosphate, deoxyguanosine triphosphate, thymidine triphosphate, 20 units RNAsin ribonuclease inhibitor, 0.1 μg oligo(dT)15, and 50 units Moloney MuLV reverse transcriptase at 37°C for 60 minutes. The reaction mixture was heat inactivated for 10 minutes at 95°C, and 80 μl of 10 mmol/l Tris, pH 8.4, 50 mmol/l KCl, 1.25 mmol/l MgCl2 (final magnesium concentration was 2 mmol/l) containing 10 μmol each of the 5′ and 3′ primers was added. The reaction mixture was heated to 95°C for five minutes; 2.5 units of Taq polymerase was subsequently added. PCR samples were analysed in the presence and absence of reverse transcriptase. The temperature profile of the amplification and electrophoresis have been mentioned earlier. The PCR analysis of mRNA was performed using 35 cycles of denaturing, annealing, and polymerisation as described earlier. PCR samples were analysed in the presence and absence of reverse transcriptase.
SOUTHERN BLOT ANALYSIS
After electrophoresis, the gel was placed in 0.25 mmol/l HCl for 2 × 15 minutes and neutralised with 1 mmol/l NH4acetate, and 0.02 mmol/l NaOH for 30 minutes. Ammonium acetate was used to neutralise and transfer the DNA to nitrocellulose. Nitrocellulose filters were air dried and UV crosslinked. A HindIII/EcoRI fragment of RSV-LacZ was radiolabelled by the random oligomer primed method using a 32P-dCTP (3000 Ci/mmol; Amersham) to a specific activity >109 counts per minute (cpm)/mg. The filters were prehybridised for 16 hours at 65°C in 6× SSPE, 50× Denhardt’s solution, 100 μg/ml salmon sperm DNA, and 0.1% SDS. Hybridisation was for 16 hours at 65°C in the presence of 1 × 106 cpm/ml of the labelled probe, followed by washing at 57°C in 2× SSC containing 0.1% SDS for 2 × 20 minutes, in 0.5× SSC containing 0.1% SDS, and in 0.1× SSC containing 0.1% SDS for 20 minutes, respectively. The blots were exposed at –80°C to Kodak XAR-5 film with intensifying screens.
Results
LUMINAL GENE TRANSFER
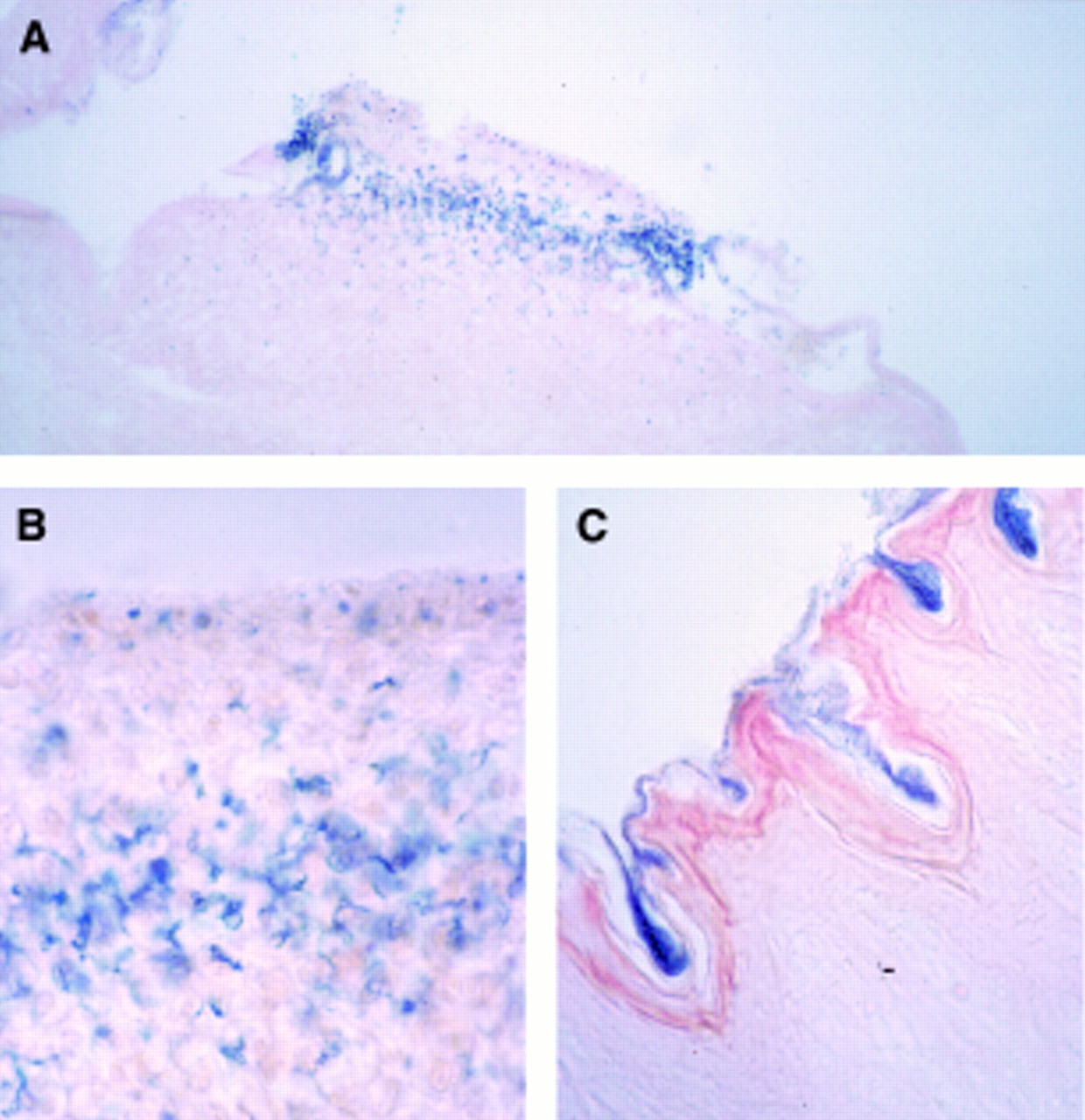
LipofectAMINE was mixed with the RSV-LacZ reporter plasmid and introduced into an oesophageal segment using a double balloon catheter. After sacrifice of animals at different time points the oesophagus was removed and tissue was incubated with X-gal chromagen to identify transgene expression. After luminal gene transfer staining revealed individual areas of blue coloration indicative of β-galactosidase activity in areas where the keratin layer was ruptured (figs 2A and2B). The blue stain was exclusively detectable in epithelial cells and limited to cells luminal of the basal membrane. No evidence of similar luminal staining was observed in areas where the keratinised layer was intact suggesting that this layer is a sufficient barrier for gene transfer using liposomes (fig 2C). After 14 days β-galactosidase staining was eliminated (data not shown). These data are consistent with the transient expression of the marker gene in oesophageal epithelial cells.

: Microscopic view of sections from transduced oesophageal epithelium using the double balloon catheter system. (A) Representative area positively stained for β-galactosidase activity (original magnification ×50); (B) an oesophageal section three days after luminal gene transfer, indicating transfected epithelial layers (original magnification ×250); (C) an oesophageal section with an intact keratin layer which acts as a barrier for gene transfer (original magnification ×100). Sections were counterstained with eosin.
GENE TRANSFER BY DIRECT INJECTION
After gene transfer by injecting DNA-liposome complexes directly into the oesophageal wall, β-galactosidase staining was seen mostly in fibroblasts in the submucosa suggesting preferential uptake of DNA-liposome complexes in these cells (fig 3). β-Galactosidase staining was preferentially detected below the basal membrane and only occasionally evident in epithelial cells in the basal layer. Light microscopy revealed β-galactosidase predominantly in fibroblasts in the submucosa including connective tissue between muscle cells (fig3B). In the muscularis mucosa smooth muscle cells were also stained (figs 3B and 3C). Gross observation of oesophageal segments that were exposed to control DNA liposome mixtures or liposomes alone showed no blue staining. In a series of experiments the time course of expression was investigated. β-Galactosidase activity diminished over time but was detectable in fibroblasts in 3/6 animals for up to 100 days (data not shown). The levels of β-galactosidase activity in sections from transduced oesophageal segments were quite variable. In the epithelium only individual cells were positive for β-galactosidase activity. Interestingly, cells stained with X-gal chromagen revealed β-galactosidase activity only in a small portion of the cytoplasm. No homogeneous staining pattern of the cytoplasm was observed. In contrast when using a monoclonal antibody against β-galactosidase, transfected cells displayed a brown, homogeneous precipitate throughout the entire cytoplasm of individual positive cells. The combination of both detection methods revealed cells with simultaneously blue (X-gal) and brown (antibody) precipitate. In addition, a number of cells were only reactive with the β-galactosidase antibody and did not show any positivity on X-gal staining. We therefore conclude that the transfection efficiency might be underestimated when X-gal staining alone is used to detect β-galactosidase activity in the oesophageal wall (fig 4).

: Microscopic view of sections from oesophagus transduced by injection. β-Galactosidase activity was detectable in the submucosa (A and B) and in the connective tissue of the muscular layer (B). Occasionally epithelial cells were positive (C). Original magnification ×100 (A), ×100 (B), ×250 (C). All sections were counterstained with eosin (A) or eosin and haematoxylin (B and C).
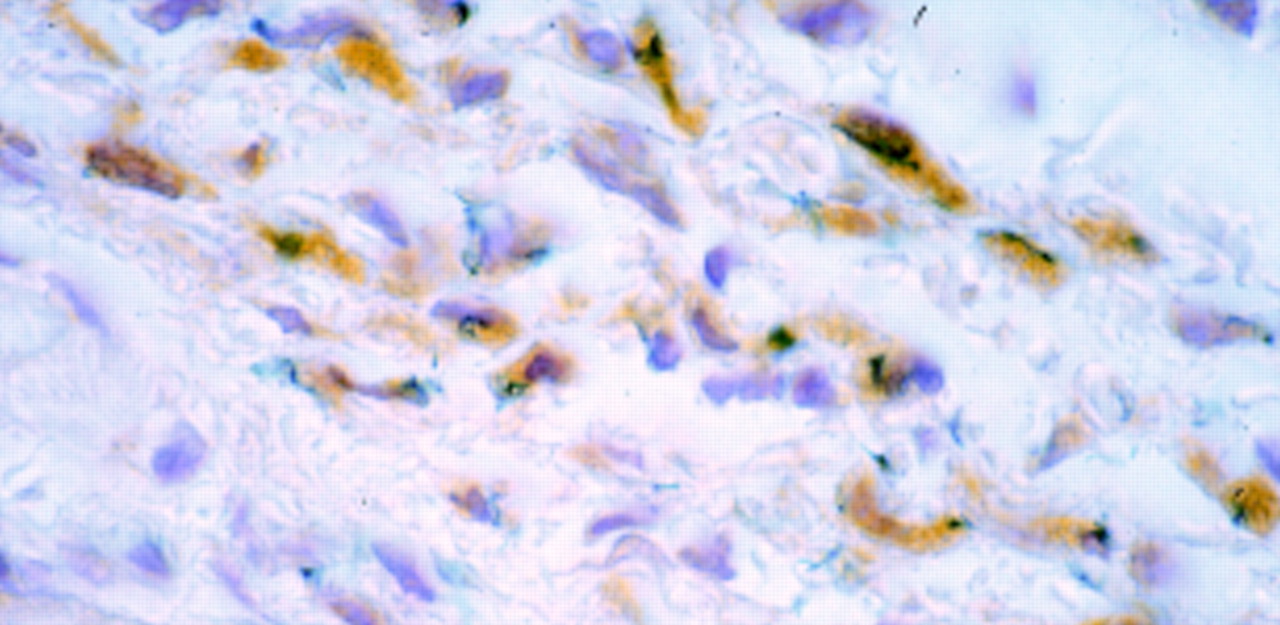
: Microscopic view of sections from oesophagus transduced by injection. Sections were incubated with a monoclonal anti-β-galactosidase antibody followed by a secondary biotin conjugated rabbit antimouse antibody and streptavidin-peroxidase complexes with DAB. β-Galactosidase activity is presented as a blue precipitate, whereas detection by the anti-β-galactosidase antibody is displayed as brown precipitate throughout the cytoplasm. The section was counterstained with haematoxylin. Original magnification ×250.
HISTOLOGICAL ANALYSIS
Histological examination of the oesophagus of animals treated with the double balloon catheter revealed minor histological changes consistent with superficial trauma due to irritation of the oesophagus epithelium with the catheter. Interruption of the superficial keratin layer exposed epithelial cells to the lumen and allowed gene transfer to take place. In these areas no major signs of acute inflammation were detectable (fig 2).
Following direct injection into the oesophageal wall, all treated animals showed signs of inflammation at the site of injection (n=20). In these areas necrotic material, extravasation of blood, and infiltration with polymorphonuclear cells were evident and reached the submucosa (fig 5A). The centre of the site of injection was most prominently affected by these changes. After 28 days minor abnormalities such as an increase in small submucosal lymphoid aggregates in the submucosa remained (figs 5B and 5C). There were no histological differences detectable between animals treated with RSV-LacZ, control plasmid, or liposomes alone.
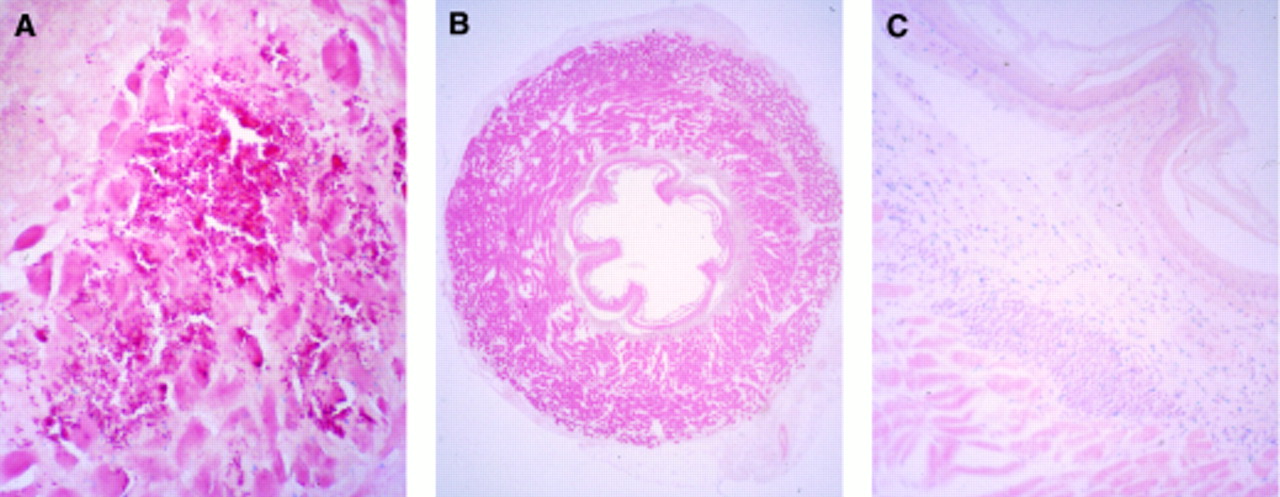
: Microscopic view of a section from the oesophagus injected with 200 μl DNA-liposomes. Three days following gene transfer, signs of inflammation, necrosis, and bleeding were visible at the area where the injection took place. Sections were fixed in 4% paraformaldehyde and stained with eosin (A) (original magnification ×50). Twenty eight days after gene transfer no signs of trauma, necrosis, or major inflammation were visible (B) (original magnification ×6.25). In these areas a local increase in small lymphoid aggregates remained in the submucosa (C) (original magnification ×50). Sections were counterstained with eosin (A) or with eosin in combination with haematoxylin (B and C).
DETECTION OF DELIVERED DNA
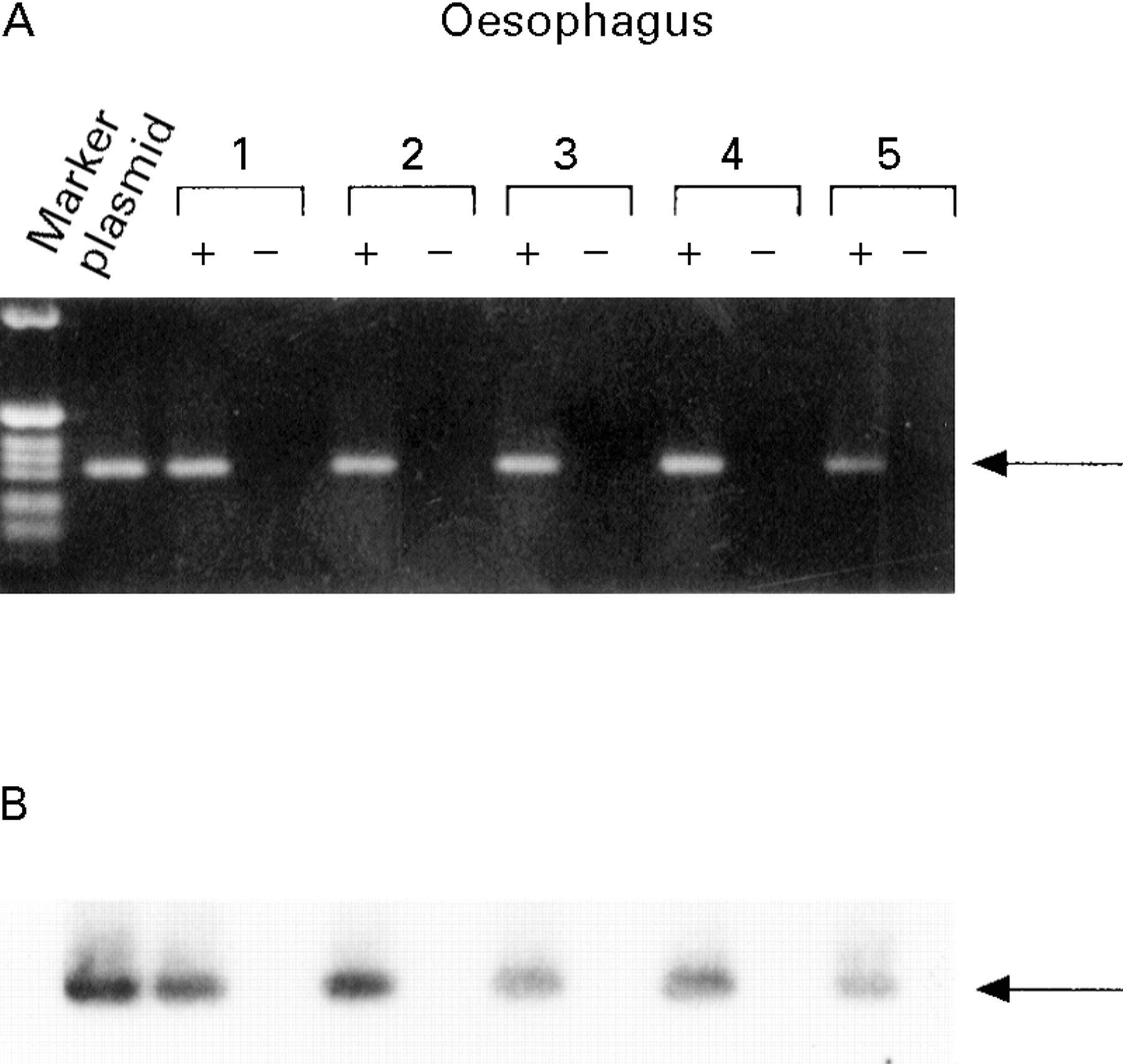
The uptake of plasmid was confirmed by PCR of total DNA prepared from oesophageal tissue, using primers which contain LacZ sequences and are thus specific for the plasmid RSV-LacZ. Animals were sacrificed three days after gene transfer using the double balloon catheter or by direct injection of the DNA/liposome mixture into the oesophageal wall. A specific 318 bp band was detected in three animals following gene transfer by injection, but not in animals receiving the control plasmid (fig 6). The amount of DNA delivered varies as seen in the intensity of the amplified band using both routes of application. Signals were also detected in animals following luminal gene transfer with the double balloon catheter (data not shown). The delivered DNA was exclusively localised in the oesophagus and not detectable in other tissues by PCR using both routes of gene transfer (fig 7). To detect traces of gene transfer to other organs, Southern blots of the PCR products were performed and hybridised to a LacZ specific probe. No band was detected in other tissues indicating site specific gene transfer (fig7B).

: Detection of plasmid DNA in oesophageal tissue three days after direct gene transfer with DNA- liposome complexes by PCR. A specific band of 318 bp was detected after transduction with RSV-LacZ using direct injection (RSV-LacZ lanes 1–3), but not in controls, where Bluescript SK with liposomes was injected (control lanes 1–3). The 1 kB ladder (Gibco) was used as a marker.

: Detection of plasmid DNA in tissue samples after gene transfer. Three days after injection of oesophageal segments with RSV-LacZ DNA-liposomes genomic DNA was isolated. Genomic DNA (1 μg) was subjected to PCR analysis; 1 ng RSV-LacZ or 293 cells transfected with 5 μg RSV-LacZ were used as positive controls. One tenth of the PCR reaction was electrophoresed on a 1.6% agarose gel, stained with ethidium bromide, and photographed. Recombinant RSV-LacZ DNA was not detected as a specific 318 bp band in all tissues except in the oesophagus (A). Positive and negative controls are shown along with DNA markers. To enhance the signal Southern blot analysis was performed (B). The gel was blotted onto nitrocellulose and hybridised to a LacZ specific probe. No signals were detected in the negative control or in any tissue except the oesophagus.
EXPRESSION OF DELIVERED DNA
Expression of the LacZ mRNA from the introduced plasmid was demonstrated using reverse transcriptase PCR (RT-PCR), applied to RNA prepared from oesophagus from treated and sham treated animals. The RT-PCR assay was performed in the presence and absence of reverse transcriptase to exclude false positive signals due to DNA contamination. Figure 8 shows that a specific band was only detected when reverse transcriptase was added to the reaction, which hybridised to the LacZ probe. As expected no PCR product was obtained in untreated animals (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
: Expression of β-galactosidase mRNA in the oesophagus in vivo. A specific band of 318 bp was detected in RNA from oesophageal segments when reverse transcriptase was used (+) (A). The 1 kB ladder (Gibco) was used as a marker and 1 ng RSV-LacZ as a positive control.
Discussion
Cancer of the oesophagus is a malignancy with a very poor clinical prognosis. Once complete surgical resection is no longer possible either pharmacological or chemotherapeutic treatment strategies are not effective means of confining tumour spread and invasion. Recent advances in recombinant DNA technology and gene transfer methods have opened entirely new avenues of influencing the molecular characteristics of malignant tumour cells. A number of strategies have been developed and shown to be effective in vitro or in tumour models using transplanted tumour cells. So far the treatment of tumours in experimental models at the site of their development has been rather limited. This is due to the lack of suitable tumour models and also because of difficulties in site specific drug application. Here we demonstrate the feasibility of introducing a marker gene directly into the rat oesophagus in vivo. We used a double balloon catheter to transduce the oesophagus wall with the marker gene, RSV-LacZ. Gene expression was detected in areas where the luminal keratin layer was ruptured and epithelial cells were exposed to the DNA-liposomes. β-Galactosidase staining was clearly detected at day 3 after transfection; it decreased over time and was eliminated after 14 days. The transduced marker gene was only detected in the oesophagus but not in other tissues using PCR analysis.
The keratinised oesophageal epithelium of rats as well as other rodents represents a natural barrier for gene transfer; this route of application is therefore limited in these species. In contrast, in humans the mucosa consists of a non-keratinising stratified squamous epithelium which would probably allow luminal liposomal transfection much more efficiently.
An alternative approach to delivering a gene into the oesophageal wall is direct injection of DNA-liposome complexes. In the rat model, DNA-liposomes were injected into the distal oesophageal wall with a needle. β-Galactosidase activity was detected using X-gal staining throughout the oesophageal wall predominantly in fibroblasts, but also occasionally in epithelial and smooth muscle cells. Interestingly, more cells were identified as positive for transgene expression when using a monoclonal antibody against β-galactosidase, indicating that with X-gal staining the transfection efficiency might be underestimated. In fibroblasts recombinant gene expression was detected for up to 100 days. Similar to the luminal application, the transfected DNA was detectable in the oesophagus but not in other organs by PCR.
In the present study liposomes were used to complex with DNA to transfer a recombinant gene into cells in vivo. The method utilising plasmid DNA transfer by lipofection appears to be simple, safe, and non-toxic to the animals. The use of cationic liposome formulation for gene transfer has been introduced by Felgner et al.21 Cationic liposomes are complexed to negatively charged, naked DNA by mixing liposomes and DNA. The resulting complexes have a net positive charge. Liposomes interact with the cell surface, allowing the DNA to traverse the plasma membrane of target cells. The DNA is subsequently transferred to the nucleus to direct transcription through incompletely defined mechanisms.24 Cationic liposomal delivery has several features. Firstly, it is a very simple procedure and almost 100% of the DNA is in the complexes.27 There is theoretically no limitation to the size of the DNA. Many different cell types can be transfected, although some cell types might transfect better than others.25 A major advantage is the lack of immunogenicity of liposomes which allows safe and repetitive application in vivo.28 ,29 Furthermore cationic liposomes can be significantly inactivated by serum.30 Several features of liposome mediated gene transfer facilitate its application to direct gene transfer in vivo. Unlike viral vectors, there are few constraints on vector size or transcriptional signals, and a variety of tissue specific enhancer/promoter elements may be used.
The in vivo behaviour of cationic liposomes complexed to DNA was tested after direct injection intravenously or directly into tumours.19 ,20 ,31-33 After intravenous injection of the DNA-liposome complexes the transgene was localised mainly to the lung and heart and occasionally to the kidney.32 In another study DNA-liposome complexes have been intravenously administered and efficiently transfected in virtually all tissues.33 After intratumoural injections DNA was detectable only within the tumour and not in all other tissues tested.19 ,32 Most importantly, no harmful effects were observed using clinical biochemical assays and histological examinations, including examinations for inflammation. No DNA was localised to gonadal tissues.32
Studies which have investigated the feasibility of delivering recombinant genes to the gastrointestinal tract are very limited.34 Most studies have focused on gene transfer into the liver; however, there are some reports dealing with liposome mediated gene transfer into the colon.28 ,29 To our knowledge gene transfer to the oesophagus has not been studied before.
Although the present model of gene delivery into the oesophagus may be useful both for the study of gene expression in vivo and for the development of gene therapies for various diseases, there are potential problems with the model that merit consideration. Firstly, the level of gene transfer was a substantial variable both in the balloon as well as in the injection technique. The number of β-galactosidase positive cells varied widely. One likely cause for the observed range of gene transfer efficiencies using the luminal application route is variability in the degree of rupture of the keratin layer in the rat oesophagus. Little or no transfection occurs when the keratin layer is intact. Rupture of the keratinised epithelium was not uniform. Efforts to improve the uniformity of disrupting or methods to permeabilise the keratin layer may prove helpful in decreasing experimental variability. A potential difficulty of injecting DNA-liposomes is the observed inflammation which may result from exposure of cells to liposomes. Cytopathogenicity has been reported after exposure of cells to high concentrations of liposomes.
However, this technique represents a novel approach to the study of gastrointestinal biology. Genes expressed in different gastrointestinal tissues can produce local effects by their ability to influence the function and integrity of cells. Using this technique it will now be feasible to test different non-viral and viral transfer systems to overexpress different genes in the oesophageal wall without the need to introduce transgenes into the germline of animals. This offers the possibility of achieving specific expression at anatomically localised sites even when tissue specific promoters or enhancers are not available. If cell type specific promoters are available, it might be feasible to transduce specific cell lineages in a given tissue. In terms of tumour therapy, tumour specific promoters could be used to express genes in tumour cells.
A future prospect of this technique will certainly be the development of new treatments for malignancies of the oesophagus. Despite the increasing understanding of the molecular basis of oesophageal cancer, this malignancy remains resistant to established traditional forms of treatment. Hopefully, advances in gene therapy techniques including genetic immunomodulation, vaccination, inhibition of mitogenic signal transduction pathways, and suicide genes will improve the prospects for the treatment of oesophageal cancer through delivery of recombinant genes in vivo.
In summary, our data have shown that recombinant gene expression can be achieved by liposome mediated gene transfer in the oesophageal wall. Gene expression was observed in different cell types located throughout the oesophageal wall. Although direct gene transfer technology is still in its infancy, expanding laboratory efforts and the lack of toxicity of this form of treatment suggest that it will become increasingly useful for the genetic treatment of oesophageal disease.
Acknowledgments
The authors wish to thank E Wolff and G Leiacker for their expert technical assistance, and M M Lerch for critical reading of the manuscript. This work was supported by SANDOZ-STIFTUNG für Therapeutische Forschung. Part of this work was presented to the Americal Gastroenterological Association at the Digestive Disease Week, San Diego, 1995.